

Abstract
Resources being amassed for genome-wide association (GWA) studies include “control databases” genotyped with a large-scale SNP array. How to use these databases effectively is an open question. We develop a method to match, by genetic ancestry, controls to affected individuals (cases). The impact of this method, especially for heterogeneous human populations, is to reduce the false-positive rate, inflate other spuriously small p values, and have little impact on the p values associated with true positive loci. Thus, it highlights true positives by downplaying false positives. We perform a GWA by matching Americans with type 1 diabetes (T1D) to controls from Germany. Despite the complex study design, these analyses identify numerous loci known to confer risk for T1D.
Introduction
Systematic GWA studies are critically dependent on the availability of very large and well-characterized control populations. With a different degree of structure in modern populations, ideally, multiple, diverse, and large control populations will be used. As platforms for GWA become standardized, numerous sources of pregenotyped control individuals are becoming available. Typically, many more controls are available than cases, and we believe these controls can be advantageous for discovering risk loci and for controlling the false-positive rate. For example, the data analyzed here include 416 Americans of European descent diagnosed with T1D (MIM 222100) and a control database of 2159 individuals from different regions of Germany.
Ancestry matching based on nongenetic variables1 and SNP genotypes2 for genetic-association studies has been proposed previously. Our approach, which we call genetic matching or GEM, goes further in that we show how to systematically obtain favorable matching by using a panel of genetic markers and how to determine outlying individuals as well as individuals that cannot be successfully matched to others in the available registry. By simulations, we will contrast matching to a commonly used method for controlling the confounding of ancestry, namely the use of eigenvector analysis3 via Eigenstrat4 to identify predictors of ancestry; for the real data, we contrast matching to both Eigenstrat and identification of common ancestry, such as European American.
We propose matching on the basis of genetic similarities derived from eigenvector decomposition (EVD), making our initial analyses similar to that taken in Eigenstrat.4 The best known form of matching is matched pairs (pMatch); however, assuming the criterion for matching are sufficient to remove the effects of unmeasured confounding, an alternative to matched pairs known as full matching (fMatch) is optimal.5 Consider a scenario in which three cases (a, b, and c) and three controls (x, y, and z) fall into two distinct ancestral clusters (a, x, and y) and (b, c, and z). Matching pairs creates three strata, (a and x), (c and z), and (b and y), but the pair (b and y) does not define a homogeneous strata. Alternatively, fMatch minimizes the total distance between individuals within strata with the constraint being that each stratum includes a single case and one or more controls, or vice versa, i.e., clusters (a, x, and y) and (b, c, and z). Of the two, fMatch is optimal because case and control samples are unlikely to have identical distributions of ancestry, and in this situation, forcing each case to match a unique control leads to suboptimal matches. (pMatch can be very useful, however, in designing follow-up studies that require preselection of case and control samples.)
In large association studies, the sample typically includes some individuals with widely varying ancestry. EVD is highly sensitive to outlying observations. A few points lying far from the majority of the data can determine multiple principal axes of the representation. Indeed, outliers can obscure the discovery of axes that potentially separate the data into distinct types. For this reason, individuals having highly unusual measures on any of the major eigenvectors are removed.4 Likewise, with matching it is necessary to determine which strata span an unusual distance leading to “unmatchable individuals.” If the controls are more numerous than the cases, they typically span a larger range of ancestries than cases, and it should be possible to find one or more controls similar to each case. Conversely, some cases may have to be removed to account for the effects of structure. In this work, we formalize the notions of outlying and unmatchable individuals and propose a method to discover the key axes that describe the population structure.
Material and Methods
A Sketch of the Matching Procedure Employed by GEM and Displayed in Figure 1
The illustration (Figure 1) shows the steps involved in matching genotyped cases and controls. To begin, create an L SNPs and N individuals matrix of scaled allele counts from which the EVD is computed (see Appendix). The top D eigenvectors form a D dimensional map describing the “ancestry” of each individual, i.e., the mapping of the ith subject in each dimension is determined by the ith element the in dth eigenvector. The dth eigenvalue determines the importance of the dth dimension in the new representation of the data. Individuals of similar ancestry map to similar values in the eigenvectors associated with large eigenvalues (Figure 1). Eigenvectors associated with small eigenvalues have little or no genetic interpretation.
Figure 1.
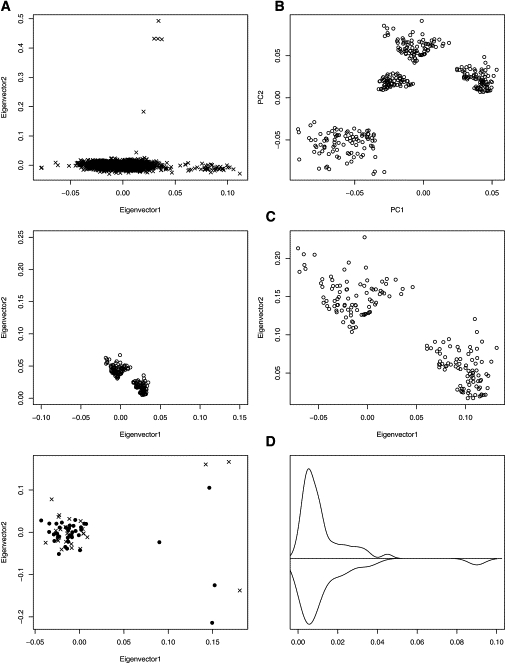
Flowchart for Genetic-Matching Algorithm Illustrated with Portions of the T1D Data
Distances between individuals are determined by the major axes of variation in the EVD representation. Outlier removal, illustrated by (A), is critical for revealing the subtle variability between individuals of similar ancestry. After major outliers are removed, clustering is used for discovery of homogeneous clusters; four distinct clusters are displayed here (B), plotted as principal component axes. Two of these clusters are displayed before ([C], left) and after ([C], right) rescaling of axes. Some observations are not outliers, but they are unmatchable ([D], left); for example, the isolated case in the center of the plot. Rescaled distances are compared to distances expected in homogeneous samples ([D], right) to identify cases and controls that can not be successfully matched. Association analysis is performed on matched strata so that the effects of population structure could be removed (not shown).
For a homogeneous population, the largest eigenvalues provides the basis for a significance test for population structure (see Patterson6 and Appendix). Applying this test with significance level α = 0.01, we determine the number of dimensions D to be used in the eigenvector representation. The EVD determines the distance between individuals on the basis of the top D eigenvectors, serving as coordinates or dimensions, and eigenvalues serving as weights to exaggerate differences in dimensions of greater importance (see Appendix).
