

Abstract
Brittle cornea syndrome (BCS) is an autosomal-recessive disorder characterized by a thin cornea that tends to perforate, causing progressive visual loss and blindness. Additional systemic symptoms such as joint hypermotility, hyperlaxity of the skin, and kyphoscoliosis place BCS among the connective-tissue disorders. Previously, we assigned the disease gene to a 4.7 Mb interval on chromosome 16q24. In order to clone the BCS gene, we first narrowed the disease locus to a 2.8 Mb interval and systematically sequenced genes expressed in connective tissue in this chromosomal segment. We have identified two frameshift mutations in the Zinc-Finger 469 gene (ZNF469). In five unrelated patients of Tunisian Jewish ancestry, we found a 1 bp deletion at position 5943 (5943 delA), and in an inbred Palestinian family we detected a single-nucleotide deletion at position 9527 (9527 delG). The function of ZNF469 is unknown. However, a 30% homology to a number of collagens suggests that it could act as a transcription factor involved in the synthesis and/or organization of collagen fibers.
Main Text
Brittle cornea syndrome (BCS [MIM 229200]) is an autosomal-recessive disorder characterized by a thin and fragile cornea that tends to perforate spontaneously or as a result of minor trauma to the eye. Keratoconus, keratoglobus, and a blue sclera are common associated findings.1–6 Despite the use of protective measures, patients suffer from progressive visual deterioration that often leads to blindness.1–3,7,8 Systemic manifestations include joint hypermobility with occasional dislocations, hyperlaxity of the skin, kyphoscoliosis, a progressive conductive-hearing defect, dental abnormalities, and an increased incidence of hernias.5,6,9–11 Electron microscopy studies performed on skin biopsies from BCS patients showed 20- to 60-micron-wide “holes,” or fiber-free spaces, filled with amorphous material, distributed over its whole thickness,11 but the molecular basis of this disease remained unknown.
Since the original description by Stein et al.,1 more than 60 BCS patients have been reported. In Israel, BCS has been described mainly among Jews of Tunisian origin. Interestingly, all but one of the Tunisian Jewish BCS patients had red hair. This is in contrast to patients from other ethnic origins that show a normal distribution of hair color. Based on this observation, Zlotogora et al.6 suggested linkage disequilibrium between the BCS gene and a gene responsible for hair color in Tunisian Jews.
In a previous study, we assigned the BCS gene to a 4.7 Mb interval on chromosome 16q24, very close to the location of the hair-color gene, MC1R12 [MIM 155555]. In this report, we present evidence showing that BCS is caused by mutations in the Zinc-Finger 469 gene (ZNF469) [GI: 113426575].
The study was approved by the institutional review board at the Sheba Medical Center, Israel, and all participants granted informed consent. The five BCS patients of Tunisian Jewish ancestry included in this study have been previously reported.12 Recently, we have encountered an additional highly inbred Palestinian family with six affected members. The patients in this family manifested typical disease features (Figure 1), but none of them had red hair. Twenty milliliters of heparinized blood were drawn from all family members, and DNA was extracted via a commercial kit (Gentra System, Minneapolis, MN, USA). In silico data mining of genomic clones within the linkage interval at 16q24.1–24.3 identified two new polymorpic markers, which were annotated as D16S3436 and D16S3437. We assumed that all the Tunisian Jewish patients are descendents of a common founder and therefore are all homozygous for the same chromosomal segment in the vicinity of the gene. Genotyping results for the five Tunisian Jewish patients are shown in Figure 2, with the ancestral chromosome at the top. In the Tunisian Jewish patient no. 5, a deviation from the ancestral chromosome set the new centromeric boundary to D16S3436. A previously described recombination event in this patient on the telomeric side12 defined the gene interval in the Tunisian Jewish patients between the markers D16S3436 and D16S3425. Genotyping of the Palestinian family with these markers revealed full segregation with the disease (Figure 3), and lod-score calculations for the marker D16S3420 yielded a maximum lod score of 4.01 at θ = 0.00, thus confirming linkage to this locus. A recombination event in patient II:1 is evident, but more interesting is the analysis of individual II-4. For the markers D16S3423, D16S3437, D16S3436, D16S3424, and D16S3422, this unaffected family member has inherited two copies of the carrier chromosome. Given that he is unaffected, the gene must be located telomeric to D16S3422 (D16S3432 was not informative in this family). This recombinant chromosome, which was passed to two other unaffected individuals, patients III-1 and III-6, sets D16S3422 as the centromeric boundary. No recombinations were detected in the Palestinian family on the telomeric side, but combination of the data from the Tunisian patients reduced the disease-gene interval to 2.8 Mb between the markers D16S3422 and D16S3425. Not surprisingly, the carrier haplotype of the Palestinian patients was completely different from that of the Tunisian one.
