

Abstract
Missing genotype data arise in association studies when the single-nucleotide polymorphisms (SNPs) on the genotying platform are not assayed successfully, when the SNPs of interest are not on the platform, or when total sequence variation is determined only on a small fraction of individuals. We present a simple and flexible likelihood framework to study SNP-disease associations with such missing genotype data. Our likelihood makes full use of all available data in case-control studies and reference panels (e.g., the HapMap), and it properly accounts for the biased nature of the case-control sampling as well as the uncertainty in inferring unknown variants. The corresponding maximum-likelihood estimators for genetic effects and gene-environment interactions are unbiased and statistically efficient. We developed fast and stable numerical algorithms to calculate the maximum-likelihood estimators and their variances, and we implemented these algorithms in a freely available computer program. Simulation studies demonstrated that the new approach is more powerful than existing methods while providing accurate control of the type I error. An application to a case-control study on rheumatoid arthritis revealed several loci that deserve further investigations.
Introduction
Thanks to comprehensive catalogs of human genetic variation1,2 and precipitous drops in genotyping costs, case-control association studies have become the primary tool in searching for genetic determinants of complex diseases. There are missing genotype data in all these studies. Even in a well-designed study with high-quality genotyping, some individuals will have missing genotypes at certain single-nucleotide polymorphism (SNP) sites because of assay failures. Genotype data may also be missing by design. For example, it is cheaper to genotype a subset of study subjects on a high-density platform and the rest on a low-density platform. Also, it may be economically feasible to completely sequence a small fraction of individuals, rather than all individuals, in a large study.
There has been an enormous recent interest in untyped SNPs, i.e., the SNPs that are not even on the genotyping platform used in the study. This is an extreme form of missing genotype data in which the SNPs of interest are missing on all study subjects. Conducting association analysis at untyped SNPs can facilitate the selection of SNPs to be genotyped in follow-up studies. This kind of analysis is also highly desirable if we wish to validate the findings of one study on some other studies that use different genotyping chips or to perform meta-analysis by combining data from association scans that use different SNP sets.
The prevailing approach to dealing with missing genotype data is imputation,3–6 which predicts the missing genotypes from the observed genotypes at neighboring SNPs and then uses the predicted values in downstream association analysis. This strategy, although very intuitive and useful, is suboptimal. Imputing missing data for cases and controls together can lead to a bias toward the null hypothesis of no association and therefore a loss of power, whereas imputing missing genotypes for cases and controls separately can inflate type I error rates.3,7,8
An alternative, less ambitious approach is to use the haplotype frequencies of neighboring SNPs to estimate the allele frequencies of the untyped SNP.9–11 This strategy is easy to implement but is restricted to the comparison of allele frequencies between cases and controls. The estimation of the allele frequencies may be inaccurate, especially for cases,10 so the power of the corresponding association test may be compromised. In addition, some of the variance estimators for the estimated allele frequencies require haplotype data.
In this article, we provide a general likelihood-based framework for handling any form of missing genotype data. We derive the observed-data likelihood that properly reflects the biased nature of the case-control sampling and that incorporates appropriate external data, such as the HapMap data. The maximization of the observed-data likelihood leads to valid and efficient analysis of genetic effects and gene-environment interactions. We demonstrate through simulation studies that our approach is more powerful than the two existing approaches mentioned above while providing correct control of the type I error. We illustrate the new method through an application to a case-control study on rheumatoid arthritis (RA [MIM 180300]). The software implementing the new method can be downloaded from our lab website.
Material and Methods
We consider a set of M SNPs that are in linkage disequilibrium (LD). Each SNP is biallelic with allele values 0 and 1. The SNP genotypes may be missing. We use the known genotypes of the SNPs that are in LD with the SNP with missing genotypes to infer its unknown values. To this end, we consider the joint distribution of the M SNPs. Let G denote the multilocus genotype of the M SNPs and H the corresponding diplotype. Suppose that the M SNPs have a total of K haplotypes, each of which is a unique sequence of 0s and 1s. We denote the K haplotypes by h1, ···, hK, with frequencies π1, ···, πK. We write H = (hk, hl) if the diplotype consists of haplotypes hk and hl. Note that H = (hk, hl) implies that G = hk + hl, where the summation is taken component-wise.