If the data have many outliers, D will be relatively large, and the principal eigenvectors will be poorly estimated.7 Outliers (Figure 1A) can be removed with visual diagnostics or the criterion from Eigenstrat;4 namely, remove any individuals with ancestry coefficients greater than 6 SDs in at least one of the D eigenvector axes. After removing outliers, the EVD should be recomputed. If the estimated dimension, D, is still greater than two or three, we suggest finding the distance between nearest pairs of controls and cases. A subject with ancestry that does not lend itself to matching will appear as an outlier via this criterion and should be removed (see the T1D example).
To determine how to match and which individuals are unmatchable, we rely on the distribution of distances between individuals in a homogeneous population. For a homogeneous sample, the distribution of distances will depend on sample size N and the number of loci L. By using simulations, we can find the distribution of distances for a homogeneous population. These simulations also yield the distribution of eigenvalues for a homogeneous sample of size N.
Real populations are heterogeneous but can be modeled as mixtures of relatively homogeneous subpopulations (Figure 1B). We wish to represent these subpopulations so that the between-subject distances within a homogeneous subpopulation are comparable to expectation if the entire sample were homogeneous. To do so, we need to model the underlying population substructure and adjust real data so that they are scaled properly (Figure 1C); otherwise, the between subpopulation variance will cause distances between individuals to be poorly calibrated (Figure 1C). We do this via a two-stage algorithm involving clustering and scaling. In stage one, we cluster individuals that appear to have common ancestry. This is done iteratively, by addition of clusters and then testing for structure (see Appendix for testing) until each cluster is homogeneous. We use Ward's algorithm8,9 to form hierarchical groups of mutually exclusive subsets based on the first D axes of the EVD. We need a stopping rule for choosing K, the number of clusters. Start with K = 2 and apply the test for population structure on each of the clusters (α = 0.001). Homogeneous clusters, as judged by the significance test, are set aside, and Ward's algorithm is applied only to the remaining data. Repeat this process, increasing K until all the clusters are homogeneous or consisting of too few observations (∼20). Finally, we rescale interindividual distances as described in the Appendix so that they are comparable to distances found in a homogeneous population. At this rescaling step, unmatchable individuals are uncovered and removed (Figure 1D).
After outliers and unmatchable individuals are removed from the sample, recalculate the EVD and determine D. Reverting back to unscaled eigenvectors, find the distance between cases and controls on the basis of the Euclidean distance with D dimensions as described in the Appendix. Match strata with either full match or pair match. Software implementing matching algorithms is widely available (e.g., we use the optmatch function in the statistical package R). Then, the data can be analyzed for disease and SNP association by conditional logistic regression. Other covariates can be entered into the model at this point.
T1D Analyses
Purified samples of genomic DNA were obtained from the Genetics of Kidneys in Diabetes (GoKinD) study10 and from T1D patients recruited at the Children's Hospital of Pittsburgh (CHP) and University of Pittsburgh Medical Center. The study employed a human gene-chip microarray (Affymetrix, Santa Clara, CA) for evaluation of genetic variants with DNA samples from T1D (case) participants with genetic typing data obtained from the KORA11 and PopGen12 “control” cohorts.13 Genotyping results were obtained with the same Affymetrix 500K SNP typing array; however, assays for case and control cohorts were performed independently. Case participants (n = 416) were recruited in the U.S., with self-declared European ancestry and T1D; control participants (n = 2159) were citizens of Germany recruited independent of phenotype (Table 1). Recruitment of participants at CHP was governed by the human subjects protocol approved by the University of Pittsburgh Institutional Review Board (IRB #011052: New Advanced Technology to Improve Prediction of Type 1 Diabetes). CHP patients (n = 28) consented to providing 10 ml blood obtained by vein puncture as well as a brief medical history relating to onset of T1D. The GoKinD cohort (n = 394) was recruited independently from the CHP cohort by collaborative efforts of the Juvenile Diabetes Research Foundation, National Institutes of Health, and U.S. Center of Disease Control.10 Material from the GoKinD cohort was provided as solutions of DNA, purified from lymphoblastoid cell lines or from whole blood. DNA solutions were provided as 50 μl aliquots containing ∼100 ng/μl DNA per aliquot dissolved in 20 mM NaCl and 1 mM EDTA (pH 7.5). DNA from the CHP samples were obtained from whole blood with methods described in Ringquist,14 and genotyping was performed by Affymetrix Services Laboratory (Affymetrix) with GeneChip 500K arrays. All of the genotype data from GoKinD samples generated by this project will be submitted to an accessible database, such as dbGaP or T1Dbase (see Web Resources).
Table 1.
Characteristics of Case and Control Participants
| Case Participants |
Control Participants |
|||
|---|---|---|---|---|
| Demographic Characteristics | CHP | GoKinD | KORA | POPGEN |
| Number of singletons | 28 | 394 | 1644 | 500 |
| Nominal European American (%) | 100% | 100% | — | — |
| German residents (%) | — | — | 100% | 100% |
| Male gender (%) | 50% | 46.7% | 49.5% | 51.8% |
| History of Diabetes | ||||
| Type 1 diabetes (%) | 100% | 100% | — | — |
| Mean age at T1D Diagnosis (yr) | 12.7 ± 7.9 | 12.2 ± 7.1 | — | — |
All T1D samples had a sufficient completion rate (>95%) for inclusion, as did almost all KORA and PopGen samples. Initially, genotypes for all three samples were called with the BRLMM algorithm.15 By using three criteria for genotype QC per SNP—greater than 90% genotype calls, test statistic for Hardy-Weinberg yields p value > 0.005, and minor allele frequency ≥0.05—we removed ∼140,000 SNPs and retained 360,000 for the T1D sample, similar to other studies. When we contrasted the T1D samples to the control samples, we noted SNPs with very different allele frequencies that were not in or near known T1D loci. Inspection of the allele frequencies showed that the control allele frequencies were remarkably similar to HapMap frequencies (see Web Resources), but the corresponding genotype clusters for the T1D samples had undesirable features.
We tried various ways to improve the genotype calls. First, we looked for substantial differences between the calls by using the two algorithms employed by Affymetrix, namely DM and BRLMM. Although some discrepancies were noted, we did not see a material improvement in the data by eliminating this small set of loci. Next, because we had the Affymetrix “cel” files for the PopGen control sample, we called all of these genotypes for PopGen and T1D together by using both the DM and BRLMM algorithms. Again, this process eliminated some problematic loci, but the results were not compelling. Finally, we tried the new Bayesian calling algorithm, CHIAMO.16 This algorithm led to a marked improvement for the genotype calls, as determined by inspection of the genotype clusters. For our data, we found that analyzing the PopGen and T1D data together (batch) yielded slightly better results than analyzing the two data sets as complementary strata, so these were the data we reported. Because we had greater confidence in the BRLMM calls for chromosome X, we reported those calls for X-linked SNPs. Because the KORA sample came to us only with genotypes called by the BRLMM algorithm, we used those genotypes for that data set.