Figure 1.
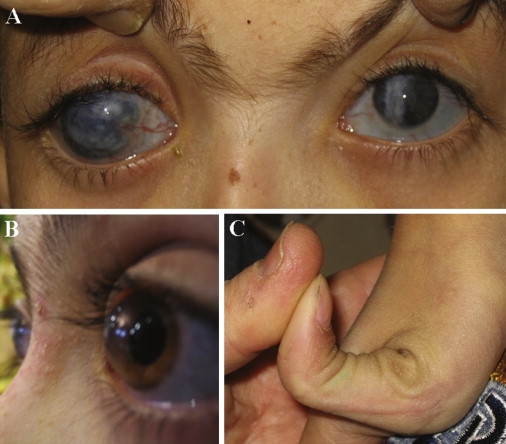
Clinical features of BCS Patients
(A) Corneal opacities due to scars.
(B) Keratoglobus.
(C) Hyperlaxity of the joints.
Figure 2.
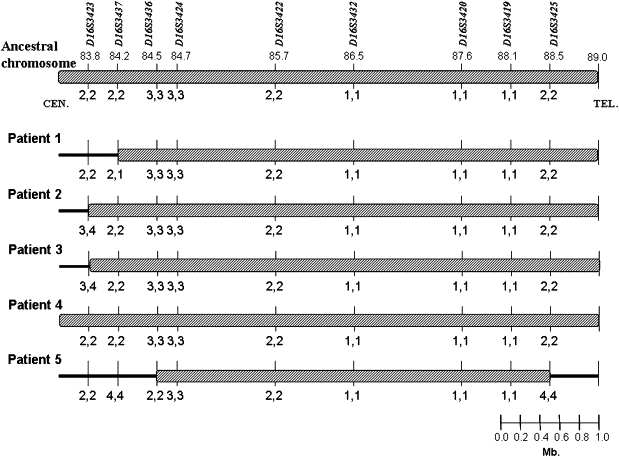
Haplotypes of Chromosome 16q24 in Tunisian Jewish Patients
Top: Ancestral chromosome.
In patient no. 5, deviation from the ancestral chromosome at the markers D16S3425 and D16S3436 on the telomeric and centromeric sides, respectively, set the disease gene interval to 4 Mb in the Tunisian patients.
Figure 3.
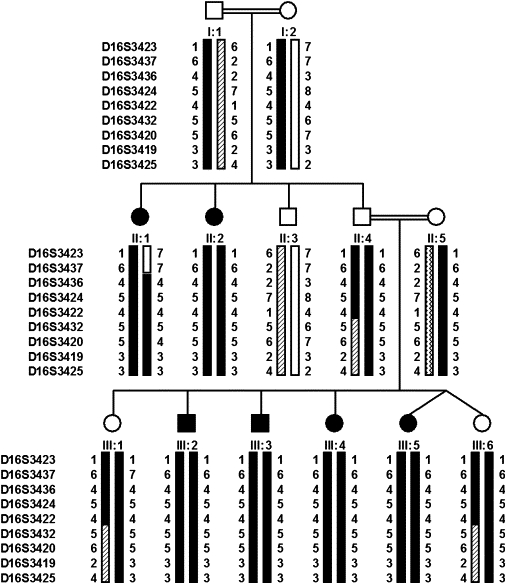
Palestinian Family Pedigree and Chromosome 16q24 Haplotypes
Recombination event in D16S3422 in individual II:4 sets this marker as the centromeric boundary. Allele 4 in D16S3420 in individual II:1 and allele 7 in D16S3437 in individual III:1 reflect mutations in these markers.