Let Y denote the disease status (1 = disease, 0 = no disease). The effects of SNP genotypes on the risk of disease can be formulated through the following logistic regression model:
| (1) |
where α is an intercept term, β pertains to log-odds ratios, and Z(hk, hl) is a (possibly vector-valued) genotype score induced by the diplotype (hk, hl). In this article, all vectors are column vectors, and aT denotes the transpose of a. If we are interested in the additive effect of a single SNP, then we set Z(hk, hl) to be the value of (hk + hl) at that SNP position; if we are interested in the recessive effect, then Z(hk, hl) indicates whether the value of (hk + hl) at the SNP of interest is equal to 2 or not; dominant and codominant effects can be similarly modeled. We can define Z(hk, hl) to formulate the joint effects of all M SNPs or any subset of them.
Suppose that we have a case-control study with a total of n subjects. For i = 1, ···, n, let Yi and Gi denote the values of Y and G for the ith subject. The values of Gi may be missing at any positions. To reflect the biased nature of the case-control sampling, we adopt the retrospective likelihood Πni = 1P(Gi|Yi). Under Model 1 with rare disease and Hardy-Weinberg equilibrium, this likelihood takes the form
| (2) |
where θ = (βT, πT)T, π = (π1, ···, πK)T, the summations in both the numerator and denominator are taken over k = 1, …, K and l = 1, …, K, and (hk, hl) ∼Gi means that the diplotype (hk, hl) is compatible with the observed value of genotype Gi (i.e., hk + hl = Gi at all SNP sites with no missing values). We maximize LS(θ) to obtain the maximum-likelihood estimator (MLE) of θ. The maximization can be carried out through the Newton-Raphson algorithm described in Appendix A.
The standard approach to the problem of missing genotypes is to remove the subjects with missing values. This strategy can be highly inefficient, especially when there is substantial missingness and different subjects are missing on different SNPs. The proposed MLE method does not remove any subjects and uses all the available data to perform efficient analysis.
To reduce cost, we may purposely set some genotypes to missing. In a large study, for instance, it is cost effective to genotype a subset of individuals with a high-density platform and the rest with a low-density one. Likewise, it may be economically feasible to determine complete sequence variation for only a small fraction of individuals rather than all individuals. The MLE approach is particularly suited to such situations, allowing efficient analysis at all the SNPs of the high-density platform and for complete sequence variation.
If one of the M SNPs is untyped (i.e., not present on the genotyping platform used for the study) or missing on all study subjects, then there is no information in the study data to determine the joint distribution of the M SNPs. We can ascertain the joint distribution from an external reference database, such as the HapMap.1 Naturally, the case-control study and the reference panel are assumed to be generated from the same underlying population. We denote the likelihood for π based on the reference database by LR(π).
To be specific, we consider the HapMap trio data. Suppose that we have a total of trios, which is 30 for the Centre d'Etude du Polymorphisme Humain (CEU) sample. For , the genotype data for the jth trio consist of Gj = (GFj, GMj, GCj), where GFj, GMj, and GCj denote the genotypes for the father, mother, and child, respectively. In this case,
| (3) |
where the summation is taken over k, l, k′, l′ = 1, …, K, and (hk, hl, hk′, hl′)∼Gj means that (hk, hl) is compatible with GFj, (hk′, hl′) is compatible with GMj, and (hk, hk′), (hk, hl′), (hl, hk′), or (hl, hl′) is compatible with GCj. The likelihood for unrelated individuals is a special case of Equation 3 with missing genotypes for all parents.
The likelihood for θ that combines the study data and reference database is
We maximize LC(θ) through the Newton-Raphson algorithm described in Appendix B. The resulting MLE of θ is approximately unbiased and normally distributed. Furthermore, the MLE is statistically efficient in that it has the smallest variance among all valid estimators and the corresponding test of association is the most powerful among all valid tests based on the same data and same assumptions.
The above framework allows association analysis at all the SNPs in the reference database. To maximize efficiency, we choose a set of (M − 1) SNPs that provides the best prediction of the missing SNP genotype. The accuracy of prediction is measured by Rs2 of Stram12 or equivalently by MD of Nicolae.13 For any SNP of interest, we find the set of (M − 1) SNPs within 100 kb, for example, that yields the largest value of Rs2. If Rs2 is close to 1, then the analysis will be nearly as efficient as if the SNP of interest is measured on all study subjects.
Performing association tests at untyped SNPs yields a wider range of SNPs to be considered for genotyping in follow-up studies. Another application is to validate the findings of one study on other studies that use different genotyping chips. Indeed, it is desirable to combine data across studies so as to increase power to detect small genetic effects. To perform this kind of meta-analysis, we include in LS(θ) all the subjects from the studies of similar populations and multiply LC(θ) over different types of populations.
