

Abstract
Hereditary spastic paraplegias (HSPs) are genetically and phenotypically heterogeneous disorders. Both “uncomplicated” and “complicated” forms have been described with various modes of inheritance. Sixteen loci for autosomal-recessive “complicated” HSP have been mapped. The SPG15 locus was first reported to account for a rare form of spastic paraplegia variably associated with mental impairment, pigmented maculopathy, dysarthria, cerebellar signs, and distal amyotrophy, sometimes designated as Kjellin syndrome. Here, we report the refinement of SPG15 to a 2.64 Mb genetic interval on chromosome 14q23.3-q24.2 and the identification of ZFYVE26, which encodes a zinc-finger protein with a FYVE domain that we named spastizin, as the cause of SPG15. Six different truncating mutations were found to segregate with the disease in eight families with a phenotype that included variable clinical features of Kjellin syndrome. ZFYVE26 mRNA was widely distributed in human tissues, as well as in rat embryos, suggesting a possible role of this gene during embryonic development. In the adult rodent brain, its expression profile closely resembled that of SPG11, another gene responsible for complicated HSP. In cultured cells, spastizin colocalized partially with markers of endoplasmic reticulum and endosomes, suggesting a role in intracellular trafficking.
Introduction
The hereditary spastic paraplegias (HSPs) are inherited neurological disorders characterized by progressive lower-limb spasticity,1,2 with an estimated prevalence of 1.27–9.6/100,000.3,4 The disease is characterized pathologically by axonal degeneration in the long ascending and descending tracts of the spinal cord, especially in their terminal portions, associated with syndrome-specific degeneration of other structures.5 Although the corticospinal tracts are predominantly affected, the columns of Goll and the spinocerebellar tracts are also involved.
HSP is classified according to (1) the absence (“uncomplicated” or “pure” HSP) or presence (“complicated” or “complex” HSP) of additional neurological symptoms, including mental retardation, dementia, behavioral disturbances, deafness, cerebellar ataxia, distal amyotrophy, syndactyly, epilepsy, dysarthria, ichthyosis, optic atrophy, peripheral neuropathy, retinitis pigmentosa, and cataract; and (2) by the mode of inheritance, which can be autosomal dominant, autosomal recessive (AR), or X-linked.6
Patients with HSP generally share the same core clinical features: spastic gait, lower-limb hypertonicity, pyramidal weakness, hyperreflexia, and extensor-plantar responses (Babinski signs). There is considerable variation, however, in age at onset and in the severity of spasticity. Furthermore, in “complicated” HSP, the accompanying neurological problems are highly variable both within and among families.2,6
Concerning their molecular bases, at least 38 HSP and related genes have been localized or identified.6 Sixteen loci are associated with “complicated” AR-HSPs, but only five of the genes have been identified: SPG7 on chromosome 16p24.3, encoding paraplegin (MIM 602783);7 SPG11 on chromosome 15q21.1, encoding spatacsin (MIM 604360);8 SPG20 on chromosome 13q12.3, encoding spartin (MIM 607111);9 SPG21 on chromosome 15q22, encoding maspardin (MIM 608681);10 and SACS on chromosome 13q12.12, encoding sacsin (MIM 270550).11 The molecular mechanisms leading to axonal degeneration are probably as diverse and complex as the genetics of HSPs, but abnormalities in intracellular trafficking and mitochondrial function have been suggested to contribute to the dysfunction or degeneration of the corticospinal tract in several of these diseases, particularly through the analysis of SPG4 and SPG7 mouse models.6,12,13
SPG15, a “complicated” AR-HSP, was initially identified in association with pigmentary maculopathy (Kjellin syndrome; MIM 270700)14,15 in two Irish families and mapped to a region of 16 Mega bases (Mb) on chromosome 14q22-q24 between the D14S1038 and D14S61 loci.16 Very recently, we refined the SPG15 locus to 5.3 Mb between markers D14S981 and rs8688, and we estimated that it accounts for 15% of AR-HSP,17 a frequency quite similar to that of SPG11.18 In addition, we showed that the clinical features varied among patients and families but cognitive impairment and distal amyotrophy were frequent (Table 1). Peripheral neuropathy, maculopathy, cerebellar ataxia, thin corpus callosum, and white matter abnormalities on MRI were also observed but were not constant clinical features of the disease.17,19
Table 1.