The candidate interval contained 50 known genes. With the assumption that the BCS gene is expressed in skin fibroblasts, all 32 genes from the candidate interval known to be expressed in fibroblasts were fully sequenced. A homozygous single-base-pair deletion at position 5943 (delA) in ZNF469 was detected in one of the Tunisian Jewish patients (Figure 4A). The deletion results in a frameshift and a putative stop codon 16 amino acids downstream. The sequence variation was confirmed, then extended to other patients and family members by restriction digestions performed on PCR-amplified DNA with the use of primer pairs 5′-GTGTGCAGGTGACAACTCTCC-3′ and 5′-GCGAGGTAAGTGGGTCTTCAC-3′. In the patients, an SmaI site was formed. Two hundred control chromosomes from individuals of Tunisian Jewish origin and 100 control chromosomes from various other ethnic backgrounds were screened for this deletion, but only the wild-type allele was found (Figure 4C). In a patient from the Palestinian family, we detected a single-base-pair deletion at position 9527 (delG) (Figure 4B) resulting in a premature stop codon 22 amino acids downstream. This mutation was confirmed, then extended to other family members by visualization of the 1 bp difference on an automated ABI Prism 3100 Genetic Analyzer (Perkin Elmer, Waltham, MA, USA) (Figure 4D). Genomic DNA was amplified with the fluorescent primers 5′-GGATGTACAACGAGCACCTG-3′ and 5′-TTCTTGGCTTCTCCCTCTTG-3′, with conditions previously described.13 We found full segregation of this mutation in the family, and we did not find it in 240 control chromosomes of Palestinian ancestry. ZNF469 was previously found to be expressed in a wide variety of tissues, including thyroid, bone, heart, mammary gland, lymph node, placenta, lung, connective, prostate, eye, liver, uterus and brain tissues. Using RT PCR (Thermo Scientific Verso c-DNA Synthesis Kit) (primer pair no. 19 detailed in Table S1, available online), we show specific expression of ZNF469 in mRNA extracted from a normal human cornea (Figure 5).
Figure 4.
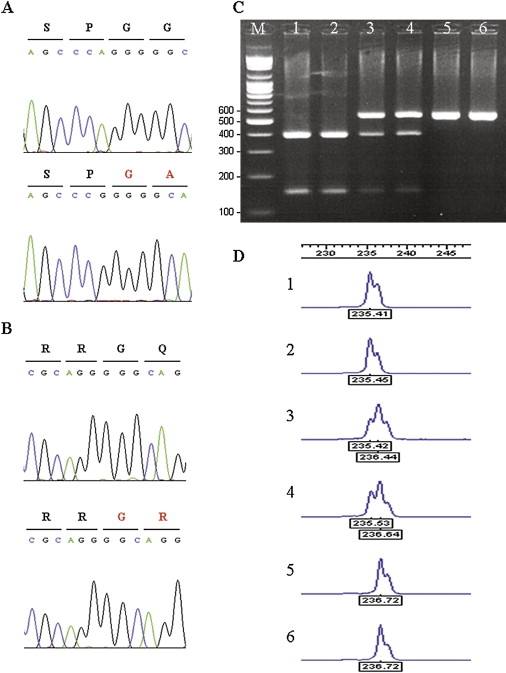
Mutations in ZNF469 Cause BCS
(A) Sequence chromatogram of a control subject (top) and a Tunisian patient with a homozygous deletion of A at position 5943 (bottom).
(B) Sequence chromatogram of a control subject (top) and a Palestinian patient with a homozygous deletion of G at position 9527 (bottom).
(C) SmaI restriction digests of the Tunisian Jewish mutation. In the carrier chromosome, the 533bp product is cleaved to yield 388 bp and 145 bp fragments. Molecular-weight marker (M), homozygotes (lanes 1 and 2), heterozygotes (lanes 3 and 4), and control subjects (lanes 5 and 6).
(D) ABI 3100 assay for the detection of the Palestinian mutation. The 1bp difference is evident in homozygotes (1 and 2) compared to controls (5 and 6). Heterozygotes appear as two peaks (3 and 4).