Preliminary quality control consisted of a six-step process that reduced the number of cases to 415, controls to 2112, and SNPs to 284,216. Step 1: Removed a case who was a clear outlier. Step 2: Removed 32 controls who had greater than 5% missing genotypes. Step 3: Removed 90,732 SNPs with >5% noncall rate in at least one of the three samples. Step 4: Removed 105,658 SNPs with minor allele frequencies less than 0.05 in either control sample. Step 5: Removed 1972 SNPs with (estimated for the two German control samples). Step 6: Removed 18,427 SNPs that violated Hardy-Weinberg equilibrium (p < 0.005) in either of the control samples.
Results
Simulations
We compare three approaches to correct for the effects of structure: Eigenstrat and GEM with fMatch and pMatch. Although we compare their size (i.e., rate of false positives) and power, these approaches are not direct competitors. The GEM methods are designed to limit analysis to strata that are chosen a priori, whereas Eigenstrat aims to remove the effects of structure in the analysis stage.
Allele frequencies for the subpopulations were generated with the “Balding-Nichols” model17 (see Appendix), with allele frequencies varying uniformly between 0.05 and 0.5. To correct for structure, L reference SNPs were generated. Of these SNPs, 99% had a minor amount of variability across subpopulations (), and 1% had substantial differentiation (). Null or causal candidate SNPs of three levels of were generated: Model (1) strongly differentiated SNPs, ; Model (2) moderately differentiated SNPs with ; and Model (3), modestly differentiated SNPs with .
Ten panels of independent reference SNPs, with L ranging from 96 to 100,000, were generated. For each of these panels, we simulated 1000 independent causal SNPs and 1000 independent null candidate SNPs. We repeated this analysis for models (1), (2) and (3) and for six choices of L. Causal SNPs with relative risk R = 2 were generated with the approach described in Price4 for power calculations.
Our first battery of simulations is based on SNPs sampled from two subpopulations, with 200 individuals per subpopulation. Case status was assigned to 80 and 20 of the individuals from subpopulations 1 and 2, respectively. The remaining individuals were assigned control status. For the matched-pairs analysis, we paired each case to the closest control until we obtained 100 matched pairs. For the other two methods of analyses, we analyzed all 400 individuals. Each method readily detects population substructure and achieves the desired type I error rate as L increases (Table 2). pMatch and fMatch successfully remove the effect of structure with a smaller panel of reference SNPs than Eigenstrat does (Figure 2A). Indeed, when a large panel of reference SNPs is available, the GEM procedures are overly conservative; consequently, Eigenstrat is slightly more powerful than both matching procedures (Table 2) under these conditions. For SNPs with less information about population membership than present in our simulated reference panels, greater numbers of SNPs would be required to remove the effects of structure.4
Table 2.
Size and Power of Tests at Level 0.05
| Eigenstrat with FST |
pMatch with FST |
fMatch with FST |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Statistic | Design | No. of Markers | 0.01 | 0.03 | 0.1 | 0.01 | 0.03 | 0.1 | 0.01 | 0.03 | 0.1 |
| Size | |||||||||||
| Two Populations | |||||||||||
| 96 | .069 | .106 | .211 | .062 | .100 | .202 | .065 | .101 | .206 | ||
| 386 | .055 | .061 | .085 | .044 | .045 | .051 | .047 | .049 | .054 | ||
| 1536 | .052 | .054 | .055 | .046 | .045 | .045 | .047 | .047 | .046 | ||
| 6144 | .053 | .052 | .051 | .044 | .045 | .045 | .047 | .047 | .046 | ||
| 12000 | .053 | .052 | .052 | .044 | .044 | .045 | .047 | .045 | .047 | ||
| 24000 | .053 | .050 | .051 | .043 | .042 | .041 | .046 | .046 | .045 | ||
| Gradient | |||||||||||
| 96 | .069 | .109 | .221 | .049 | .067 | .109 | .061 | .097 | .201 | ||
| 386 | .054 | .058 | .071 | .043 | .046 | .048 | .048 | .052 | .063 | ||
| 1536 | .052 | .051 | .050 | .045 | .044 | .044 | .047 | .047 | .046 | ||
| 6144 | .052 | .051 | .050 | .045 | .045 | .044 | .046 | .045 | .047 | ||
| 12000 | .052 | .052 | .052 | .044 | .044 | .045 | .047 | .047 | .047 | ||
| 24000 | .052 | .052 | .052 | .044 | .045 | .044 | .047 | .046 | .046 | ||
| Power | |||||||||||
| Two Populations | |||||||||||
| 96 | .783 | .710 | .683 | .693 | .635 | .620 | .754 | .685 | .659 | ||
| 386 | .767 | .701 | .682 | .682 | .632 | .621 | .735 | .673 | .653 | ||
| 1536 | .766 | .702 | .677 | .683 | .635 | .622 | .736 | .674 | .653 | ||
| 6144 | .765 | .694 | .676 | .684 | .630 | .623 | .735 | .671 | .653 | ||
| 12000 | .765 | .697 | .676 | .684 | .633 | .624 | .735 | .673 | .653 | ||
| 24000 | .763 | .696 | .677 | .684 | .632 | .624 | .734 | .671 | .653 | ||
| Gradient | |||||||||||
| 96 | .939 | .917 | .833 | .886 | .872 | .804 | .922 | .900 | .814 | ||
| 386 | .924 | .891 | .796 | .877 | .857 | .782 | .902 | .869 | .775 | ||
| 1536 | .917 | .876 | .774 | .876 | .850 | .775 | .894 | .856 | .754 | ||
| 6144 | .913 | .874 | .768 | .873 | .849 | .773 | .892 | .853 | .747 | ||
| 12000 | .915 | .873 | .771 | .874 | .849 | .774 | .891 | .850 | .749 | ||
| 24000 | .912 | .874 | .768 | .873 | .849 | .771 | .892 | .852 | .747 | ||
Columns depict the results as FST varies (0.01, 0.03, and 0.1) in the candidate markers. Results are shown for two scenarios: a two-population mixture and a nine-population gradient. For the size, the expected number of p values smaller than 0.05 is 50.
Figure 2.
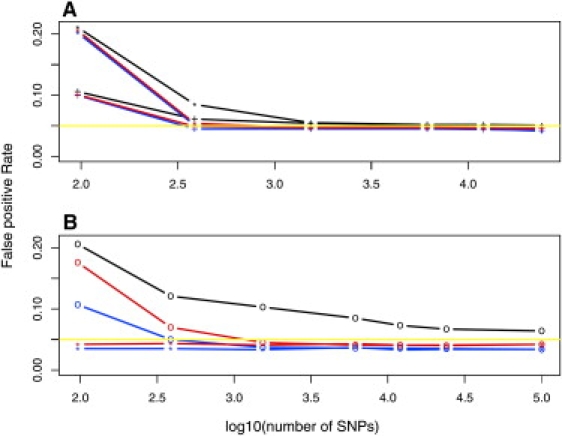
False-Positive Rate versus Log of the Number of Markers Available for Estimating Structure
Results are for Eigenstrat (black), pMatch (blue), and fMatch (red). The desired nominal rate of 0.05 is plotted as a yellow line. In (A), a sample derived from two simulated populations is shown. Results are displayed for markers with two levels of differentiation (∗) and 0.03 (+). The former exhibits a higher rate of errors than the latter for small numbers of markers. In (B), a sample derived from a gradient of simulated populations is shown. Results are displayed for the full sample (plotting character “o”) and with unmatchable individuals removed (plotting character “∗”; this applies to the matching methods only).