Clinical and Paraclinical Features of 22 SPG15 Patients
| Family/Patient | Sex/Age at onset (yrs) | Disease Duration (yrs) | Functional Handicap (Scale) | LL/UL Reflexes | Cognition |
Cerebellar Signs | Visual Function | Other Signs | ENMG | Brain MRI | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mental Retardation | Cognitive Deterioration | ||||||||||
| 16/161 | M/12 | 20 | Severe (6/7) | Brisk/Brisk | + (Moderate) | No | No | Normal | Pes cavus, pseudo bulbar dysarthria, severe UL and LL amyotrophy | Axonal PNP | Normal |
| 30/274 | F/11 | 17 | Severe (6/7) | Very brisk/Brisk | + (Moderate) | No | No | Normal | Pes cavus, scoliosis, pseudo bulbar dysarthria, moderate LL amyotrophy | Axonal PNP | Normal |
| 30/275 | F/8 | 19 | Severe (5/7) | Very brisk/Brisk | + (Moderate) | No | No | Normal | Severe LL amyotrophy | ND | ND |
| 30/279 | F/8 | 1 | Mild (2/7) | Very brisk/Normal | + (Mild) | No | No | Normal | None | ND | Normal |
| 761/3 | M/12 | 26 | Severe (6/7) | Very brisk/Very brisk | No | + (Severe) | + | Reduced visual acuity | Pseudo bulbar dysarthria; oral and hand dystonia- Sensorineural hearing deficit | Axonal PNP | TCC, WMA, marked cortical atrophy, mild cerebellar atrophy |
| 761/5 | F/12 | 22 | Severe (6/7) | Very brisk/Normal | No | + (Mild) | + | Normal | Pseudo bulbar dysarthria. Sensorineural hearing deficit | Axonal PNP | TCC, cortical and cerebellar atrophy |
| 761/9 | M/9 | 12 | Severe(5/7) | Very brisk/Brisk | No | + (Mild-Moderate) | No | Pigmentary retinopathy | Pseudo bulbar dysarthria and nystagmus Sensorineural hearing deficit | Axonal PNP | TCC and WMA |
| 761/10 | M/5 | 12 | Moderate (4/7) | Very brisk/Normal | + | No | No | Normal | Pseudo bulbar dysarthria | Normal | TCC |
| 17/168 | F/14 | 6 | Moderate (3/7) | Brisk/Normal | No | No | No | Normal | Pes cavus | ND | TCC, cortical and mild cerebellar atrophy |
| 1007/3 | F/13 | 20 | Severe (6/7) | Brisk/Brisk | + | + | + | Macular pigmentation | Epilepsy, distal amyotrophy, bladder dysfunction | Normal | Diffuse cerebral atrophy |
| 1007/4 | M/14 | 18 | Severe (6/7) | Brisk/Brisk | + | + | + | Macular pigmentation | Bladder dysfunction, pseudo bulbar dysarthria, distal amyotrophy, | Normal | ND |
| 1007/5 | M/16 | 14 | Severe (6/7) | Brisk/Brisk | + | + | + | Normal | Focal dystonia, distal amyotrophy, bladder dysfonction | Normal | ND |
| 353/3 | F/12 | 20 | Severe (6/7) | Brisk/Brisk | No | + | + | Normal | Decreased vibration sense, distal amyotrophy, pes cavus, bladder dysfunction | Axonal PNP | ND |
| 353/4 | M/10 | 20 | Severe (6/7) | Brisk/Brisk | No | No | + | Normal | Decreased vibration sense, distal amyotrophy, pes cavus | ND | ND |
| 353/6 | F/10 | 15 | Moderate (4/7) | Brisk/Normal | No | No | No | Normal | Decreased vibration sense | ND | ND |
| 353/10 | M/10 | 6 | Moderate (3/7) | Brisk/Normal | No | No | + | Normal | Pes cavus, | Axonal PNP | ND |
| 444/7 | F/12 | 15 | Severe (7/7) | Brisk/Brisk | No | + | + | ND | Decreased vibration sense | ND | ND |
| 444/6 | M/12 | 15 | Severe (5/7) | Brisk/Brisk | No | No | + | ND | Decreased vibration sense | ND | ND |
| 444/9 | F/16 | 2 | Moderate (3/7) | Brisk/Brisk | No | No | No | ND | Pes cavus, | ND | ND |
| 671/10 | F/<10 | >13 | Moderate (4/7) | Very brisk/Brisk | No | + | No | Normal | Decreased vibration sense, distal amyotrophy, bladder dysfunction | ND | ND |
| 671/4 | F/18 | 6 | Moderate (4/7) | Very brisk/Brisk | No | + | No | Normal | Raynaud phenomenon, high-arched palate, wide interdental spaces, distal amyotrophy, bladder dysfunction | Axonal PNP | TCC, WMA, mild cortical atrophy |
| 671/5 | F/19 | 4 | Moderate (3/7) | Brisk/Brisk | No | + | No | Normal | Hands extrapyramidal rigidity, mild hand tremor, decreased vibration sense, distal amyotrophy, | ND | TCC, WMA |
Here, we report the refinement of the SPG15 locus to a 2.64 Mb genetic interval on chromosome 14q23.3-q24.2 and the identification of truncating mutations in the zinc-finger FYVE domain containing 26 gene (ZFYVE26), also known as KIAA0321, that segregate with “complicated” AR-HSP, with or without maculopathy, in eight families.
Subjects and Methods
Patients and Controls
We analyzed seven previously described SPG15-linked AR-HSP families16,17,19 and one large kindred partially reported20 that we found significantly linked to SPG15 with a significant multipoint LOD score of +3.3 (data not shown). Affected patients and their relatives were recruited with their informed and written consent, as prescribed by the law on bioethics of the European Community and after approval by the local ethics committee (approval No. 03-12-07 granted to A.B. and A.D. by the “Comité Consultatif pour la Protection des Personnes et la Recherche Biomédicale,” Paris-Necker). Genomic DNA from 300 unrelated healthy individuals (Europeans, n = 200; North Africans, n = 100) was used as a control panel for molecular studies.
Linkage Analysis
To further reduce the SPG15 interval, indirect genetic studies were undertaken in the three families reported by Elleuch et al.17 Primers flanking new polymorphic nucleotide repeats (supplementary table) from the Working Draft of the Human Genome available at UCSC were designed. Genotyping was performed via PCR with fluorescently labeled primers, electrophoresis on an ABI-3730 sequencer, and analysis with GeneMapper software 4.0 according to the manufacturer's recommendations (Applied Biosystems). Haplotypes were reconstructed manually by minimization of the number of recombination events. Genetic distances between markers were those of the Marshfield Centre for Medical Genetics, and map positions were verified on the draft of the Human Genome sequencing (UCSC and Ensembl centers).
Mutation Screening
Four genes were screened for mutations in one affected member of each family, with the use of primers flanking the exon and intron-exon boundaries: ZFYVE26 (for zinc-finger FYVE domain containing 26, GenBank accession number NM_015346; 42 exons; see Table S1, available online, for the primers); PLEK2 (encoding pleckstrin 2, NM_016445; 9 exons); PLEKHH (for pleckstrin homology domain containing, family H [with MyTH4 domain] member 1, NM_020715; 9 exons); and WDR22 (encoding WD repeat domain protein 22, NM_003861; 9 exons). PCR products were sequenced with the use of the Big Dye Terminator Cycle Sequencing Kit v3.1 (Applied Biosystems) on an ABI-3730 automated sequencer. Nucleotides were numbered relative to the A of the start codon (ATG) of the ZFYVE26 cDNA sequence (NM_015346).