Figure 5.
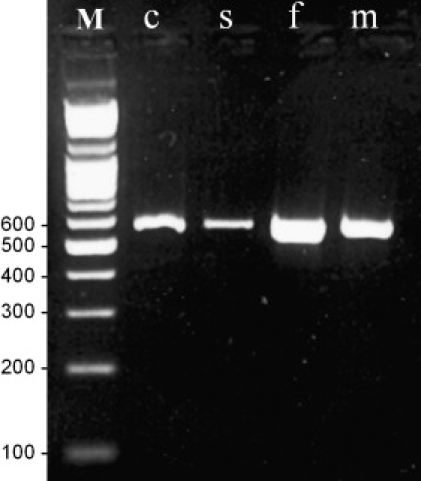
ZNF469-Expression Studies
The 559bp RT-PCR product in cornea (c), sclera (s), skin fibroblasts (f) and striated muscle (m). M denotes molecular-weight marker.
ZNF469, also known as KIAA1858, is composed of two very large exons. The predicted mRNA spans 13,229 bp and encodes a protein of 3925 amino acids. ZNF469 has orthologues in Pan troglodytes (95.7%), Macaca mulatta (90.4%), Mus musculus (56.5%), Canis familiaris (56.2%) and Rattus norvegicus (53.7%), but not in C. elegans and S. cerevisiae, suggesting a relatively late evolutionary development. The protein contains three zinc-finger domains (type C2H2) located toward its 3′ end. Zinc fingers (ZNFs) are extremely abundant in eukaryotes and have been classified into several groups according to their structure. Once considered to function exclusively as sequence-specific DNA-binding motifs, ZNFs are now known to have additional activities, such as the recognition of RNAs and proteins. The C2H2 domains classify this protein as a classical ZNF.13 In addition to their DNA-binding properties, classical ZNFs have been associated with protein-to-protein interactions, and therefore they may be found outside the nucleus.
We found that ZNF469 has a 30% homology to the helical parts of COL1A2 [MIM 120160], COL4A1 [MIM 120130], and COL1A1 [MIM 120150] (Figure 6), all of which are highly expressed in the cornea. Organized as orthogonal lamellae with small, homogeneous diameters and regular packing, collagen fibrils are a major component of the normal cornea.14 The transparency and strength of the cornea require the maintenance of this structural organization, as well as the precise regulation of fibril and matrix assembly. ZNF469 could act either as a nuclear transcription factor or as an extra-nuclear regulatory molecule involved in the synthesis and/or organization of these collagen fibers.
Figure 6.

Partial Homology of ZNF469 to COL1A1
The Smith-Waterman algorithm was used to calculate local alignment (EMBOSS).
Top line: ZNF469.
Mutations in collagen genes and collagen-associated genes might lead to various corneal diseases. These could involve different corneal layers, result in mild or severe visual impairments, be stationary or progressive, and can affect corneal shape. For example, significant corneal thinning has been documented in both classic Ehlers-Danlos syndrome (EDS) type I [MIM 130000] and type II [MIM 130010] patients and in mouse models with COL5A1 [MIM 120215] and COL5A2 [MIM 120190] alterations.15 As many as nine clinically distinct dystrophies affecting the middle corneal layer (stroma) result from mutations in the TGFBI gene, which is involved in collagen metabolism and arrangement.16 Posterior-corneal-layer dystrophies, such as Fuchs endothelial [MIM 136800] and posterior polymorphous [MIM 609140] dystrophies, are associated with COL8A2 mutations.17,18
Of special interest is autosomal-recessive EDS type VI [MIM 225400], which is often confused with BCS. This disease, characterized by rupture of the eye globe, joint hypermobility, and lax skin, is caused by mutations in the lysyl hydroxylase gene, located on chromosome 1p36.22. Use of polymorphic markers very close to the lysyl hydroxylase locus enabled us to rule out this gene as the cause of the disease in the Palestinian family (data not shown), clearly differentiating BCS from EDS VI.