Our second battery of simulations is based on nine subpopulations distributed along a gradient, designed to simulate a cline such as the north to south cline observed in western Europe. The 100 cases are distributed with 2, 4, 6, 7, 9, 12, 15, 20, and 25 individuals in populations 1–9, respectively. The 300 controls are distributed randomly across the nine subpopulations. Results from this simulation are qualitatively similar to those shown in Figure 2A (Table 2). The first two batteries of simulations illustrate that when the case and control samples are drawn from the same subpopulations, but with different frequencies, the effects of substructure can be removed with any of the three methods described. Even the effects of highly differentiated SNPs can be removed provided the reference panel is sufficiently informative.
Our third battery of simulations is also based on a nine population gradient; however, the cases and controls are apportioned in a manner that simulates the complexity of human populations and GWA designs. As in the previous simulation, we simulate nine populations and draw 300 controls randomly. In contrast, all 50 of the cases are drawn from populations 6–9. Because of the nature of this third battery, namely the presence of unmatchable observations, we analyze the data in two ways: Unmatchable observations are removed as described previously; or unmatchable observations are retained. In choosing only a single control for each case, pMatch includes only 50 of the controls in the study regardless of the treatment of outliers. Provided the reference panel is sufficiently informative, many of these controls will be derived from populations 6–9. Eigenstrat, on the other hand, uses all of the data, as will fMatch when unmatchable observations are retained. For fMatch, this means that cases drawn from population 6 will tend to have many controls in their strata sampled from populations 1–5. The remaining cases will tend to have only one or two controls in their strata. By grouping the outlying observations, fMatch attempts to minimize the effect of unmatchable observations. Eigenstrat must account for controls sampled from populations 1–5 with regression techniques, which are well known to suffer adverse consequences when they are extrapolating beyond the range of the data.
When unmatchable observations are retained, pMatch corrects for the effects of substructure with fewer reference SNPs than the other two methods (Table 3 and Figure 2B). Indeed, Eigenstrat fails to remove the effects of population substructure. By comparing pMatch and fMatch, we see that the latter has greater power. This makes sense because fMatch is using more of the data (Table 3).
Table 3.
Size and Power of the Tests before and after Removing Outliers, with Eigenstrat, pMatch, and fMatch
| Outliers Present |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Eigenstrat with FST |
pMatch with FST |
fMatch with FST |
||||||||
| Statistic | No. of Markers | .01 | .03 | .1 | .01 | .03 | .1 | .01 | .03 | .1 |
| Size | ||||||||||
| 96 | .064 | .097 | .206 | .044 | .056 | .107 | .056 | .082 | .176 | |
| 386 | .057 | .068 | .121 | .038 | .040 | .050 | .045 | .051 | .070 | |
| 1536 | .056 | .062 | .103 | .037 | .037 | .037 | .043 | .042 | .045 | |
| 6144 | .056 | .061 | .085 | .037 | .037 | .035 | .041 | .042 | .041 | |
| 12000 | .058 | .058 | .073 | .037 | .036 | .036 | .042 | .040 | .041 | |
| 24000 | .057 | .058 | .067 | .037 | .037 | .035 | .042 | .042 | .040 | |
| 100000 | .055 | .057 | .064 | .037 | .037 | .034 | .043 | .043 | .042 | |
| Power | ||||||||||
| 96 | .804 | .753 | .650 | .590 | .579 | .511 | .770 | .726 | .623 | |
| 386 | .784 | .731 | .630 | .583 | .566 | .489 | .721 | .686 | .583 | |
| 1536 | .771 | .716 | .615 | .581 | .567 | .482 | .671 | .642 | .548 | |
| 6144 | .762 | .711 | .604 | .583 | .566 | .485 | .639 | .620 | .531 | |
| 12000 | .751 | .704 | .595 | .582 | .564 | .485 | .637 | .615 | .528 | |
| 24000 | .746 | .699 | .593 | .584 | .565 | .484 | .637 | .613 | .529 | |
| 100000 | .748 | .694 | .592 | .588 | .565 | .484 | .639 | .612 | .528 | |
| Outliers Removed | ||||||||||
| Size | ||||||||||
| 96 | .061 | .090 | .195 | .037 | .036 | .035 | .044 | .043 | .042 | |
| 386 | .057 | .060 | .095 | .036 | .036 | .035 | .043 | .041 | .043 | |
| 1536 | .054 | .053 | .057 | .035 | .038 | .033 | .040 | .044 | .041 | |
| 6144 | .054 | .053 | .056 | .040 | .037 | .037 | .044 | .044 | .043 | |
| 12000 | .052 | .053 | .054 | .038 | .035 | .033 | .041 | .042 | .041 | |
| 24000 | .052 | .052 | .053 | .036 | .039 | .034 | .041 | .045 | .041 | |
| 100000 | .052 | .052 | .053 | .037 | .035 | .034 | .042 | .042 | .042 | |
| Power | ||||||||||
| 96 | .906 | .931 | .927 | .706 | .713 | .656 | .772 | .776 | .719 | |
| 386 | .873 | .885 | .870 | .698 | .713 | .656 | .771 | .769 | .726 | |
| 1536 | .856 | .857 | .834 | .700 | .716 | .660 | .771 | .774 | .727 | |
| 6144 | .849 | .850 | .829 | .703 | .716 | .666 | .771 | .774 | .729 | |
| 12000 | .843 | .843 | .818 | .703 | .713 | .663 | .769 | .767 | .726 | |
| 24000 | .840 | .840 | .817 | .701 | .715 | .667 | .771 | .775 | .726 | |
| 100000 | .835 | .834 | .813 | .700 | .719 | .669 | .772 | .776 | .728 | |
Columns depict the results as varies (.01, .03, and .1) in the candidate markers. The simulated data are a gradient with nine subpopulations; controls are drawn from 1–9 and cases are only from 6–9.
On the basis of the clustering and rescaling process, most of the controls from populations 1–5 are unmatchable, and such a result is desirable because cases were only drawn from populations 6–9. In this instance, the size of the matched analyses is now closer to the nominal level even when L is small, as expected. Interestingly, there is the considerable enhancement in power for fMatch and pMatch when unmatchable individuals are removed, as recommended by our methods, as opposed to when they are forced to be retained (Table 3). This occurs because removal of the outliers leads to improved performance of the EVD and hence superior choices of matches in the analysis. In addition, for fMatch the removal of controls from populations 1–5 leads to a more homogeneous sample that tends to increase power.