We can estimate the allele frequencies for any SNPs of interest by using the MLE of π. To infer missing genotypes, we calculate the posterior probabilities of individual diplotypes
where I(·) is the indicator function and the unknown parameters β and π are evaluated at their MLEs. By taking appropriate sums of these posterior probabilities, we can obtain the posterior probabilities for the genotypes of interest.
We can extend our framework to study gene-environment interactions. Let X represent environmental factors. We expand Model 1 as follows:
where Z(hk, hl, x) is a specific vector function of (hk, hl) and x. The retrospective likelihood involves the unknown distribution of X, which is high-dimensional. We use the profile-likelihood arguments of Lin and Zeng14 to eliminate the distribution of X and replace Equation 2 with the following profile likelihood:
| (4) |
where θ = (μ, βT, πT)T, μ is an unknown constant, and the summation in the denominator is taken over k, l = 1, …, K and y = 0, 1. The maximizations of this likelihood and the corresponding combined likelihood LC(θ) are discussed in Appendices A and B.
Whereas our approach integrates inference of missing genotypes and estimation of odds ratios into a single likelihood framework, the imputation approach first imputes missing genotypes (without reference to phenotype information) and then assesses the association between the imputed genotypes and the phenotype. There are various ways to impute missing genotypes.3–6 An attractive recent method5 generates each individual's genotype from a hidden Markov model in which the hidden states are a sequence of pairs of the haplotypes observed in the reference panel and in which mutations and recombinations are allowed. Given the imputed genotypes, one can use the most likely genotype, the expected genotype counts, or the probability distribution of the genotype for each individual in the ensuing association test. Marchini et al.5 recommended the use of the probability distribution because it accounts for more of the uncertainty in imputed genotypes. Because it disregards phenotype information when imputing missing genotypes and ignores case-control sampling in association analysis, the imputation approach may not provide unbiased estimation of odds ratios at causal SNPs.
Nicolae10 estimated the allele frequency for the untyped SNP by a weighted sum of the haplotype frequencies of the (M − 1) genotyped SNPs, with the weights determined by the haplotype frequencies of the M SNPs in the reference panel, and he dubbed the corresponding association test TUNA (testing untyped alleles). Zaitlen et al.11 proposed a class of tests based on the weighted sum of haplotype frequencies, which includes Nicolae's test and the single-haplotype test of de Bakker et al.9 as special cases, and they found the set of weights used by Nicolae10 to be optimal. The variance estimators provided by Zaitlen et al.11 are based on the multinomial distribution of haplotypes and thus require the use of haplotype data rather than genotype data. According to the documentation for the TUNA software, there are numerical difficulties with the testing procedure originally suggested by Nicolae.10 The TUNA software estimates the variance of the test statistic by two methods: an asymptotic interpretation of MD and bootstrap. There is no explanation of the first method, and the second method is computationally intensive. We propose to estimate the variance of the weighted sum of (estimated) haplotype frequencies by using the information matrix for the haplotype frequencies based on (unphased) genotype data. This variance estimation is statistically valid and computationally efficient.
Results
Simulation Studies
We used Monte Carlo simulation to evaluate the new and existing methods. We simulated genotypes for various sets of SNPs according to the haplotype distributions observed in the CEU sample of the HapMap project.1 We generated the disease status from Model 1 with a potentially causal SNP. For each scenario, we set the overall disease rate to approximately 5% and obtained 10,000 simulated data sets with 1,000 cases and 1,000 controls.
We first studied the problem of genotyped SNPs with missing data. We were particularly interested in SNPs 60–64 on chromosome 18 of the CEU sample in the HapMap genome-wide data. This set of SNPs was previously considered by Lin and Huang,8 who provided its haplotype frequencies. The LD among these five SNPs is not particularly strong. We set SNP 60 to be causal with an additive effect. We let the genotypes of the five SNPs be missing independently with the same probability and performed multi-SNP analysis by including all five SNPs in the logistic model. We compared the new method to the imputation method based on the output of fastPHASE.4
Figures 1 and 2 display, respectively, the type I error of the association tests at SNP 61, which is null, and the power of the association tests at SNP 60, which is causal. Clearly, the new method maintains its type I error around the nominal significance level. The imputation method based on fastPHASE has inflated type I error, the inflation worsening as more genotypes are missing and as the odds ratio of the causal SNP increases. The new method is more powerful than fastPHASE. The improvement of the new method over the standard complete-case analysis is substantial. The loss of power—caused by missing genotypes—for the new method is rather moderate, even when there is substantial missingness and the LD among the SNPs is weak.
