

Abstract
Linkage analysis and DNA sequencing in a family exhibiting an X-linked mental retardation (XLMR) syndrome, characterized by microcephaly, epilepsy, ataxia, and absent speech and resembling Angelman syndrome, identified a deletion in the SLC9A6 gene encoding the Na+/H+ exchanger NHE6. Subsequently, other mutations were found in a male with mental retardation (MR) who had been investigated for Angelman syndrome and in two XLMR families with epilepsy and ataxia, including the family designated as having Christianson syndrome. Therefore, mutations in SLC9A6 cause X-linked mental retardation. Additionally, males with findings suggestive of unexplained Angelman syndrome should be considered as potential candidates for SLC9A6 mutations.
Main Text
In 2001, 33 X-linked mental retardation (XLMR) genes had been cloned, of which 26 related to syndromal conditions (syndromal X-linked mental retardation [MRXS]).1 Since then, a steadily increasing number of novel genes have been identified, both in MRXS and in nonsyndromal (nonsyndromic X-linked mental retardation [MRX]) forms, with the most up-to date information available via two XLMR-related websites (see Web Resources). At present, 82 cloned XLMR genes are known; 38 MRXS, 16 MRX, and 28 related to neuromuscular conditions.2 In the present work, we show that mutations in SLC9A6 give rise to an X-linked mental retardation syndrome in which the clinical picture overlaps with that of Angelman syndrome (OMIM 105830).
Severe developmental delay with absent or minimal use of words, ataxia, and a happy demeanor with disposition for frequent smiling or spontaneous laughter are typical findings in Angelman syndrome.3 Seizures, usually before three years of age, are seen in more than 80% of cases, and an abnormal electroencephalogram (EEG) with characteristic patterns of slow- and high-voltage wave rhythms are seen in almost all cases.4,5 An open mouth with frequent drooling, abnormal food-related behavior, and flexed arms occur commonly. Clinical symptoms are usually not present at birth; delayed growth in head circumference often results in microcephaly by age two years.
The prevalence of Angelman syndrome has been estimated to vary between 1:15,0006,7 and 1:40,000.8,9 Although the principal genetic mechanisms interfere with the expression of UBE3A on chromosome 15q11-q13, approximately 10%–15% of cases with Angelman syndrome are apparently unrelated to changes in the 15q11-q13 region.10 In some of these cases, other conditions mimicking Angelman syndrome have been diagnosed;11 such as Rett syndrome, for example.12 On the basis of the findings given below, we suggest that mutations of SLC9A6, encoding the Na+/H+ exchanger protein NHE6, should be added to the list of alternative genetic mechanisms causing an Angelman syndrome-like phenotype.
We examined three affected males in the same family (Figure 1A), all of whom exhibited profound developmental delay, seizures, ataxia, flexed arms, hyperkinetic behavior, and a happy demeanor with frequent smiling and occasional spontaneous laughter. Initially, they had been suspected of having Angelman syndrome, but relevant genetic examinations regarding this diagnosis had been negative. Normal methylation and negative linkage to the critical 15q11-q13 region, as well as a normal sequence of UBE3A, suggested a genetic cause unrelated to chromosome 15q. Because the pedigree was consistent with X-linked inheritance, multipoint parametric linkage analysis was performed with the three affected males and seven unaffected relatives. A panel of 48 polymorphic microsatellite markers, spanning the X chromosome with an average distance of 3.9 cM, was genotyped. Linkage analysis was performed with GENEHUNTER-PLUS13 assuming fully penetrant X-linked recessive inheritance and a frequency of 0.0001 of the disease allele. A maximum LOD score of 1.50 was observed in the Xq24-q27.3 interval, and haplotype analysis identified that the closest recombination breakpoints limited the interval to a 23.31 cM (26.7 Mb) region containing 141 known genes.
Figure 1.

Inheritance of Phenotypes Mimicking Angelman Syndrome Attributed to SLC9A6 Mutations
(A) Pedigree of a Norwegian family linked to Xq24-q27.3. Affected males are indicated by filled squares, carrier females by circles containing a dot. The pictures show individual III-5 (left), aged two years, and individual III-4 (right), aged eight years, demonstrating tendency towards flexed arms, open mouth with drooling, and a happy disposition.