Eigenstrat defines outliers without specific reference to cases and controls; thus, none of the observations are unmatchable observations. Nevertheless, if the regression approach is applied after removal of those observations declared unmatchable by the fMatch procedure, the type I error is successfully controlled, and the power is slightly greater than it is for fMatch (Table 3). This hybrid approach to analysis has some potential for further development.
GWA of Type 1 Diabetes Data with fMatch
We analyzed 416 cases of T1D,18 derived from the GoKinD10 cohort (n = 394) and T1D patients recruited from the Children's Hospital of Pittsburgh (n = 28). Samples were genotyped with the Affymetrix 500K GeneChip. All identified their ancestors as European. The mean age of onset for T1D was 12.2 and 12.7 years of age for the GoKinD and Pittsburgh cohorts, respectively. Controls genotyped by the same chip were obtained from the PopGen and KORA repositories, which consist of 500 individuals from north Germany (PopGen) and 1644 individuals from southern Germany (KORA).11–13,19 The four cohorts were recruited independently of one another. The relevant characteristics of these cohorts are summarized in Table 1.
Stringent quality control reduced the number of SNPs to 284,216 and the number of controls to 2112 (samples were removed if the rate of missing genotypes exceeded 5%). To reconstruct ancestry, we chose 23,552 independent or “tag” SNPs by using the H-clust algorithm20 with an r2 cutoff value of 0.04. Both case and control individuals exhibit complex population heterogeneity. For example, individuals were included in the PopGen and KORA registry on the basis of residence rather than known German ancestry. We removed one case individual who had very different ancestry from the other 415. For 415 cases and 2112 controls, D = 22 dimensions were required to explain the significant axes of genetic variation. Many of these axes exhibited extreme outliers (Figure 1A). After removing 53 controls, only three important axes of variation remained. On the basis of the first two eigenvectors, a cluster of cases that differs in ancestry from the control sample was clearly evident (Figure 3A). To identify unmatchable individuals more completely, we computed the distance between each case and the nearest control and vice versa on the basis of three axes of the EVD map. The resulting distribution of distances indicated that 21 cases could not be matched to a control with similar ancestry (Figure 4). By repeating this process of finding the significant eigenvalues and the corresponding minimum distances between cases and controls in the corresponding axes, we subsequently removed an additional one case and 15 controls. After excluding these outliers, only two significant eigenvalues remain when a significance level of 0.01 was used.
Figure 3.
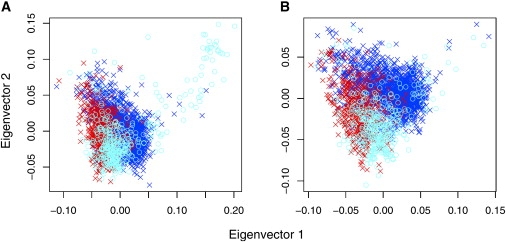
Plots of the First Two Eigenvector Axes for T1D Data before and after Removing the Unmatchable Individuals after Clustering and Rescaling of the Data
Each case (light blue) has a matched control (dark blue = South Germany, red = North Germany) in a close neighborhood after removal of unmatchable individuals; compare before (A) with after (B).
Figure 4.
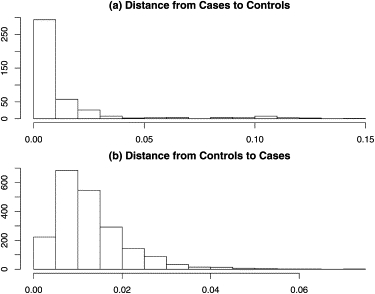
The Distance between Each Case and the Nearest Control and Vice Versa Based on Three Principal Components Are Computed
The distributions differ, and we eliminate 34 cases with distances to the nearest control greater than 0.075. (A) shows the histogram of distances between each case and the nearest control. (B) shows the histogram of distances between each control and the nearest case.
Next, with cluster analysis to identify homogeneous strata, 2136 individuals were clustered into 26 strata, each with 20 or more elements and no significant structure within cluster (p > 0.001). The remaining 301 individuals were clustered into 24 small clusters. On the basis of these strata, the data were rescaled and the distance between cases and matched controls was determined. Those that were considered unmatchable individuals on the basis of the simulation results were removed (see Appendix). With this process, an additional 20 cases and 48 controls are removed from the dataset for fMatch. The resulting distance between the remaining cases and controls in fMatch is consistent with expectations for cases and controls matched within homogeneous strata (data not shown). In the reduced fMatch sample, two principal axes separate the German control samples by region and define a space, spanned by both cases and controls, that facilitates matching (Figure 3B). These dimensions presumably map onto genetic gradients on the European continent; e.g., the horizontal axis is likely to be related to a north-south gradient because it tends to separate the German samples by north (PopGen)12 and south (KORA)11 origin.21,22 In the pMatch sample, one additional axis is needed to explain important variation (data not shown).
After final removal of outliers and unmatchable individuals for fMatch, cases and controls were stratified on the basis of their genetic ancestry into 298 strata. Most of the strata (159) contain a single case matched to several controls. A single case matched to a single control occurred in 111 strata. A minority of strata (28) contain a single control matched to multiple cases. For example, in the most extreme strata, a single case was matched to 71 controls and a single control was matched to 13 cases. When a single case is matched to a large number of controls (or vice versa), the information gain from the strata is essentially equivalent to that obtained from a single case matched to a moderate number of individuals. Nevertheless, conditional logistic regression is valid regardless of the lack of balance in the strata. In all, 373 cases were contrasted with 1996 controls by conditional logistic regression (Figure 5, top panel). The results highlight the HLA region, which contains numerous SNPs achieving GWA significance. Variation in the HLA region is well known to account for a large fraction of the risk for T1D.23–26 No other location in the genome contains SNPs with test statistics meeting reasonable criteria for GWA significance (≤10−7) after ensuring quality genotype calls by visual inspection of the genotype clusters (see Figures S1 and S2 available online for examples). It should be noted, however, that visual inspection of genotype clusters is essential to interpret this Affymetrix “first-generation” genotype data, a feature other GWA studies with this genotyping platform also report.27
Figure 5.