Figure 1.
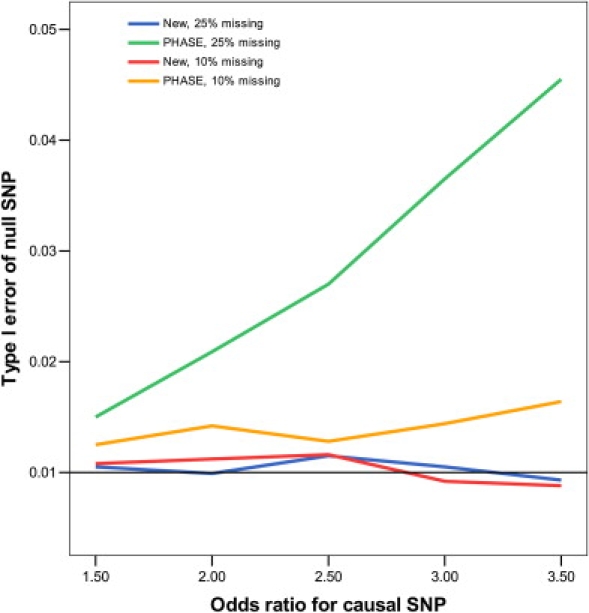
Type I Error of Association Tests on SNP 61 at the 1% Nominal Significance Level When SNP 60 Has an Additive Effect on the Risk of Disease
The analysis includes SNPs 60–64, which are missing independently with the same probability.
Figure 2.
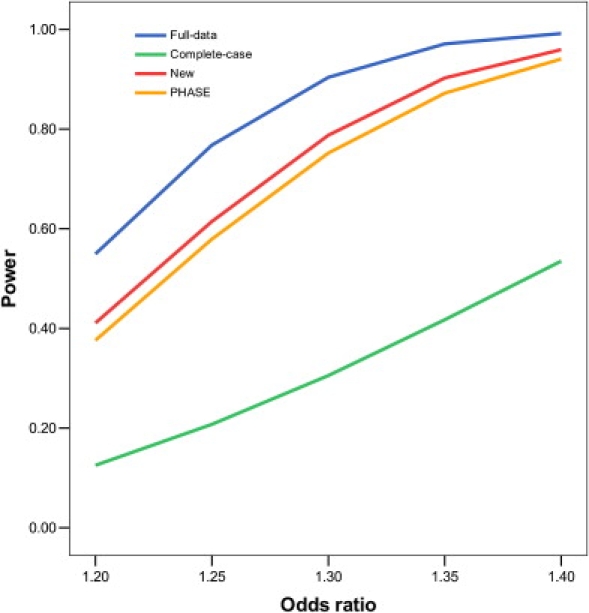
Power of Association Tests on SNP 60, which Has an Additive Effect on the Risk of Disease, at the 1% Nominal Significance Level
The analysis includes SNPs 60–64, which are missing independently with the same probability. For complete-case analysis, all subjects with missing data are removed. For full-data analysis, the missing genotypes are replaced by their true values.
We extensively studied the problem of untyped SNPs. We considered the two regions shown in Tables 1 and 2 of Nicolae,10 as well as various subsets of the first 100 SNPs on chromosome 18 of the CEU sample in the HapMap genome-wide data. For each region, we set a potentially causal SNP to be untyped and performed single-SNP analysis on that SNP. In addition to the case-control sample, we generated a reference panel with 30 trios. All the case-control subjects had missing values at the untyped SNPs, whereas the trios had known genotypes at all SNPs.
We evaluated the new method as well as the two existing approaches mentioned earlier: imputation of missing genotypes and estimation of allele frequencies. For the imputation approach, we used the EM algorithm to estimate the haplotype frequencies of the M SNPs from the trio data and then determined the probability distribution of the untyped SNP. (Other imputation methods are expected to yield similar estimates when M is small.) We then used the probability distribution in the corresponding association test, as recommended by Marchini et al.5 For the allele-frequency estimation approach, we used the method of Nicolae,10 together with our proposed variance estimator.
In all simulation studies, the new method estimated the odds ratio with little bias (see Supplemental Data available online). The variance estimator for the estimated odds ratio accurately reflects the true variation. The corresponding Wald test had proper type I error, and the confidence interval had correct coverage; see Supplemental Data. The Nicolae method (with our variance estimator) also had appropriate type I error. The imputation method did not always preserve the type I error. For Table 1 of Nicolae,10 the imputation method had type I error of approximately 3% (under the additive model) at the targeted significance level of 1%.