Quantification of ZFYVE26 mRNA in Human Tissues by RT-PCR
ZFYVE26 mRNA expression was analyzed semiquantitatively via RT-PCR in adult human tissues, normalized with respect to vimentin (VIM) expression. One microgram of commercially available RNA (Human Total RNA Master panel II, Clontech) was reverse-transcribed with the High Capacity cDNA Archive Kit (Applied Biosystems) primed with Oligo d(T)16 (Applied Biosystems) in accordance with the supplier's recommendations. A 470 bp fragment from exon 2 to exon 5 was amplified with forward primer 5′-AGGGGATATCCCAAAGAGGG-3′ and reverse primer 5′-CCTTTCGAATGAGGTCCACC-3′, and a 507 bp fragment including exons 38 to 42 was amplified with forward primer 5′-TCACCACTTTGCCTCTGCCA-3′ and reverse primer 5′-GCCACTGGGCACAGATGTCT-3′. A 274 bp fragment of VIM (GenBank accession number NM_003380) containing exons 1 to 4 was amplified with forward primer 5′-ACCAGCTAACCAACGACAAA-3′ and reverse primer 5′-TGCTGTTCCTGAATCTGAGC-3′, as a reference.
Expression of Rat ZFYVE26 mRNA Detected by in situ Hybridization
Adult (P68, 200 g) Sprague-Dawley rats (Charles River) were killed by decapitation, and their brains were rapidly extracted and frozen in isopentane at −50°C. Sections were cut every 600 μm on a cryostat (−20°C) from the medulla to the striatum (+1.7 mm from the bregma, according to the rat brain coordinates of Paxinos and Watson), thaw-mounted on glass slides, and stored at −80°C. Whole rat embryos (E14.5) were fixed in PFA 4% for 24 hr, rinsed in PBS, dehydrated in graded ethanols (70% to 100%), rehydrated with the reverse procedure, cryoprotected in 15% sucrose for 24 hr, then frozen in isopentane at −35°C. Sixteen micron slices were cut every 250 μm and stored.
Rat ZFYVE26 mRNA expression was probed with three 34 bp antisense oligonucleotides recognizing exon 15 or the 5′ or 3′ portions of the large exon 21, designed with Helios ETC oligo design software (Helios Biosciences, Paris, France) from the mRNA sequence (XM_234335.3) of Rattus norvegicus. Each oligonucleotide or a mix of the three oligonucleotides gave identical results. A mix of three sense oligonucleotides was used as negative control. In brief, the oligonucleotides were labeled with [35S]-dATP with terminal transferase (Amersham Biosciences) to a specific activity of 5 × 108 dpm/μg. The day of the experiment, slices were fixed in 4% formaldehyde in PBS, washed with PBS, rinsed with water, dehydrated in 70% ethanol, and air-dried. Sections were then covered with 140 μl of hybridization medium (Helios Biosciences, Paris, France) containing 3–5 × 105 dpm of the labeled oligonucleotide mix. Slices were incubated overnight at 42°C, washed, and exposed to a BAS-SR Fujifilm Imaging Plate for 5–10 days. The plates were scanned with a Fujifilm BioImaging Analyzer BAS-5000 and analyzed with Multi Gauge Software (Fuji).
Immunohistochemistry in Rat Brain
Brains were processed as for in situ hybridization. Sections were fixed in 4% paraformaldehyde/PBS, preincubated in PBS containing 6% goat serum and 0.1% triton, then incubated in the same buffer with antibodies against NeuN (Chemicon International, 1/250, mouse) or GFAP (Dako, 1/500, rabbit), followed by biotinylated horse anti-mouse or rabbit IgG and ABC reagents (Vector Laboratories, Burlingame, CA). Labeling was revealed by autoradiography.
Expression of Epitope-Labeled Spastizin In Cultured Cells
The ZFYVE26 cDNA from clone DKFZp781H1112Q (RPDZ) was PCR-amplified with Easy-A polymerase (Stratagene) and primers 5′-GGCTCAAACATGGCTGCGCT-3′ and 5′-CTTCTTGGAGCCTGGGCCA-3′, and the PCR product was introduced in phase with the V5 and HIS tags in the pcDNA-3.1/V5-HIS-TOPO cloning vector, as recommended by the supplier (Invitrogen). The construction was verified via sequencing after ligation, transformation, and plasmid extraction—with the use of standard procedures—and correction of an initial nonsense mutation by directed-site mutagenesis (Quick-Change Site-Directed Mutagenesis Kit, Stratagene).
COS-7 cells, maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, penicillin (100 UI/ml) and streptomycin (100 μg/ml), were plated on coverslips coated with collagen and transfected 24 hr later with 2 μg plasmid DNA per well, in 6-well plates with DMRIE-C, according to the manufacturer's instructions (Invitrogen). The cells were fixed for 15 min in 4% formaldehyde, 48 hr post-transfection, and immunocytochemistry was performed via classical procedures with the following primary antibodies: rabbit anti-giantin (1/2000, Abcam), rabbit anti-calreticulin (1/400, Stressgen), mouse anti-EAA1 (1/1000, BD Biosciences), rabbit anti-Cox2 (1/400, gift of A. Lombes), mouse anti-Lamp2 (1/200, Abcam), mouse anti-V5 (1/200, Invitrogen) and rabbit anti-V5 (1μg/ml, Sigma). Secondary antibodies were alexa-488 anti-mouse and anti-rabbit (1/1000, Molecular Probes) and Cy3 anti-mouse and anti-rabbit (1/1000, Sigma). Cells were counterstained with DAPI (1 μg/ml, Sigma) and mounted with Fluoromount-G (Southern Biotech). Images were acquired with a Leica SP1 confocal microscope and Leica software.