(B) Pedigree of the Swedish family. Individual III-1, aged six years, demonstrates a happy disposition and an open mouth with drooling. (C) Pedigree of the UK family with picture of individual II-5, aged three years, demonstrating an open mouth with drooling and II-1, aged 20 years, with severe kyphoscoliosis.
In (B) and (C), only individuals marked with an asterisk were tested as part of this investigation.
Candidate-gene selection was aided by four freely available computer programs designed to guide prioritization.14 A higher priority was assigned to genes identified by more than one method. Coding exons and exon-intron boundaries were sequenced for the following genes: GRIA3, GLUD2, UBE2A, SLC9A6, RBMX, FGF13, THOC2, ODZ1, and ZNF449 with an Applied Biosystems (Foster City, CA) 3730 DNA Analyzer. A 6 bp deletion within the coding sequence of the SLC9A6 gene, localized to Xq26.3, was found to segregate with affected males and carrier females. This deletion results in the loss of two highly conserved amino acid residues (p.E255_S256 del) from the Na+/H+ exchanger domain of the encoded NHE6 polypeptide (see Figure S1, available online), which shows tissue-wide expression.15 A hydrophobicity plot of the NHE6 polypeptide places the deleted residues in a region of only moderate transmembrane potential (data not shown), but homology to other experimentally studied homologous NHE isoforms would suggest that the deletion falls within a transmembrane domain.16 Importantly, the deleted residues have been shown to be essential for ion transport in the related Na+/H+ exchangers NHE1 and NHE8.17–19 Examination of X chromosomes from 278 normal individuals (males and females) for the presence of the 6 bp deletion was negative, indicating that this sequence variant was not a common polymorphism.
Further Families Included for Study. In addition to the original Norwegian family, we sequenced DNA for mutations in SLC9A6 in male probands from five Norwegian, 14 Swedish, one UK, and 50 American families. The Norwegian, Swedish, and UK cases had an Angelman syndrome-like phenotype and had tested negative for 15q findings and MECP2 mutations. The criterion for inclusion of the American cases was that they had tested negative for 15q involvement related to Angelman syndrome. An unknown number had also been tested for MECP2 mutations. A further XLMR family was obtained from the IGOLD (International Genetics of Learning Disability; see Web Resources) sequencing study, an independent XLMR project.
An SLC9A6 mutation was found in a male patient from Sweden (Figure 1B). The mutation creates an Opal stop codon that prematurely truncates the predicted protein (p.R468X), removing the final transmembrane domain and C terminus. Alternatively, the stop codon could also initiate a nonsense-mediated mRNA decay.20 A splice-site mutation causing skipping of exon 3 (p.V144_R169 del) was identified in a UK family (Figure 1C) from the IGOLD sequencing study. This mutation removes the entire predicted fourth transmembrane domain of the protein,15 and will therefore likely affect the folding of the protein within the membrane.
We also examined DNA from two affected males of a South African family of Afrikaans-speaking origin previously described and published by Christianson et al.21 (Christianson syndrome, OMIM 300243), and we identified a 2 bp deletion. This family was included because it showed linkage of an overlapping interval of the Xq24-Xq27 region and shared several clinical features with the Norwegian family. The deletion is predicted to introduce a frameshift, truncating the NHE6 polypeptide at residue 171 (p.H171fs) and introducing 59 residues of nonsense-derived sequence. All the SLC9A6 mutations are detailed in Figure 2. In addition, an alignment showing the mutations in the SLC9A6 protein in the context of orthologous multiple-sequence conservation is presented in Figure S1. Primer sequences used to amplify and sequence SLC9A6 exons can be provided on request.
Figure 2.
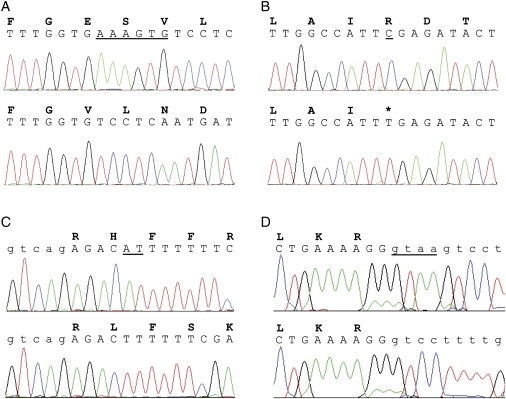
Mutations Found in SLC9A6
Sequence traces of wild-type (upper) and patient (lower) DNA, with corresponding amino acid single-letter codes. Sequences shown are from affected male patients from (A) Norwegian (Figure 1A), (B) Swedish (Figure 1B), (C) South African (Christianson et al, 199921), and (D) UK (Figure 1C) families. Intronic DNA sequences are shown in lower case. Deleted or altered bases are underlined.