Figures 3 and 4 contrast the power curves of the three competing methods for four regions on chromosome 18 of the HapMap CEU sample under the additive and recessive models, respectively. The new method is more powerful than the two existing methods, especially when Rs2 is small. The power differences are much more profound under the recessive models than under the additive models. The imputation method tends to be more powerful than Nicolae's method.
Figure 3.
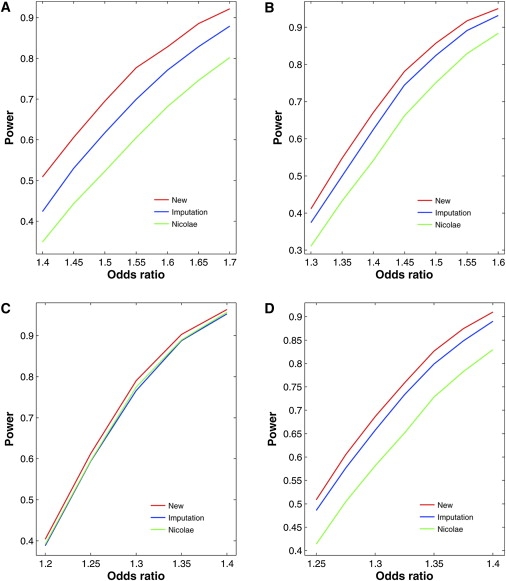
Power of Association Tests for Untyped SNPs at the 1% Nominal Significance Level under Additive Models for Four Regions of Five SNPs on Chromosome 18 of the HapMap CEU Sample
(A) SNP 21 in the region of SNPs 20–24 with MAF of 0.40 and Rs2 of 0.24.
(B) SNP 22 in the region of SNPs 20–24 with MAF of 0.25 and Rs2 of 0.42.
(C) SNP 26 in the region of SNPs 24–28 with MAF of 0.25 and Rs2 of 0.85.
(D) SNP 65 in the region of SNPs 63–67 with MAF of 0.37 and Rs2 of 0.62.
Figure 4.
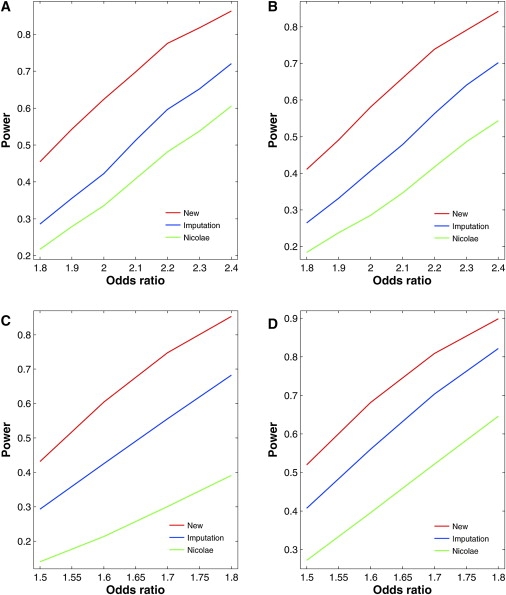
Power of Association Tests for Untyped SNPs at the 1% Nominal Significance Level under Recessive Models for Four Regions of Five SNPs on Chromosome 18 of the HapMap CEU Sample
(A) SNP 21 in the region of SNPs 20–24 with MAF of 0.40 and Rs2 of 0.24.
(B) SNP 22 in the region of SNPs 20–24 with MAF of 0.25 and Rs2 of 0.42.
(C) SNP 26 in the region of SNPs 24–28 with MAF of 0.25 and Rs2 of 0.85.
(D) SNP 65 in the region of SNPs 63–67 with MAF of 0.37 and Rs2 of 0.62.
Rheumatoid Arthritis Data
The North American Rheumatoid Arthritis Consortium conducted a case-control study to identify genetic factors that predispose for rheumatoid arthritis (RA). RA is a complex disease with a moderately strong genetic component. The recurrence-risk ratio for siblings is estimated at around 5 in Caucasians. The prevalence in Caucasians is approximately 0.8%. Females tend to be at higher risk than males, with an approximately 3 to 1 preponderance. The mean age of disease onset is in the fifth decade, with considerable variability.