Results
Refinement of the SPG15 Locus
We recently refined the SPG15 locus in three large Arab families with AR-HSP and mental retardation but not maculopathy.17 To further restrict this interval, we analyzed additional polymorphic markers found in the Human Genome sequence draft (Table S1). Obligatory recombination events between loci VNTR25TG and D14S1069 (patient 444-6) and between loci D14S588 and D14S1029 (Individual 353-11, who is still unaffected at age 18 and assumed to be a noncarrier of the disease gene)17 refined the centromeric and telomeric boundaries of this locus to a 2.64 Mb interval on chromosome 14q23.3-q24.2 (Figure S1) containing 23 known genes and five putative new genes (Figure 1).
Figure 1.
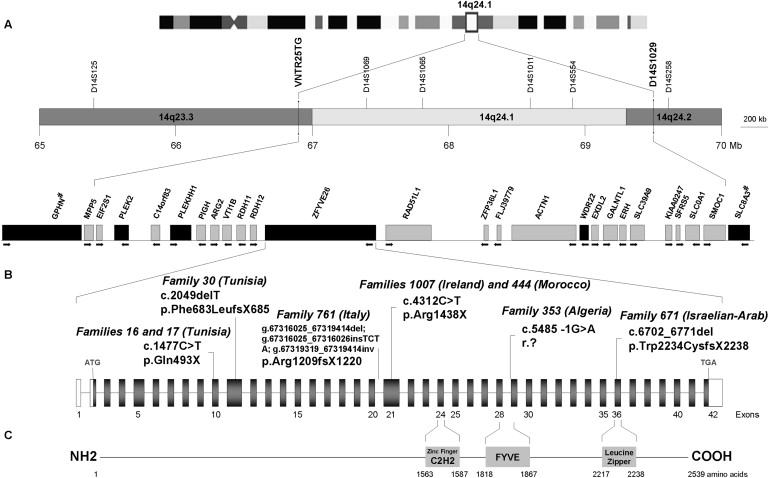
Critical Region of the SPG15 Locus, Structure of the ZFYVE26 Gene, and Mutations Identified in Eight SPG15 Families
(A) Physical and genetic map of human chromosome 14q23.3-q24.2, with markers defining the reduced SPG15 candidate interval in bold. Location and direction of transcription (arrow) of the known genes are schematically represented. The candidate genes analyzed in this study and in a previous report17 [indicated with “#”] are indicated by black boxes. Distances on chromosome 14 are indicated according to the Ensembl and UCSC Genome Browser databases.
(B) Structure of the ZFYVE26 gene (GenBank NM_015346) and location of the six different disease-causing mutations. The gene, located on chromosome 14q24.1, is transcribed from telomere to centromere and consists of 42 exons covering a genomic region of 70,063 bp. The full-length transcript is 9,688 bp long, with a coding sequence (exons 2–42) of 7,620 bp (mRNA NM_015346.2). The coding region is indicated in gray, and untranscribed regions (UTRs) (5′ and 3′) are indicated in white. The mutations are numbered according to the nomenclature of the Human Genome Variation Society, such that +1 is the A of the start codon (ATG) of the cDNA sequence.
(C) Putative functional domains (boxes) present in spastizin (according to Predictprotein).
Candidate-Gene Analysis
Mutations causing other HSPs have been reported to affect not only cellular processes such as intracellular trafficking and mitochondrial function but also myelination and development of corticospinal tract.2,6,12,13 This information provided us with criteria for selecting candidate genes located in the 2.64 Mb SPG15 interval (Figure 1). We first sequenced the exon and intron-exon boundaries of PLEK2, PLEKHH1, and WDR22. No disease-causing alterations, only already reported single-nucleotide polymorphisms (reported at UCSC, available on request), were found in several affected patients of the eight families.
We then analyzed the gene encoding ZFYVE26, which appeared to be a good candidate, given that missense and nonsense mutations in two other genes of the same family were recently identified in patients with SPG33 (ZFYVE27 on chromosome 10q24.2; MIM 610244)21 and Charcot-Marie-Tooth disease 4H (FGD4 on chromosome 12p11.21; MIM 611104).22,23 Six different homozygous truncating ZFYVE26 mutations were detected in the index patients of the eight families (Figures 1 and 2) linked to SPG15.16,17,19 Two were nonsense mutations: c.1477C→T in two consanguineous Tunisian families (F16 and F17) with similar flanking haplotypes, suggesting a common ancestral event (data not shown); and c.4312C→T in patients from two apparently unrelated consanguineous families from Morocco (F444) and Ireland (F1007), in which the mutation probably resulted from deamination of cytosine 4312 located in a CpG pair, a documented mechanism of mutation.24 Two mutations were frameshift deletions: a 70 base-pair deletion (c.6702_6771 del) in a large consanguineous family of Arab origin from Israel (F671) and c.2049 delT in a Tunisian kindred (F30). One mutation (c.5485-1G→A), homozygous in patients of the Algerian family 353, affected the splicing acceptor site and was shown, in silico (BDGP and Cold Spring Harbor Laboratory web sites), to strongly alter the splice score from +3.1 to −7.8. Finally, one mutation was a complex indel-inversion rearrangement that deleted exons 21–23 in four patients from a consanguineous Italian family (F761) and lead to a premature stop codon (Figure 2). The six mutations segregated with the disease in all eight families (Figure 2) and they were not detected in a panel of 600 chromosomes from unrelated controls (Caucasians, n = 400; North Africans, n = 200).
Figure 2.