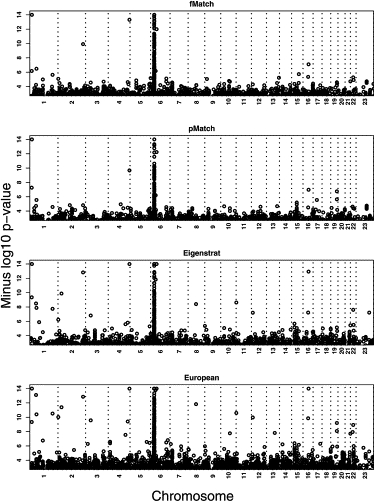
Transformed p Values after Conditional Logit Regression Was Performed on the Data Stratified with fMatch
Transformed p values (negative of the log, base 10). Results from conditional logistic regression on the data stratified with fMatch (top panel) and pmatch (second panel) are shown. Results obtained with Eigenstrat are shown in the third panel. Results obtained when removing observations with very divergent ancestries (inferred with the Eigenstrat rule for outliers) from the bulk of the sample, which was European, are shown in the bottom panel.
Results from fMatch agree with our expectations. Other GWA studies have established that all genetic variation thus far uncovered, aside from variation in the HLA region, account for a modest portion of the risk for T1D.28,29 For detecting loci of modest effect with good power, either sample sizes must be substantial (i.e., thousands of cases and controls genotyped) or a staged study design must be employed. The staged design typically sets a significance level between 0.01 and 0.001 in stage 1, then genotypes all loci meeting this significance level (and quality-control criteria) in a second, larger sample.30,31 Treating our study as stage 1 with a significance level of 0.007,30 results from fMatch would include SNPs for genotyping in stage 2 from six out of ten loci now believed to confer risk to T1D.29 Of the remaining four loci, only one had more than a few SNPs in the region.
Aside from the HLA region, SNPs in or near PTPN22 (MIM 600716), IL2RA (CD25 [MIM 147730]), and CTLA4 ([MIM 123890]; window = gene location ± 40 Kb) showed enough signal to be passed to stage 2. The smallest p value for each gene was 0.000706 (rs2488457), 0.000995 (rs10905669), and 0.000336 (rs231726). The smallest p values for SNPs close to “risk SNPs” rs2292239 and rs12708716 were 0.00667 (rs2292239) and 0.00539 (rs11647011) for window = SNP location ± 50 Kb. Could it be that the signals in these regions occurred by chance? To answer this question, we performed a simulation experiment. We randomly select from the genome ten intervals that correspond to the same size as the original ten windows (for the HLA region, we assumed a window of 3 Mb). Then, we count the number of intervals in which one or more SNPs have p < 0.007 and would thus be genotyped in stage 2. We perform this random selection 106 times, counting how many times six or more intervals would have SNPs genotyped in Stage 2. By this experiment, we determined that our results would rarely occur by chance, roughly one in ten thousand times.
A few other observations from these analyses are worth noting. Within the HLA region, Todd29 cites rs3129934 as the replicated SNP; our independent data and analyses yield a p value for association of 7.2 × 10−10 with this SNP; for the replicated SNP identified in CTLA4, rs3087243, our data and analyses yield a p value for association of 0.013, and, as noted above, the replicated SNP rs2292239 produced a p value of 0.00667 from our data. Although the HLA region needs no more support, our results provide further evidence for replication in CTLA4 and at rs2292239. For genotype cluster plots for the cited SNPs, see Figure S1. In addition, for all of the loci cited above, we have compared our data to that reported by the Wellcome Trust Case Control Consortium.27 For these loci, the allele in excess in cases is the same for both data sets (data not shown).
Four loci did not pass stage 1 criteria. None of these SNPs reported by Todd et al.29 as risk loci were on our Affymetrix genotyping array. Of these four risk SNPs, only rs1893217 in 18p11 was covered well in terms of genotyped SNPs in substantial linkage disequilibrium (LD) with it. This SNP is in almost complete LD with rs2542151 according to HapMap; it passed our QC, but it shows no evidence for association in our data (p = 0.51). For the proinsulin precursor gene, INS (MIM 176730), only two SNPs on the array pass QC and fall in the region, but HapMap contains no information about their LD with the reported risk SNP, rs689, and they show no association (p > 0.35). For the gene encoding interferon-induced helicase C domain-containing protein 1, IFIH1 (MIM 606951), the reported risk allele shows modest LD with a SNP we genotyped, namely rs7608315, which shows no association (p = 0.38). Finally, for the 12q24 region, rs3184504 is identified as the risk SNP. One SNP in this region passed QC for our data, and it is modestly associated with risk for T1D (p = 0.046).
The vast majority of the SNPs from this or any relevant GWA are independent of risk for T1D. Many SNPs from the HLA regions of chromosome 6 are associated, however. After eliminating HLA SNPs, ∼5% of the association tests are expected to have p values <0.05. Of the 284,216 tests, 7.0% were significant at α = 0.05 for fMatch. A moderate excess of false positives occurs for any reasonable choice of α. Given the success of the GEM method in the simulations, in terms of controlling the false-positive rate, we wondered whether the source of additional false positives could be poor-quality genotype calls. Indeed, by assessing genotype clusters for all SNPs producing p values ≤10−4, we find a rate of poor calls of 60%–67% (Figure S2). The rate of poor-quality genotype calls increases as the p value decreases. Predominantly, the problematic calls occur for the T1D sample. On the basis of our estimated rate of poor-quality genotype calls, we believe the excess false-positive rate is attributable to data-quality issues, not the method.
We also analyzed GWA data by using pMatch and Eigenstrat and by ignoring population substructure after discarding outliers with the Eigenstrat rule (see Figure 5). As expected, pMatch shows the lowest rates of positive findings, whereas ignoring structure yields the most. Like fMatch, it appears the excess of false positives for pMatch is due to poor-quality genotype calls. The same is predominantly true for Eigenstrat, but we note that an ample number of SNPs producing small p values are not attributable to poor quality, and this problem is amplified by ignoring structure. At significance level 0.0001, after visual inspection of genotypes fMatch has half the false-positive rate of Eigenstrat.
To further validate GEM, we tried a null experiment. We randomly labeled half of the KORA data as cases and half as controls and repeated the matching analysis. Removal of 72 outliers reduced the number of significant eigenvalues required to explain the variation from 24 to 2. After this simplification, only 12 unmatchable individuals remained. All three methods of analysis (Eigenstrat, Pmatch, and fMatch) produced type I error rates that were on target.
Discussion
Our GWA analyses of T1D are meant to accomplish two goals. First, they illustrate the utility of ancestry matching in the face of a very difficult problem, that being when cases are sampled in a region quite different from the region of the controls. In our case, the T1D sample comes from any American of nominal European ancestry, whereas the controls were recruited among residents of Germany. Such constellations can also arise even if cases and controls are sampled from the same geographical region. We would expect the example to be especially salient for American samples. Second, we wished to use the results to evaluate reported T1D risk loci and, in later analyses, discover new loci. The results show that genetic or ancestry matching can be an important ingredient in the toolbox of researchers who are performing GWA analyses. Moreover, our results do lend support for previous GWA findings for T1D.28,29
We do not yet know whether our analyses have identified any new risk loci for T1D. Although it seems unlikely given the modest sample of cases, a substantial number of controls have been analyzed. Moreover, for a rare disease like T1D, using unscreened instead of screened controls has almost no impact on power.32 We plan various kinds of stage 2 analyses to assess the association signals from our GWA results. In addition, by agreement the data generated by our project will be reported back to the GoKinD database, and GoKinD will make the data available to qualified investigators. Thus, these data will shortly be available to the research community, and we will be pleased to share detailed results upon request.