Phenotypic Features. Table 1 summarizes the phenotypic features and the corresponding mutations in the four families. In the Norwegian family, the three affected males in Figure 1A (III-2, born 1998; III-4, born 1996; and III-5, born 2002) appeared normal at birth. Deceleration of head growth occurred in the first year of life. At the last examination, their occipitofrontal head circumferences (OFCs) ranged between 2 and 4 cm below the 2.5th percentile. Epilepsy occurred between nine and 26 months. All three cases exhibited a happy demeanor with frequent smiling and episodes of unprovoked laughter, sometimes lasting for several hours, particularly at night. A video film (of cases III-2 and III-5) displaying ataxia, hyperkinetic movements, and smiling is provided online as Supplemental Data. The deep tendon reflexes were hyperactive, and the plantar responses were inverted. Other notable features included an open mouth, profuse drooling, swallowing difficulties, and frequent regurgitations due to gastroesophageal reflux. They had a thin body habitus; weight in relation to height ranged from the 10th percentile to below the 2.5th percentile. Case III-4 was successfully treated for standard-risk acute lymphatic leukemia between 6 and 8½ years of age. Case III-2 was transiently treated for systemic hypertension of unknown cause. He has been investigated for hirsutism and hyponatremia, suggesting an underlying endocrinological defect such as congenital adrenal hyperplasia. Recently, he was found to have a V281L mutation in CYP21A2, the gene most commonly associated with 21-hydroxylase deficiency (OMIM 201910). At present, we do not know if he is compound heterozygote or if only one allele is affected. In the latter case, we could speculate that the SLC9A6 mutation precipitated his endocrinological symptoms.
Table 1.
Phenotypic Findings in Four Families with SLC9A6 Mutations
| Affected Males: Total Number (Examined) | Family 1 Norway: 3 (3) | Family 2 Sweden: 2 (1) | Family 3 UK: 3 (3) | Family 4 South Africa21: 16 (4) |
|---|---|---|---|---|
| Development and Behavior | ||||
| Profound delay | + | + | + | + |
| Verbal language absent | + | + | + | + |
| Easily provoked laughter | + | + | + | − |
| Sleep disturbance | 1/3 | + | 1/3 | NR |
| CNS Findings | ||||
| Epilepsy | + | + | + | + |
| Ataxia | + | + | + | + |
| Hyperkinetic movements | 2/3 | − | + | − |
| Squint | + | + | + | + |
| Physical Findings | ||||
| Microcephaly | + | 2.5th percentile | + | 3/4 |
| Open mouth + drooling | 2/3 | + | + | NR |
| Swallowing difficulty | 2/3 | + | 1/3 | 1/4 |
| Flexed arms | + | NR | 1/3 | + |
| Electroencephalography | ||||
| Epileptiform activity | + | + | + | + |
| Background frequency | 10-11 Hz | 1.5-3 Hz | 4-7 Hz | 3-6 Hz to 11-14 Hz |
| Brain MRI/Autopsy | ||||
| Cerebellar atrophy | 1/3 | NR | NR | 2/4 |
| Increased glutamate | 1/3 | NR | NR | NR |
| Mutation | p.E255_S256del c.764_769delAAAGTG | p.R468X c.1402C→T | p.V144_R169del c.507 + 1delGTAA | p.H171fs c.512_513delAT |
All DNA mutations relate to cDNA reference sequence NM_006359.
+ indicates “present in all examined cases”; “−“ indicates “not present”; MRI denotes “magnetic resonance imaging”; NR denotes “not recorded.”
In the Swedish family (Figure 1B), only case III-1 was available for clinical examination. He was born at term, with normal weight and height. His general development was delayed, and he never established proper eye contact or obtained an expressive verbal language. He learned to walk independently at 18 months. Epilepsy was diagnosed at 16 months, with multiple seizure types: generalized tonic clonic seizures (GTCs), tonic seizures, and attacks of complex motor seizures with hand and jaw movements, accompanied by autonomic nervous system symptoms such as dilatation of the pupils. Other features include a happy demeanor with frequent smiling and outbursts of laughter (day and night), an open mouth and frequent drooling, and mild kyphosis. Although he can move relatively quickly on a flat surface, his gait is unsteady because of ataxia. Deep tendon reflexes were normal, and there was no spasticity.