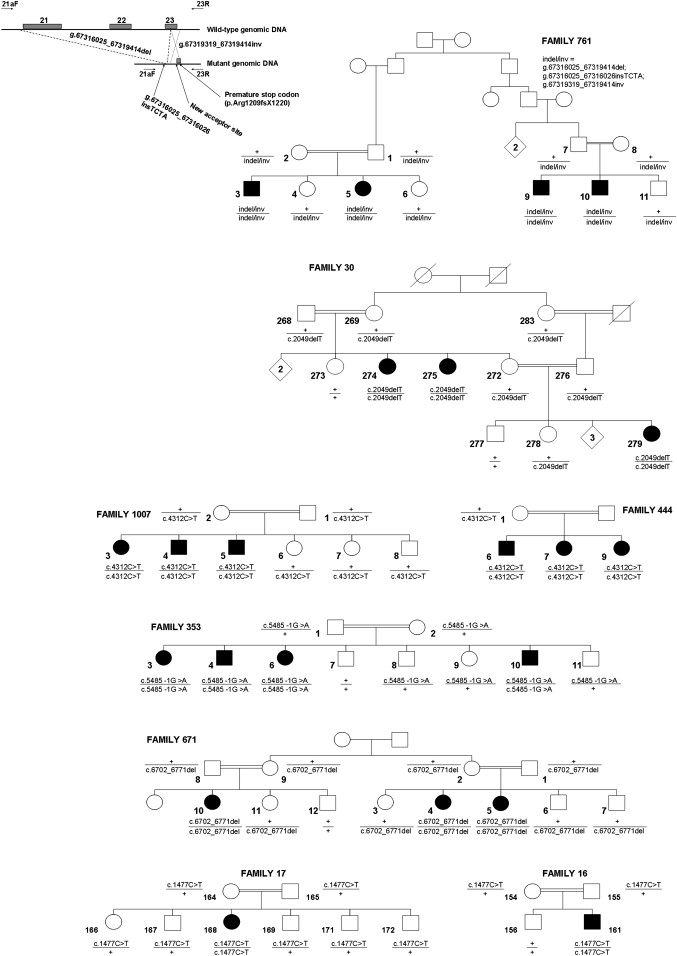
Pedigrees Showing Segregation of Disease-Causing Mutations in ZFYVE26 in Eight Families Linked to SPG15
Square symbols represent men, circles represent women. Affected subjects are represented with filled symbols. The numbers are an internal reference for each sampled individual. The genotypes are indicated below the analyzed individuals. “+” indicates wild-type allele. For the detection of the genomic rearrangement in family 671, primers 21aF and 23R (Table S1) were used, and they generated a 238bp-fragment in mutation carriers only.
Phenotype of SPG15 Mutation Carriers and Phenotype-Genotype Correlations
Some of the clinical features of the patients were reported previously and are summarized in Table S1.16,17,19,20 The overall phenotype associated with SPG15 mutations in 22 affected patients was early-onset spastic paraplegia (range: 5 to 19 years) associated with additional neurological symptoms that varied among patients and families: cognitive deterioration or mental retardation (73%, 16/22), axonal neuropathy (67%, 8/12), mild cerebellar signs (36%, 8/22), and, less frequently, a central hearing deficit, decreased visual acuity, or retinal degeneration. A thin corpus callosum and white matter hyperintensities were found on brain MRI in 64% (7/11) and 36% (4/11) of the patients, respectively, independent of disease duration.
All mutations resulted in the loss of the c-terminal putative leucine zipper domain, and they resulted in the loss of the FYVE domain as well in all but one family (F671). The phenotype in this family did not differ significantly from that of the other families. Given that all mutations resulted in premature stop codons, the RNA is probably degraded by nonsense-mediated mRNA decay, but this could not be confirmed because cells of SPG15 patients were not available.
Analysis of the Expression of the SPG15 Gene
In order to obtain insight into the function of the SPG15 gene, we first analyzed expression of ZFYVE26 mRNA by RT-PCR on total RNA from various human tissues. It was widely expressed, but most strongly in the adrenal gland, bone marrow, adult brain, fetal brain, lung, placenta, prostate, skeletal muscle, testis, thymus, and retina (Figure S2). Intermediate levels were detected in other structures, including the spinal cord.
When the expression of ZFYVE26 was investigated by the use of in situ hybridization in adult rat brain (P68), mRNA levels were generally low but strong signals were observed in the pineal gland, the edges of the lateral ventricles, the granular layer of the cerebellum, and the hippocampus (Figure 3 and Figure S3). Interestingly, expression was wider and stronger in rat embryos (E14.5) than in adult brain (Figure S3), particularly in the spinal cord and the cortical, cerebellar, thalamic, and hippocampal neuroepithelia.
Figure 3.
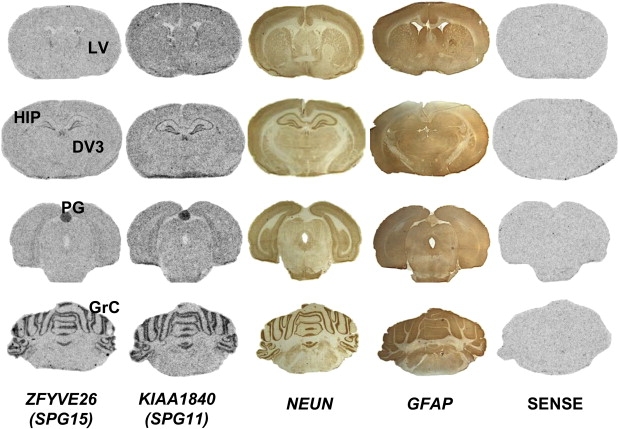
Expression Profile of ZFYVE26
Comparison of ZFYVE26 (SPG15) and KIAA1840 (SPG11) mRNA expression in the adult rat brain (P68) via in situ hybridization with a pool of three sense or antisense probes. Both ZFYVE26 and KIAA1840 expression resembled expression of the neuronal marker NeuN more than expression of the glial marker GFAP labeled on adjacent slices. No specific staining was observed with the sense probes. Expression of ZFYVE26, like KIAA1840, was low throughout the brain except in the following structures: HIP, hippocampus; PG, pineal gland; GrC, granular cell layer of the cerebellum; and the edges of the ventricles (DV3, third ventricle; LV, lateral ventricles). Similar results for both genes were obtained on adjacent slices with use of the pool of three probes or each probe independently.