A total of 460 cases were selected from throughout the United States. Confirmation of RA diagnosis was obtained from patients' rheumatologists. Radiographs of the hands and wrists were also obtained to document the presence and extent of joint involvement. A total of 460 unrelated controls from Long Island, New York City were frequency-matched to the cases by age and sex. All study subjects are non-Ashkenazi Caucasians.
A dense panel of 2,297 SNPs were genotyped by Illumina for an approximately 10 Mb region of chromosome 18q that showed evidence for linkage in the U.S. and French linkage scans. The SNPs were a custom set selected from dbSNP “double hit” SNPs on the basis of their distribution and favorable assay design characteristics. The 2,297 SNPs represent the SNPs successfully typed with minor allele frequency greater than 5% out of the 3,072 SNPs attempted.
We applied the new method to this study, with the HapMap CEU sample as the reference panel. As an illustration, we show in Figure 5 the results in a 315 kb region containing the ferrochelatase gene (FECH [MIM 177000]). This region covers 100 SNPs genotyped in the RA study and 210 untyped HapMap SNPs with minor allele frequency (MAF) > 5%. For each untyped SNP, we found a set of four genotyped SNPs within 30 kb that yields the largest Rs2. Only four untyped SNPs had Rs2 < 0.25, all of which had MAF < 7%. There were 94% of the SNPs with Rs2 > 0.5, 78% with Rs2 > 0.8, 67% with Rs2 > 0.9, and 50% with Rs2 = 1.
Figure 5.
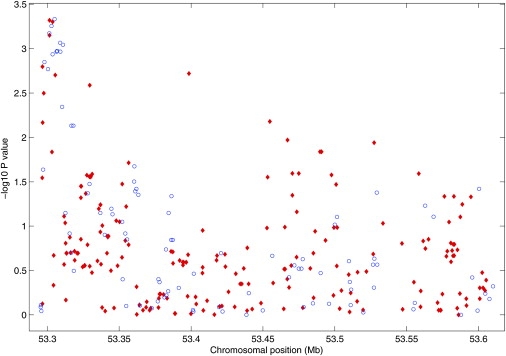
Results of Association Tests for Additive Effects in the Region of the FECH Gene from the RA Data
−log10(p values) for the genotyped and untyped SNPs are shown in blue circle and red diamonds, respectively.
The figure displays the results for both the genotyped and untyped SNPs. The inclusion of the results at the untyped SNPs enables us to have a much more detailed view of the region and provides stronger evidence of association than those of the genotyped SNPs alone. The strength of association signal from the untyped SNPs is similar to that of the genotyped SNPs at the beginning and the end of the region. In the middle of the region, most of the signal comes from the untyped SNPs. Among the ten most significant untyped SNPs, two had estimated odds ratios of 1.65 and 1.54, and the rest had estimated odds ratios of about 1.4. The ten most significant genotyped SNPs all had estimated odds ratios of about 1.4.
Discussion
We have presented a simple and coherent framework for dealing with missing genotypes. Our approach fully accounts for the uncertainty in predicting the unknown variants, so that the estimated odds ratios are attached with appropriate standard-error estimates and the corresponding association tests have correct type I error, even if the unknown variants are predicted with poor accuracy. For genotyped SNPs with missing values, our approach is likely to be more useful when genotypes are missing by design rather than by chance. With the continuing improvements in genotyping technologies, missing data for genotyped SNPs have been reduced rapidly; however, it may not be economically feasible to genotype all study subjects on a high-density platform or to completely sequence a large number of individuals.
For untyped SNPs, it is necessary to use external data to determine the joint distribution of the untyped and typed variants. For genotyped SNPs with partial missing data, it is not necessary to use external data, so greater robustness can be achieved by employing the likelihood based solely on the study data. For untyped SNPs, Nicolae's method tends to be less powerful than the new method and the imputation method. However, Nicolae's method is expected to be more robust to the choice of the reference panel because the genotypes of the reference panel enter into the test statistic only as weights.
The first step of our method is very similar to that of Nicolae's in that both methods identify a small number of genotyped SNPs that provides the best prediction for the untyped SNP. By contrast, Marchini et al.5 used information from all markers in LD with the untyped SNP in a way that decreases with genetic distance from the untyped SNP. The latter approach avoids the decision to choose a set of markers, but requires an approximate population-genetics model. Although our approach uses a small set of markers to predict the unknown variants, that set is chosen to provide the best prediction among all relevant sets of markers in LD with the untyped SNP. This strategy yields a very accurate prediction for most HapMap SNPs, as demonstrated in the RA example.