In the UK family, all three half siblings had similar facial features, with a long face, pointed jaw with profuse dribbling, absent speech, poor growth parameters, and variable ambulatory capacity (Figure 1C). They exhibited a happy demeanor with frequent smiling. The oldest patient (case II-1), age 25 years old, had developed a severe kyphoscoliosis. He was thin, with little subcutaneous fat. Muscle strength and tendon reflexes were regarded as normal.
Of the 16 affected males in the pedigree published by Christianson et al.,21 a clinical appraisal was presented for four affected males aged 10, 20, 24, and 37 years. Their features included profound mental retardation, microcephaly (n = 3), epilepsy, ataxia, absence of expressive verbal language, impaired eye movements (ophthalmoplegia), and squint. They had some craniofacial dysmorphism (long narrow face, long nose, and prognathic jaw) and decreased muscle bulk. A slow regression had been noted; for example, the ability to walk had been lost with advancing age. The youngest, the only patient who was ambulatory, displayed a friendly demeanor.
EEG and Neuroradiological Examinations. EEG examinations were performed in all affected individuals in the Norwegian family (Table 1). Apart from frequent epileptiform activity, the EEG recordings showed repeated runs of approximately 10 Hz activity (data not shown) before two years of age, an abnormally fast rhythm considering their young age.22
For Norwegian patient III-2 (see Figure 1A and Table 1), a magnetic resonance imaging (MRI) examination was performed at four and seven years of age. The examinations included sagittal T1-weighted spin-echo, transverse and coronal T2-weighted, inversion recovery T1-weighted, and fluid-attenuated inversion recovery images. Progressive cerebellar atrophy was evident during the period that elapsed between the examinations (Figures 3A and 3B). Proton magnetic resonance spectroscopy (MRS) was also performed at seven years of age in Norwegian patient III-2. Single-voxel MRS of the brain was performed with a point-resolved spectroscopy sequence with chemical-shift water suppression (repetition time [TR] 1500 ms, echo time [TE] 30 ms). Volumes of interest were located in the left parietal white matter and in the right basal ganglia. The interpretation of the MRS study was limited because consent was only given for examination of a single patient and we were unable to use absolute metabolite concentrations. Nonetheless, it suggested increased levels of glutamate-glutamine complex in the basal ganglia (Figures 3C and 3D).
Figure 3.
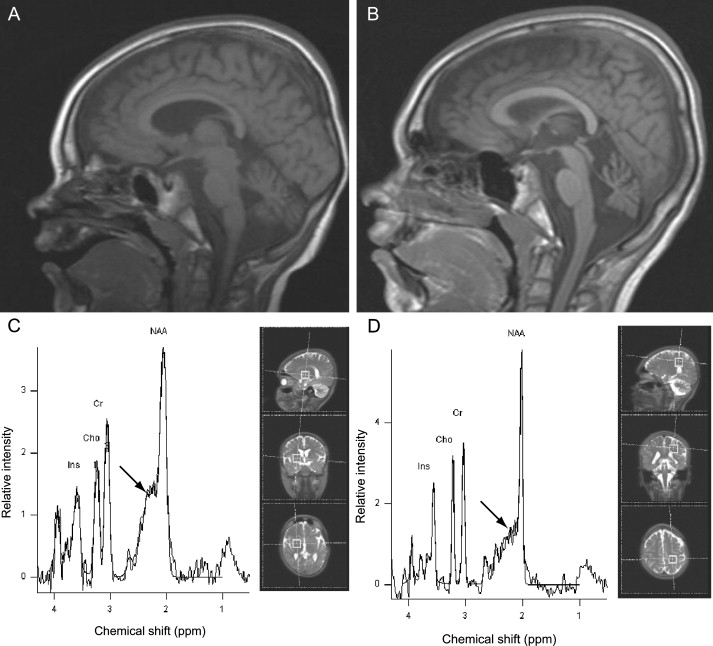
Neuroradiological Examinations
(A and B) Magnetic Resonance Imaging (MRI). Saggital T1-weighted MRI examination of the brain in Norwegian patient III-2 (see Figure 1A) at age four years (0.5 Tesla) (A) and seven years (1.5 Tesla) (B), showing progressive cerebellar atrophy.