The ZFYVE26 protein, which we suggest calling “spastizin” (SPASTIcity due to the ZFYVE26 proteIN), belongs to the FYVE-finger family, which includes the early endosome antigen 1 (EAA1, MIM 605070), involved in endocytic membrane trafficking.25–27 Because there is yet no antibody against endogenous spastizin, we explored its subcellular location with overexpression of a spastizin-HIS-V5 fusion protein in COS-7 cells. Epitope-tagged wild-type spastizin expressed in COS-7 cells and immunolabeled with antibodies against the V5 tag was partially colocalized in small dots or vesicles with immunolabeled endosomal marker EAA1 and calreticulin (CALR, MIM 109091), a marker of the endoplasmic reticulum, but not with giantin (GOLGB1, MIM 602500), lysosomal-associated membrane protein 2 (LAMP2, MIM 309060), or mitochondrially encoded cytochrome c oxidase II (MT-CO2 [COX2], MIM 516040),markers of the Golgi apparatus, lysosomes, and mitochondria, respectively (Figure 4).
Figure 4.
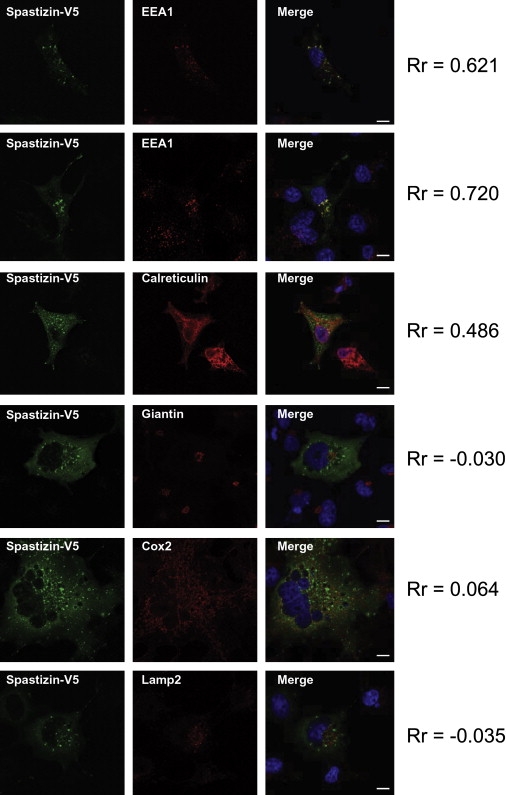
Cellular Expression of Spastizin
A spastizin-HIS-V5 fusion protein was overexpressed in COS-7 cells and labeled 48 hr after transfection with an antibody against the V5 tag (green). The Pearson coefficient (Rr) was calculated for estimation of the degree of colocalization between spastizin-HIS-V5 and the organelle markers (in red). The values range from −1.0 (not colocalized) to +1.0 (fully colocalized). Nuclei are counterstained with DAPI (blue). Images were obtained with a Leica SP1 confocal microscope (objective ×63, scale bar represents 10 μm). Spastizin partially colocalized with the endosomal marker EEA1 and the endoplasmic-reticulum marker calreticulin but did not show any significant colocalization with Golgi (anti-giantin), mitochondria (anti-Cox2), or lysosomes (anti-Lamp2).
Expression of a V5-tagged fusion protein of the expected size was verified on Western blots of cell extracts with a V5 antibody (data not shown).
Discussion
In this study, we refined the SPG15 locus to a 2.64 Mb interval on chromosome 14q23.3-q24.2, which allowed us to identify ZFYVE26 as the causative gene. The six identified mutations segregated with a complex phenotype in eight families previously linked to SPG15 (Figure 1 and 2) and were not found in a large series of control chromosomes.
This study and previous reports17,19 have demonstrated the broad clinical variability of Kjellin syndrome, which is complete in only a minority of our mutated patients. The core feature common to all our mutated cases was a severe and early-onset spastic paraplegia frequently associated with mental retardation and/or cognitive deterioration. Retinal degeneration—a major feature of this syndrome—as well as the MRI anomalies can be absent in some patients, even after long disease durations.17,19
Spastizin Functions
Spastizin, the product of the ZFYVE26 gene, is one of more than 30 different proteins with a FYVE domain in mammals.25,27 The FYVE domain is a zinc-finger-binding domain, highly conserved from yeast to humans, characterized by the presence of eight conserved cysteine residues, the third of which is flanked by characteristic basic amino acids, CX2CX9–39RRHHCRXCX4CX2–6CX4–48CX2C (X = nonconserved amino acid residues), suggested to bind the FYVE-finger proteins to endosomes.25–28 The majority of FYVE-finger proteins are involved in interactions with different forms of phosphoinositides (e.g., phosphatidylinositol 3-phosphate [PtdIns3P; PI3P]), which are found mainly in endosomes and serve as regulators of endocytic membrane trafficking.27,28 These phospholipids are components of membranes and have been implicated in the recruitment of proteins to membranes for signal transduction, membrane trafficking, cytoskeletal functions, and apoptosis.25 FYVE fingers bind with much higher affinity to membrane-associated PI3P than to its soluble analogs, explaining why most FYVE-finger proteins are associated with endosomal trafficking.27,28
The colocalization of spastizin with markers of the endoplasmic reticulum and endosomes suggests that this protein plays a role in endosomal trafficking, but examination of the endogenous protein will be necessary to confirm these results, which contribute to accumulating evidence that defects in trafficking are an important underlying cause of HSP.