Although the numerical results reported in this article were focused on main genetic effects, our approach can be used to detect gene-gene and gene-environment interactions. The imputation approach can also be used to test interactions by using the most likely genotype or the expected genotype counts, but it would be difficult to use the probability distribution of the genotype.
A unique feature of our approach is that it provides valid estimates of odds ratios for genetic effects as well as gene-environment interactions. Although the first scan of the genome is typically done by association tests, most genome-wide association studies have reported odds-ratio estimates. Our method offers such estimates, together with appropriate confidence intervals, for untyped SNPs.
Another advantage of our approach is that it is computationally very fast. It takes less than 1 s on an IBM HS21 machine to perform the association analysis at an untyped SNP for a study with 2,000 individuals. Thus, the analysis of 3 million untyped SNPs can be completed overnight with a cluster of 50 machines. The software—called SNPMStat—implementing the new method is available at the Lin lab website.
Our simulation studies were concerned with a small number of markers and did not incorporate the hidden Markov model of Marchini et al.5 It would be highly valuable to compare the performance of competing methods in various genome-wide association studies as well as in large-scale simulation studies mimicking real data. Indeed, this task is currently taken on by the imputation subgroup of the GAIN Collaborative Research Group.15
Like the existing methods, our method requires Hardy-Weinberg equilibrium. This assumption may be violated when there is population substructure. We can relax this assumption by incorporating an inbreeding coefficient into the Hardy-Weinberg proportions and modifying the numerical algorithms accordingly.14 If the study involves different race groups, then the likelihoods LC(θ) should be constructed separately for each race group and then multiplied together.
It is of interest to assess genome-wide statistical significance. Because of the strong LD among densely distributed polymorphisms, the commonly used Bonferroni correction is punitively conservative, especially when all HapMap SNPs are tested. The permutation test is not computationally feasible and may be inappropriate for detecting gene-environment interactions. We are currently exploring the use of the Monte Carlo approach of Lin,16 which is efficient and versatile.
There has been a considerable debate about whether one should use SNP-based or haplotype-based analysis. The relative power depends on several factors.9,10,17–19 This article assumes that SNP-based analysis is of primary interest. So far, the first scan of the genome has always been performed with single-SNP tests. Our method uses the haplotype distribution to infer missing genotypes and can be unified with our earlier work on the analysis of haplotype-disease association.14
This article is focused on case-control studies with reference panels consisting of trios. We are currently extending our approach to other study designs and phenotypes, as well as other types of reference panels. Indeed, our software already allows both trios and unrelated individuals as reference panels.
Appendix A
Maximization of Case-Control Likelihood
We show how to maximize the likelihood of the case-control study given in Equation 4. The maximization of the likelihood in Equation 2 is similar but simpler. In the absence of environmental factors, the maximization of the likelihood in Equation 4 yields the same estimators of β and π as that of Equation 2. Thus, Equation 2 can be treated as a special case of Equation 4 for numerical purposes.
We use the EM algorithm20,21 to obtain initial estimates of haplotype frequencies based on the control sample. To avoid numerical instabilities in the maximization of the likelihood in Equation 4, we exclude those haplotypes whose estimated frequencies are 0 or very close to 0, i.e., < max(2/n, 0.001). We redefine K as the total number of haplotypes that are retained.
To accommodate the constraints and πk ≥ 0 (k = 1, …, K), we define πk∗ = πk/πK and νk = log πk∗. Write ν = (ν1, …, νK−1)T, θ = (μ, βT, νT)T, and
Then Equation 4 can be written as
The corresponding score function and information matrix are
and
respectively, where a⊗2 = aaT. To obtain the MLE , we solve the equation US(θ) = 0 by the Newton-Raphson algorithm. The initial values of μ and β are set to 0, and the initial value of ν is based on the estimated haplotype frequencies from the control sample.
By definition,
We use the above transformations to obtain the MLE from . Let J be the Jacobian matrix of (μ, βT, π1, …, πK)T with respect to (μ, βT, ν1, …, νK−1)T. That is, the first row of J is the derivative of μ with respect to (μ, βT, ν1, …, νK−1)T, which equals (1, 0, …, 0); the other rows are calculated similarly. Then the standard-error estimates for are the square roots of the diagonal elements in the matrix .