(C and D) MR spectra (TE = 30 ms), also from Norwegian patient III-2 at seven years of age. Spectral peaks for inositol (Ins), choline (Cho), creatine (Cr), and N-acetylaspartate (NAA) are indicated. Increased amounts of the glutamate-glutamine complex (arrow) are suggested by the increased glutamate-glutamine:Cr ratio in the basal ganglia (C), whilst normal metabolite levels were seen in deep cerebral white matter (D). The illustrations to the right in the panels indicate the position of the voxels of interest (dimensions 2 × 2 × 2 cm).
Na+/H+ Exchange. The Na+/H+ exchanger (NHE) protein family is composed of at least nine members, with members NHE1-5 found in the plasma membrane and members NHE6-9 in the membranes of intracellular organelles.23 The NHE proteins consist of an N-terminal domain of 10–12 transmembrane helices mediating ion exchange, and a regulatory hydrophilic C-terminal domain. There exist at least two alternatively spliced variants of the SLC9A6 gene,24 although more appear likely according to current database annotations. NHE6 is found in early recycling endosomal membranes and transiently associates with the plasma membrane.25 In endosomes, NHE6 is suggested to have a role in regulating lumen pH.19,25 Thus, a possible consequence of NHE6 inactivity could be lowered endosomal pH and decreased monovalent ion content, both of which might affect protein folding and trafficking.26
Recycling endosome trafficking is essential for growth of dendritic spines during long-term potentiation (LTP),27 a process considered to be the molecular basis of learning and memory.28 Therefore, if mutations in SLC9A6 impair endosomal trafficking, LTP is likely to be affected. Interestingly, abnormalities in synaptic development and/or plasticity were shown to be involved in the pathogenesis of Angelman syndrome.29
Previous studies have shown that the NHE6 protein colocalizes with internalized transferrin and early endosomal antigen 1 (EEA1) in COS7 cells, suggesting that the protein is harbored in early endosomes.19 In order to explore the intracellular function of NHE6, we used cultured fibroblasts from patient III-2 (Figure 1A) and two unrelated controls. By using real-time PCR and NHE6-specific primers, we verified that NHE6 was expressed in all three cell lines at a level similar to that observed in HeLa cells (data not shown).
We used confocal immunofluorescence microscopy analysis with endosomal markers to assess endosomal morphology, size, and distribution. Fibroblast cells grown on coverslips were permeabilized with 0.05% saponin, fixed with 3% paraformaldehyde, and stained for fluorescence microscopy as described.30 Coverslips were analyzed with a Zeiss LSM 510 META confocal microscope equipped with a Plan-Apochromat X63/1.4 and Neo-Fluar X100/1.45 oil-immersion objectives. Appropriate emission-filter settings were included in order to exclude bleed-through effects. Endosomal morphology, size, and distribution were assessed with antibodies against the endosomal markers EEA1, CD63, and lyso-bisphosphatidic acid (LBPA), and aggregation of ubiquitylated proteins was assessed with antibodies against ubiquitin (FK2, Affiniti). However, we did not observe any large-scale endosomal aberrations in fibroblasts from the patient compared with controls (Figure S2). We therefore evaluated vesicular pH by means of LysoTracker, a dye which fluoresces in an acidified milieu normally found within the lumens of endosomes and lysosomes. Fibroblasts from the patient and the controls were incubated with 250 nM LysoTracker Red (Molecular Probes) for 5 min. Live cells were analyzed immediately by fluorescence microscopy to quantify the amount of LysoTracker-positive structures. Between 20 and 30 cells were quantified from each sample and scanned at fixed-intensity settings below pixel-value saturation. The images were processed with the histogram function in the Zeiss LSM software. However, no differences were visible between the patient's and the controls' cells (Figure S2). It remains possible that the fibroblasts used in our analysis might not exhibit aberrations at the endosomal level for cell-type-specific reasons, such as functional redundancy with other NHE family members expressed in fibroblasts but not in brain cells.