Additionally, the temporal and regional distribution of ZFYVE26 mRNA observed via in situ hybridization in adult rat brain was very reminiscent of the expression of the SPG11 gene,8 the clinical features of which overlap those of SPG15, suggesting that the corresponding proteins could interact or function in a common pathway (Figure 3). In addition, the stronger and wider expression of ZFYVE26 in embryos, including in spinal cord and in cerebellar, hyppocampal, thalamic, and cortical neuroepithelia, suggests a critical role of this gene during the development of the nervous system (Figure S3).
Pathogenic Mechanisms
It has been previously demonstrated that an intact FYVE domain and several conserved cysteine residues in this domain are necessary for the endosomal localization of the proteins of this family.26,28,29 Interestingly, all of the mutations identified in our SPG15 families in ZFYVE26 were truncating mutations located upstream of the putative leucine zipper domain, and all except one (F671) were found upstream of the FYVE domain. Nonsense-mediated mRNA decay, a well-documented cellular mechanism,30,31 probably contributes to the loss of function of this protein in all cases.
It is interesting to note that a missense mutation in the FYVE-domain protein encoded by ZFYVE27 was identified in an “uncomplicated” form of autosomal-dominant HSP in a single German family with SPG33.21 The ZFYVE27 protein product was found to bind spastin (SPG4), another HSP-associated protein, but the missense mutation in ZFYVE27 induces an aberrant structure which interferes with its interaction with spastin and thus with microtubules.21 Whether spastizin also binds spastin remains to be determined.
In conclusion, the identification of ZFYVE26 as the gene responsible for SPG15 has increased our knowledge of the genetic and clinical heterogeneity of HSPs and will help to orient the molecular analysis of patients in view of a diagnosis. Our in situ hybridization and colocalization studies are also a starting point for our understanding of the normal cellular mechanisms in which spastizin participates and for the elucidation of the mechanisms underlying axonal degeneration in the SPG15, which probably include endosomal trafficking and development anomalies. Elucidation of these mechanisms will be necessary for the development of effective therapeutic strategies.
Supplemental Data
Three figures and one table are available with this paper online at http://www.ajhg.org/.
Supplemental Data
Web Resources
BDGP Splice Site Prediction; http://www.fruitfly.org/seq_tools/splice.html
Cold Spring Harbor Laboratory, http://rulai.cshl.edu/new_alt_exon_db2/HTML/score.html
Ensembl Genome Browser, http://www.ensembl.org
GenBank, http://www.ncbi.nlm.nih/Genbank/
Human Genome Variation Society, http://www.hgvs.org/mutnomen/
Online Mendelian Inheritance in Man (OMIM), www.ncbi.nlm.nih.gov/omim/
PREDICTPROTEIN, http://www.predictprotein.org/
UCSC Genome Browser, http://genome.ucsc.edu/
Acknowledgments
We are grateful to the families who participated; to M. Ruberg, J.M. Rozet, and H. Azzedine for helpful discussions; to J. Truchetto, S. Dumas (Helios Biosciences), M. Dehem (Genoscreen), A. Bouhouche, N. Bouslam, V. Meiner, I. Lerer, and the IFR70 Imaging Platform for technical assistance; and to M.I. Miladi for clinical evaluations. This work was supported in part by the Verum Foundation (Germany, to A.Br.), the Agence Nationale pour la Recherche (France, to A.D. and G.S.), the Association Strümpell-Lorrain (France, to the SPATAX network and to A.Bo.), the Telethon GGP06188 (Italy, to F.M.S. and P.D.), a cooperative program between INSERM (France, to A.Br.) and DGRST (Tunisia, to C.M.), and PRIN-2006063820 (Italy, to F.M.S. and P.D.). C.G. was financially supported by the Centre Hospitalier Universitaire de Bordeaux, and E.M. received a fellowship from the Fondation pour la Recherche Médicale (France, Grant Line Pomaret-Delalande).