Interestingly, a thin and bulging cornea is a major finding in keratoconus [MIM 148300]. The inheritance of keratoconus is heterogeneous and complex, but in many families it runs as an autosomal-dominant trait.19 Tyynismaa et al.20 assigned a keratoconus locus to chromosome 16q22.3–q23.1, close to the location of ZNF469, in a panel of Finnish families. It is conceivable that some keratoconus patients could harbor mutations in ZNF469.
BCS shares a number of common clinical features with osteogenesis imperfecta (OI [MIM 166200]), including thin cornea and blue sclera, conductive-hearing loss, elastic skin, and dental anomalies. Most but not all OI patients have mutations in COL1A1 and COL1A2.21 ZNF469 is an appealing candidate in OI patients with no mutations in these two genes.
Identification of the BCS gene will provide insight into molecular pathways involved in the development of the normal cornea and connective tissues and could shed light on the pathogenesis of disorders that implicate these organs.
Acknowledgments
This research was supported by The Israel Science Foundation (grant no. 317/06). The study was performed in partial fulfillment of the requirements for the Ph.D. degree of A.A. from Bar Ilan University, Ramat Gan, Israel. We thank the patients and families for their participation, and we thank Y. Corsia from the Ministry of Welfare for his help.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/GenBank/
Ensembl, http://www.ensembl.org/
ExPASy Proteomics Server, http://www.expasy.org/
UniGene, http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene
Map Viewer, http://www.ncbi.nlm.nih.gov/mapview
Primer 3, http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi
SNP database (dbSNP), http://www.ncbi.nlm.nih.gov/projects/SNP/
NEBcutter V2.0, http://tools.neb.com/NEBcutter2/index.php
Stanford SOURCE, http://smd-www.stanford.edu/cgi-bin/source/sourceSearch
Mouse Genome Informatics (MGI), http://www.informatics.jax.org/
EMBOSS, http://bioinfo.hku.hk/EMBOSS/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Cyrillic.210, http://www.cyrillicsoftware.com/bottom.htm
The Genome Database, http://www.gdb.org/
References
- 1.Stein R., Lazar M., Adam A. Brittle cornea. A familial trait associated with blue sclera. Am. J. Ophthalmol. 1968;66:67–69. [PubMed] [Google Scholar]
- 2.Hyams S.W., Kar H., Neumann E. Blue sclerae and keratoglobus. Ocular signs of a systemic connective tissue disorder. Br. J. Ophthalmol. 1969;53:53–58. doi: 10.1136/bjo.53.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregoratos N.D., Bartsocas C.S., Papas K. Blue sclerae with keratoglobus and brittle cornea. Br. J. Ophthalmol. 1971;55:424–426. doi: 10.1136/bjo.55.6.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ticho U., Ivry M., Merin S. Brittle cornea, blue sclera, and red hair syndrome (the brittle cornea syndrome) Br. J. Ophthalmol. 1980;64:175–177. doi: 10.1136/bjo.64.3.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinhorst U., Kohlschutter A., Steinmann B., von Domarus D. Brittle cornea syndrome: A hereditary disease of connective tissue with spontaneous corneal perforation. Fortschr. Ophthalmol. 1988;85:659–661. [PubMed] [Google Scholar]
- 6.Zlotogora J., BenEzra D., Cohen T., Cohen E. Syndrome of brittle cornea, blue sclera, and joint hyperextensibility. Am. J. Med. Genet. 1990;36:269–272. doi: 10.1002/ajmg.1320360303. [DOI] [PubMed] [Google Scholar]
- 7.Tucker D.P. Blue sclerotics syndrome simulating buphthaimos. Am. J. Ophthalmol. 1959;47:345–348. doi: 10.1016/s0002-9394(14)76536-5. [DOI] [PubMed] [Google Scholar]