We have described how to use genetic matching to enhance a case-control study. We note, however, that these methods can also be used for the analysis of quantitative traits. Once homogeneous clusters are identified, they can be entered into a model as block effects, and the quantitative trait can be analyzed with standard statistical tools, such as analysis of variance.
Theory, simulations, and real-data analyses suggest that genetic matching is useful and powerful for GWA, especially when the samples of cases and controls cannot be guaranteed to be drawn from the same population. It can diminish the false-positive rate, sometimes substantially, and have only modest impact on power. Among others,33–36 methods similar to Eigenstrat4 also limit the impact of population structure, but for challenging designs, they cannot be expected to completely control the false-positive rate. Perhaps the gold standard for GWA studies should be to evaluate the data with both regression methods such as Eigenstrat and epidemiological methods such as fMatch. When the results of these methods agree, researchers have greater assurance of validity; it is when the results diverge that we should be wary.
Appendix
EVD of Allele Counts
Using allele counts for SNPs l = 1, …, L, and individuals i = 1, …, N, create an matrix X. For , the lth allele frequency, center allele counts in column l by subtracting and scale by dividing by . Find the EVD of . In the D dimensional space defined by the top D eigenvectors, the “ancestry” value for the ith subject is determined by the ith row of the eigenvectors ui1, …, uiD. The dth eigenvalue, , determines the scaling of distances in the dth dimension. These coordinates are used for matching.
Model for Population Stratification
The mean of allele frequencies from a set of populations is assumed to be the allele frequency of an ancestral population. Individual populations have each diverged from the ancestral population over time, with fixation index , a measure of population differentiation. Within a subpopulation j, suppose that allele counts are independent and identically distributed and that allele a is drawn with probability . If X is counting allele a, then . Let P be the random variable that varies across subpopulations, with as the realized value in subpopulation j: , . Assume that we have the minor allele frequencies of an ancestral population p.loci (in our simulations p.loci is uniform between .05 and .5) at L loci. From the ancestral population J, subpopulations have been formed. By knowing , for each marker l we can define and and generate the alleles as described above. When used in simulation studies, this is often called the Balding-Nichols model.17 For simulation of a cline (or a gradient), it is enough to order so that for each l.
Hypothesis Test for Population Structure
A formal significance test for population structure is based on a theoretical result for the eigenvalue distribution of a null sample covariance matrix.6,37 For a homogeneous population, the largest eigenvalue, properly normed, approximately follows the Tracy-Widom distribution37 with centering and scaling parameters that depend on both N and L, and . We can test the null hypothesis of population homogeneity against an alternative hypothesis of population heterogeneity. The sample covariance matrix S follows a Wishart distribution. The test for population structure will be applied iteratively (i.e., the leading eigenvalue, then the second and so on). If we find the first d eigenvalues to be significant, we test as though S were an Wishart matrix. If an eigenvalue is not significant, the smaller eigenvalues will not be significant either.
Removing Unmatchable Individuals
EVD determines the distance between individuals on the basis of the top D eigenvectors and eigenvalues. To stabilize the distance metric, we use the normed eigenvalues, , plus a constant a, chosen to ensure the weights are positive. The distance between individuals i and i' is calculated as .
To rescale the distances, let be the indices of individuals in the k′th cluster. Let rk be the number of individuals in the k′th cluster. For scaling subject , we use the eigenvector values but not the eigenvalues. Assume that the eigenvector representation of each individual consists of an ancestry signal plus random noise: .
For homogeneous data, because all individuals came from a common source, the ancestry signal is 0 and the representation consists simply of random noise . Our target is to identify approximately homogeneous subpopulations that have little or no diversity for ancestry. If the clustering is successful, the signal of each individual in subset can be approximated by , and the noise can be approximated by . But notice that EVD automatically scales the eigenvectors so that and . A traditional sum of squares decomposition leads to
i.e., the total sum of squares (SSTotal) equals the sum of squares attributable to random variation or error (SSError) plus the sum of squares attributable to ancestry differences (SSModel). Unit scaling of SSTotal causes the distances between individuals from heterogeneous populations to be uncomparable to distances in homogeneous populations. For example, if the sample derives from two highly differentiated populations so that SSError = 0.01 and SSModel = 0.99, then the expected distance between two individuals with common ancestry is ∼0.01/n. Alternatively, if the populations have identical ancestry, then the expected distance between two individuals is ∼1/n. For comparing to a homogeneous scaling, we wish to rescale the random noise so that SSError is 1. It follows that the data will be rescaled equivalently to homogeneous data if we set and rescale the data such that
In practice, provides a good estimate of the signal only when the cluster size is sufficiently large, say greater than 10. Hence, to compute , include only those clusters including 10 or more elements in the sum and then multiply by to account for the missing clusters. Notice that we scale differently for each of the d dimensions to stretch and shrink accordingly to get the proper scaling of the data.
In the final step, find the distances between individuals with the instead of and use the expected value of normed eigenvalues obtained from the simulation, instead of the actual eigenvalues. Match rescaled data with fMatch or pMatch and measure the distances between cases and controls. Any individuals with distances in this metric exceeding the 99.9th quartile of the null distribution of distances are declared unmatchable.
Supplemental Data
Two figures are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
CHIAMO, http://www.stats.ox.ac.uk/%7Emarchini/software/gwas/chiamo.html
GoKinD, http://www.jdrf.org/gokind
HapMap Frequencies, http://www.hapmap.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Optmatch, http://cran.r-project.org/doc/packages/
T1Dbase, http://t1dbase.org/
Acknowledgments
The Genetics of Kidneys in Diabetes (GoKinD) Study sample collection is supported by the Juvenile Diabetes Research Foundation in collaboration with the Joslin Diabetes Center and George Washington University and by the United States Centers for Disease Control and Prevention. This work was funded by the National Institutes of Health grant MH057881 awarded to B.D. and K.R. and by the Department of Defense (grant W81XWH-07-1-0619) awarded to M.T. The MONICA/KORA Augsburg studies were financed by the GSF-National Research Center for Environment and Health, Neuherberg, Germany, and supported by grants from the German Federal Ministry of Education and Research (BMBF). Part of this work was financed by the German National Genome Research Network (NGFN). The genotypes reported in this publication were generated independently from any other initiatives supported by other organizations.