Appendix B
Maximization of Combined Likelihood
We obtain initial estimates of haplotype frequencies for the M SNPs by applying the EM algorithm to the likelihood for the reference-trio data given in Equation 3. We exclude the haplotypes with estimated frequencies < max(2/n, 0.001) and redefine K as the number of retained haplotypes. As in Appendix A, we reparametrize π as ν and redefine θ = (μ, βT, νT)T. Then Equation 3 becomes
where
The corresponding score function and information matrix are
and
respectively, where
The score function and information matrix associated with the combined likelihood LC(θ) are
and
respectively. To obtain the MLE , we solve the equation UC(θ) = 0 by the Newton-Raphson method. The initial values of μ and β are set to 0, and the initial value of ν is based on the estimated haplotype frequencies of the reference database. We then transform ν to π and obtain the standard-error estimates for and in the same manner as in Appendix A.
Supplemental Data
One table is available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
The North American Rheumatoid Arthritis Consortium, http://www.naracdata.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
SNPMStat (for C code for implementing the new method), http://www.bios.unc.edu/∼lin/software/SNPMStat
Acknowledgments
This research was supported by the National Institutes of Health (NIH). We wish to thank Drs. Chris Amos, Peter Gregersen, and Elaine Remmers and North American Rheumatoid Arthritis Consortium (NARAC) for the use of data from the rheumatoid arthritis study, which was supported by the NIH grants R01AR44422 and N01AR82232. We are indebted to Dr. Donglin Zeng for very helpful discussions on closely related topics.
References
- 1.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hinds D.A., Stuve L.L., Nilsen G.B., Halperin E., Eskin E., Ballinger D.G., Frazer K.A., Cox D.R. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 3.Stephens M., Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheet P., Stephens M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 6.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing data inference for whole genome association studies using localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balding D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006;7:781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- 8.Lin D.Y., Huang B.E. The use of inferred haplotypes in downstream analyses. Am. J. Hum. Genet. 2007;80:577–579. doi: 10.1086/512201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bakker P.I.W., Yelensky R., Pe'er I., Gabriel S.B., Daly M.J., Altshuler D. Efficiency and power in genetic association studies. Nat. Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 10.Nicolae D.L. Testing untyped alleles (TUNA) – applications to genome-wide association studies. Genet. Epidemiol. 2006;30:718–727. doi: 10.1002/gepi.20182. [DOI] [PubMed] [Google Scholar]
- 11.Zaitlen N., Kang H.M., Eskin E., Halperin E. Leveraging the HapMap correlation structure in association studies. Am. J. Hum. Genet. 2007;80:683–691. doi: 10.1086/513109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stram D.O. Tag SNP selection for association studies. Genet. Epidemiol. 2004;27:365–374. doi: 10.1002/gepi.20028. [DOI] [PubMed] [Google Scholar]
- 13.Nicolae D.L. Quantifying the amount of missing information in genetic association studies. Genet. Epidemiol. 2006;30:703–717. doi: 10.1002/gepi.20181. [DOI] [PubMed] [Google Scholar]
- 14.Lin D.Y., Zeng D. Likelihood-based inference on haplotype effects in genetic association studies (with discussion) J. Am. Stat. Assoc. 2006;101:89–118. [Google Scholar]
- 15.The GAIN Collaborative Research Group New models of collaboration in genome-wide association studies: The Genetic Association Information Network. Nat. Genet. 2007;39:1045–1051. doi: 10.1038/ng2127. [DOI] [PubMed] [Google Scholar]
- 16.Lin D.Y. Evaluating statistical significance in two-stage genomewide association studies. Am. J. Hum. Genet. 2007;78:505–509. doi: 10.1086/500812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris R.W., Kaplan N.L. On the advantage of haplotype analysis in the presence of multiple disease susceptibility alleles. Genet. Epidemiol. 2002;23:221–233. doi: 10.1002/gepi.10200. [DOI] [PubMed] [Google Scholar]
- 18.Zhao L.P., Li S.S., Khalid N. A method for the assessment of disease associations with single-nucleotide polymorphism haplotypes and environmental variables in case-control studies. Am. J. Hum. Genet. 2003;72:1231–1250. doi: 10.1086/375140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaid D.J. Evaluating associations of haplotypes with traits. Genet. Epidemiol. 2004;27:348–364. doi: 10.1002/gepi.20037. [DOI] [PubMed] [Google Scholar]
- 20.Excoffier L., Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 21.Qin Z.S., Niu T., Liu J.S. Partition-ligation-expectation maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am. J. Hum. Genet. 2002;71:1242–1247. doi: 10.1086/344207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.