References
- 1.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 2.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 3.Polo J.M., Calleja J., Combarros O., Berciano J. Hereditary ataxias and paraplegias in Cantabria, Spain: An epidemiological and clinical study. Brain. 1991;114:855–866. doi: 10.1093/brain/114.2.855. [DOI] [PubMed] [Google Scholar]
- 4.Filla A., De M.G., Marconi R., Bucci L., Carillo C., Castellano A.E., Iorio L., Kniahynicki C., Rossi F., Campanella G. Prevalence of hereditary ataxias and spastic paraplegias in Molise, a region of Italy. J. Neurol. 1992;239:351–353. doi: 10.1007/BF00867594. [DOI] [PubMed] [Google Scholar]
- 5.Behan W.M., Maia M. Strumpell's familial spastic paraplegia: genetics and neuropathology. J. Neurol. Neurosurg. Psychiatry. 1974;37:8–20. doi: 10.1136/jnnp.37.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Depienne C., Stevanin G., Brice A., Durr A. Hereditary spastic paraplegias: an update. Curr. Opin. Neurol. 2007;20:674–680. doi: 10.1097/WCO.0b013e3282f190ba. [DOI] [PubMed] [Google Scholar]
- 7.Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 8.Stevanin G., Santorelli F.M., Azzedine H., Coutinho P., Chomilier J., Denora P.S., Martin E., Ouvrard-Hernandez A.M., Tessa A., Bouslam N. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 2007;39:366–372. doi: 10.1038/ng1980. [DOI] [PubMed] [Google Scholar]
- 9.Patel H., Cross H., Proukakis C., Hershberger R., Bork P., Ciccarelli F.D., Patton M.A., McKusick V.A., Crosby A.H. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- 10.Simpson M.A., Cross H., Proukakis C., Pryde A., Hershberger R., Chatonnet A., Patton M.A., Crosby A.H. Maspardin is mutated in Mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am. J. Hum. Genet. 2003;73:1147–1156. doi: 10.1086/379522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engert J.C., Berube P., Mercier J., Dore C., Lepage P., Ge B., Bouchard J.P., Mathieu J., Melancon S.B., Schalling M. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000;24:120–125. doi: 10.1038/72769. [DOI] [PubMed] [Google Scholar]
- 12.Crosby A.H., Proukakis C. Is the transportation highway the right road for hereditary spastic paraplegia? Am. J. Hum. Genet. 2002;71:1009–1016. doi: 10.1086/344206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid E. Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. J. Med. Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kjellin K. Familial spastic paraplegia with amyotrophy, oligophrenia, and central retinal degeneration. Arch. Neurol. 1959;1:133–140. doi: 10.1001/archneur.1959.03840020007002. [DOI] [PubMed] [Google Scholar]
- 15.Webb S., Patterson V., Hutchinson M. Two families with autosomal recessive spastic paraplegia, pigmented maculopathy, and dementia. J. Neurol. Neurosurg. Psychiatry. 1997;63:628–632. doi: 10.1136/jnnp.63.5.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes C.A., Byrne P.C., Webb S., McMonagle P., Patterson V., Hutchinson M., Parfrey N.A. SPG15, a new locus for autosomal recessive complicated HSP on chromosome 14q. Neurology. 2001;56:1230–1233. doi: 10.1212/wnl.56.9.1230. [DOI] [PubMed] [Google Scholar]
- 17.Elleuch N., Bouslam N., Hanein S., Lossos A., Hamri A., Klebe S., Meiner V., Birouk N., Lerer I., Grid D. Refinement of the SPG15 candidate interval and phenotypic heterogeneity in three large Arab families. Neurogenet. 2007;8:307–315. doi: 10.1007/s10048-007-0097-x. [DOI] [PubMed] [Google Scholar]
- 18.Stevanin G., Azzedine H., Denora P., Boukhris A., Tazir M., Lossos A., Rosa A.L., Lerer I., Hamri A., Alegria P. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. 2007;131:772–784. doi: 10.1093/brain/awm293. [DOI] [PubMed] [Google Scholar]
- 19.Boukhris A., Feki I., Denis E., Miladi M.I., Brice A., Mhiri C., Stevanin G. Spastic paraplegia 15: Linkage and clinical description of three Tunisian families. Mov. Disord. 2007;23:429–433. doi: 10.1002/mds.21848. [DOI] [PubMed] [Google Scholar]
- 20.Casali C., Valente E.M., Bertini E., Montagna G., Criscuolo C., De Michele G., Villanova M., Damiano M., Pierallini A., Brancati F. Clinical and genetic studies in hereditary spastic paraplegia with thin corpus callosum. Neurology. 2004;62:262–268. doi: 10.1212/wnl.62.2.262. [DOI] [PubMed] [Google Scholar]
- 21.Mannan A.U., Krawen P., Sauter S.M., Boehm J., Chronowska A., Paulus W., Neesen J., Engel W. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 2006;79:351–357. doi: 10.1086/504927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delague V., Jacquier A., Hamadouche T., Poitelon Y., Baudot C., Boccaccio I., Chouery E., Chaouch M., Kassouri N., Jabbour R. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am. J. Hum. Genet. 2007;81:1–16. doi: 10.1086/518428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stendel C., Roos A., Deconinck T., Pereira J., Castagner F., Niemann A., Kirschner J., Korinthenberg R., Ketelsen U.P., Battaloglu E. Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am. J. Hum. Genet. 2007;81:158–164. doi: 10.1086/518770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glass J.L., Thompson R.F., Khulan B., Figueroa M.E., Olivier E.N., Oakley E.J., Van Zant G., Bouhassira E.E., Melnick A., Golden A. CG dinucleotide clustering is a species-specific property of the genome. Nucleic Acids Res. 2007;35:6798–6807. doi: 10.1093/nar/gkm489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gillooly D.J., Simonsen A., Stenmark H. Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins. Biochem. J. 2001;355:249–258. doi: 10.1042/0264-6021:3550249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seet L.F., Hong W. Endofin, an endosomal FYVE domain protein. J. Biol. Chem. 2001;276:42445–42454. doi: 10.1074/jbc.M105917200. [DOI] [PubMed] [Google Scholar]
- 27.Stenmark H., Aasland R., Driscoll P. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett. 2002;20:77–84. doi: 10.1016/s0014-5793(01)03308-7. [DOI] [PubMed] [Google Scholar]
- 28.Itoh F., Divecha N., Brocks L., Oomen L., Janssen H., Calafat J., Itoh S., Dijke Pt, P. The FYVE domain in Smad anchor for receptor activation (SARA) is sufficient for localization of SARA in early endosomes and regulates TGF-beta/Smad signalling. Genes Cells. 2002;7:321–331. doi: 10.1046/j.1365-2443.2002.00519.x. [DOI] [PubMed] [Google Scholar]
- 29.Hayakawa A., Hayes S.J., Lawe D.C., Sudharshan E., Tuft R., Fogarty K., Lambright D., Corvera S. Structural basis for endosomal targeting by FYVE domains. J. Biol. Chem. 2004;13:58–66. doi: 10.1074/jbc.M310503200. [DOI] [PubMed] [Google Scholar]
- 30.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in Health and disease. Hum. Mol. Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 31.Amrani N., Sachs M.S., Jacobson A. Early nonsense: mRNA decay solves a translation problem. Nat. Rev. Mol. Cell Biol. 2006;7:415–425. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.