- 8.Arkin W. Blue sclera with keratoglobus. Am. J. Ophthalmol. 1964;58:678–682. doi: 10.1016/0002-9394(64)91389-3. [DOI] [PubMed] [Google Scholar]
- 9.Judisch G.F., Waziri M., Krachmer J.H. Ocular Ehlers-Danlos syndrome with normal lysyl hydroxylase activity. Arch. Ophthalmol. 1976;94:1489–1491. doi: 10.1001/archopht.1976.03910040323006. [DOI] [PubMed] [Google Scholar]
- 10.Al-Hussain H., Zeisberger S.M., Huber P.R., Giunta C., Steinmann B. Brittle cornea syndrome and its delineation from the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VI): report on 23 patients and review of the literature. Am. J. Med. Genet. A. 2004;124:28–34. doi: 10.1002/ajmg.a.20326. [DOI] [PubMed] [Google Scholar]
- 11.Royce P.M., Steinmann B., Vogel A., Steinhorst U., Kohlschuetter A. Brittle cornea syndrome: a heritable connective tissue disorder distinct from Ehlers-Danlos syndrome type VI and fragilitas oculi, with spontaneous perforations of the eye, blue sclerae, red hair, and normal collagen lysyl hydroxylation. Eur. J. Pediatr. 1990;149:465–469. doi: 10.1007/BF01959396. [DOI] [PubMed] [Google Scholar]
- 12.Abu A., Frydman M., Marek D., Pras E., Stolovitch C., Aviram-Goldring A., Rienstein S., Reznik-Wolf H., Pras E. Mapping of a gene causing brittle cornea syndrome in Tunisian jews to 16q24. Invest. Ophthalmol. Vis. Sci. 2006;47:5283–5287. doi: 10.1167/iovs.06-0206. [DOI] [PubMed] [Google Scholar]
- 13.Gamsjaeger R., Liew C.K., Loughlin F.E., Crossley M., Mackay J.P. Sticky fingers: zinc-fingers as protein-recognition motifs. Trends Biochem. Sci. 2007;32:63–70. doi: 10.1016/j.tibs.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Ihanamäki T., Pelliniemi L.J., Vuorio E. Collagens and collagen-related matrix components in the human and mouse eye. Prog. Retin. Eye Res. 2004;23:403–434. doi: 10.1016/j.preteyeres.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Segev F., Héon E., Cole W.G., Wenstrup R.J., Young F., Slomovic A.R., Rootman D.S., Whitaker-Menezes D., Chervoneva I., Birk D.E. Structural abnormalities of the cornea and lid resulting from collagen V mutations. Invest. Ophthalmol. Vis. Sci. 2006;47:565–573. doi: 10.1167/iovs.05-0771. [DOI] [PubMed] [Google Scholar]
- 16.Klintworth G.K. The molecular genetics of the corneal dystrophies – current status. Front. Biosci. 2003;8:d687–d713. doi: 10.2741/1018. [DOI] [PubMed] [Google Scholar]
- 17.Gottsch J.D., Sundin O.H., Liu S.H., Jun A.S., Broman K.W., Stark W.J., Vito E.C., Narang A.K., Thompson J.M., Magovern M. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of fuchs corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 2005;46:1934–1939. doi: 10.1167/iovs.04-0937. [DOI] [PubMed] [Google Scholar]
- 18.Biswas S., Munier F.L., Yardley J., Hart-Holden N., Perveen R., Cousin P., Sutphin J.E., Noble B., Batterbury M., Kielty C. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 2001;10:2415–2423. doi: 10.1093/hmg/10.21.2415. [DOI] [PubMed] [Google Scholar]
- 19.Brancati F., Valente E.M., Sarkozy A., Fehèr J., Castori M., Del Duca P., Mingarelli R., Pizzuti A., Dallapiccola B. A locus for autosomal dominant keratoconus maps to human chromosome 3p14-q13. J. Med. Genet. 2004;41:188–192. doi: 10.1136/jmg.2003.012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tyynismaa H., Sistonen P., Tuupanen S., Tervo T., Dammert A., Latvala T., Alitalo T. A locus for autosomal dominant keratoconus: linkage to 16q22.3-q23.1 in Finnish families. Invest. Ophthalmol. Vis. Sci. 2002;43:3160–3164. [PubMed] [Google Scholar]
- 21.Martin E., Shapiro J.R. Osteogenesis imperfecta:epidemiology and pathophysiology. Curr. Osteoporos. Rep. 2007;5:91–97. doi: 10.1007/s11914-007-0023-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.