References
- 1.Lee W.-C. Case-control association studies with matching and genomic controlling. Genet. Epidemiol. 2004;27:1–13. doi: 10.1002/gepi.20011. [DOI] [PubMed] [Google Scholar]
- 2.Hinds D.A., Stokowski R.P., Patil N., Konvicka K., Kershenobich D., Cox D.R., Ballinger D.G. Matching strategies for genetic association studies in structured populations. Am. J. Hum. Genet. 2004;74:317–325. doi: 10.1086/381716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang S., Zhu X., Zhao H. On a semiparametric test to detect associations between quantitative traits and candidate genes using unrelated individuals. Genet. Epidemiol. 2003;24:44–56. doi: 10.1002/gepi.10196. [DOI] [PubMed] [Google Scholar]
- 4.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 5.Rosenbaum P.R. Springer-Verlag; New York: 1995. Observational Studies. [Google Scholar]
- 6.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2 doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jolliffe I.T. Springer; New York: 2002. Principal Component Analysis. [Google Scholar]
- 8.Venables W.N., Ripley B.D. Fourth Edition. Springer; New York: 2002. Modern Applied Statistics with S. [Google Scholar]
- 9.Everitt B.S. Edward Arnold; London: 1993. Cluster Analysis. [Google Scholar]
- 10.Mueller P.W., Rogus J.J., Cleary P.A., Zhao Y., Smiles A.M., Steffes M.W., Bucksa J., Gibson T.B., Cordovado S.K., Krolewski A.S. Genetics of Kidneys in Diabetes collection available for identifying genetic susceptibility factors for diabetic nephropathy in type 1 diabetes. J. Am. Soc. Nephrol. 2006;17:1782–1790. doi: 10.1681/ASN.2005080822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wichmann H.E., Gieger C., Illig T., MONICA/KORA Study Group. KORA-gen–resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2006;67(Suppl 1):S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 12.Krawczak M., Nikolaus S., von Eberstein H., Croucher P.J., El Mokhtari N.E., Schreiber S. PopGen: Population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 13.Steffens M., Lamina C., Illig T., Bettecken T., Vogler R., Entz P., Suk E.K., Toliat M.R., Klopp N., Caliebe A. SNP-based analysis of genetic substructure in the German population. Hum. Hered. 2006;62:20–29. doi: 10.1159/000095850. [DOI] [PubMed] [Google Scholar]
- 14.Ringquist S., Styche A., Rudert W.A., Trucco M. Pyrosequencing-based strategies for improved allele typing of human leukocyte antigen loci. Methods Mol. Biol. 2007;373:115–134. doi: 10.1385/1-59745-377-3:115. [DOI] [PubMed] [Google Scholar]
- 15.Rabbee N., Speed T.P. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006;22:7–12. doi: 10.1093/bioinformatics/bti741. [DOI] [PubMed] [Google Scholar]
- 16.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies via imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 17.Balding D.J., Nichols R.A. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica. 1995;96:3–12. doi: 10.1007/BF01441146. [DOI] [PubMed] [Google Scholar]
- 18.Pasquali L., Fan Y., Trucco M., Ringquist S. Rehabilitation of adaptive immunity and regeneration of beta cells. Trends Biotechnol. 2006;24:516–522. doi: 10.1016/j.tibtech.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Lowel H., Doring A., Schneider A., Heier M., Thorand B., Meisinger C. The MONICA Augsburg surveys–basis for prospective cohort studies. Gesundheitswesen. 2005;67(Suppl 1):S13–S18. doi: 10.1055/s-2005-858234. [DOI] [PubMed] [Google Scholar]
- 20.Rinaldo A., Bacanu S.-A., Devlin B., Sonpar V., Wasserman L., Roeder K. Characterization of multilocus linkage disequilibrium. Genet. Epidemiol. 2005;28:193–206. doi: 10.1002/gepi.20056. [DOI] [PubMed] [Google Scholar]
- 21.Cavalli-Sforza L.L., Menozzi P., Piazza A. Princeton University Press; Princeton, NJ: 1994. The History and Geography of Human Genes. [Google Scholar]
- 22.Rosenberg N.A., Mahajan S., Ramachandran S., Zhao C., Pritchard J.K., Feldman M.W. Clines, clusters and effect of study design on the inference of human population structure. PLoS Genetics. 2005;1:e70. doi: 10.1371/journal.pgen.0010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todd J.A., Bell J.I., McDevitt H.O. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- 24.Morel P.A., Dorman J.S., Todd J.A., McDevitt H.O., Trucco M. Aspartic acid at position 57 of the HLA-DQ beta chain protects against type I diabetes: A family study. Proc. Natl. Acad. Sci. USA. 1988;85:8111–8115. doi: 10.1073/pnas.85.21.8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorman J.S., LaPorte R.E., Stone R.A., Trucco M. Worldwide differences in the incidence of type I diabetes are associated with amino acid variation at position 57 of the HLA-DQ beta chain. Proc. Natl. Acad. Sci. USA. 1990;87:7370–7374. doi: 10.1073/pnas.87.19.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyttinen V., Kaprio J., Kinnunen L., Koskenvuo M., Tuomilehto J. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: A nationwide follow-up study. Diabetes. 2003;52:1052–1055. doi: 10.2337/diabetes.52.4.1052. [DOI] [PubMed] [Google Scholar]
- 27.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hakonarson H., Grant S.F., Bradfield J.P., Marchand L., Kim C.E., Glessner J.T., Grabs R., Casalunovo T., Taback S.P., Frackelton E.C. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 29.Todd J.A., Walker N.M., Cooper J.D., Smyth D.J., Downes K., Plagnol V., Bailey R., Nejentsev S., Field S.F., Payne F. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H., Thomas D.C., Pe'er I., Stram D.O. Optimal two-stage genotyping designs for genome-wide association scans. Genet. Epidemiol. 2006;30:356–368. doi: 10.1002/gepi.20150. [DOI] [PubMed] [Google Scholar]
- 31.Skol A.D., Scott L.J., Abecasis G.R., Boehnke M. Optimal designs for two-stage genome-wide association studies. Genet. Epidemiol. 2007;31:776–788. doi: 10.1002/gepi.20240. [DOI] [PubMed] [Google Scholar]
- 32.Bacanu S.A., Devlin B., Roeder K. The power of genomic control. Am. J. Hum. Genet. 2000;66:1933–1944. doi: 10.1086/302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 34.Pritchard J., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devlin B., Bacanu S.A., Roeder K. Genomic control to the extreme. Nat. Genet. 2004;36:1129–1130. doi: 10.1038/ng1104-1129. [DOI] [PubMed] [Google Scholar]
- 36.Epstein M.P., Allen A.S., Satten G.A. A simple and improved correction for population stratification in case-control studies. Am. J. Hum. Genet. 2007;80:921–930. doi: 10.1086/516842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnstone I. On the distribution of the largest eigenvalue in principal components analysis. Annals of Statistics. 2001;29:295–327. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.