Putative Consequences of SLC9A6 Mutations. Ataxia, spontaneous seizures, and growth retardation have been observed in the related Nhe1 null mutant mouse.31 Notably, mutant Slc9a6 −/− female and mutant Slc9a6 −/0 male mice show motor hyperactivity and a lowered threshold for pharmacologically induced seizures (see Deltagen under Web Resources) consistent with the phenotypic abnormalities described in our patients. The complete function of this cation exchanger and how an impaired function can cause a phenotype similar to Angelman syndrome remain to be explored. Both UBE3A and SLC9A6 are involved in the intracellular protein-processing apparatus: UBE3A in protein ubiquitylation, and SLC9A6 in regulating endosomal pH and Na+ concentrations. However, how or if they interact via a common pathway is currently unclear.
Nonetheless, phenotypic features overlapping with Angelman syndrome were evident to the clinical investigators of both the Swedish patient (M. Ky.) and the Norwegian patients (P.S.). On the basis of the four families included in the present study, we propose that the “SLC9A6-opathies” encompass a clinical spectrum of feature clusters ranging from those typical of Angelman syndrome to those typical of Christianson syndrome. However, the most Angelman syndrome-like patients (III-2, III-4, and III-5 in Figure 1A; II-1 in Figure 1B; and II-5 in Figure 1C) were also the youngest (<10 years), whereas three of the patients described by Christianson et al.21 were ≥ 20 years of age. These patients had gradually lost their ability to walk. The youngest patient, a 10-year-old boy, was ambulatory with support and was the only one described to be “friendly and approachable.” This could indicate that SLC9A6 mutations cause a slow progression of symptoms and that resemblance to Angelman syndrome is more striking at a younger age. Cerebellar atrophy, demonstrated on MRI examination in one of our patients (see Figure 3), was reported on autopsy in another case described by Christianson et al.21 Developmental regression and cerebellar atrophy could indicate SLC9A6 mutation rather than 15q-related Angelman syndrome. However, the clinical picture in Angelman syndrome can progress as some adult patients lose their ability to walk and develop spinal deformities.32,33 Interestingly, these features were prominent in the oldest UK patient (II-1 in Figure 1C).
Almost all patients with SLC9A6 mutations were observed to be very thin. This is in contrast to the symptom of Angelman syndrome in which older patients tend to become obese.33 Another distinguishing feature was the rapid background frequency of 10–14 Hz on EEG observed in some patients (see Table 1). A slow frequency of 1.5–3 Hz with high voltage (300 mV), an EEG pattern observed in Angelman syndrome,34 was observed only in the Swedish patient.
We speculate that the patients' phenotype might result, at least in part, from glutamate-glutamine accumulation (see Figure 3) as an indirect consequence of SLC9A6-mutation-initiated loss of function. Glutamate is excitotoxic at high levels and has been associated with a variety of clinical neurological abnormalities, including epilepsy and cerebellar degeneration.35 Notably, changes in pH have been shown to affect glutamine transport,36 which, as discussed above, could be affected by a loss of NHE6 function. Alternatively, aberrant glutamate-glutamine transporter trafficking could underlie the elevated levels observed by proton MRS (Figure 3C).
Carrier Females. In the family described by Christianson et al.,21 there were ten female carriers of whom three were either mentally retarded or had learning problems. None of the carriers displayed selective X inactivation. In the Norwegian family, carrier females II-2 and II-4 (Figure 1A) were available for assessment. They did not display clinical symptoms. An EEG examination of II-4 was normal. X-inactivation analysis was performed on DNA from lymphocytes37 of all three carrier females in the family, and all showed X-inactivation ratios that were normal for their age. Female carrier II-5 in the Swedish family (Figure 1B) had severe dyslexia. Clinical symptoms were not reported in the carrier mother of the UK patients (Figure 1C). Therefore, the range of carrier phenotypes possible with SLC9A6 mutation could depend on the severity of the mutation, functional overlap with other solute carrier proteins, or genetic-modifier effects.
In conclusion, our study showed that mutations in SLC9A6 lead to a syndrome associated with microcephaly, seizures, ataxia, and absent speech. Some patients, particularly those who were in a younger age group, displayed symptoms resembling those of Angelman syndrome. We suggest that disruption of SLC9A6 should be considered in male patients with a non-15q11-13-related Angelman syndrome phenotype, particularly when X-linked inheritance in the family is suspected.
Acknowledgments
We thank the patients and their families for their cooperation. O. Flesvig and L. Tveten (Department of Habilitation, Innlandet Hospital, Norway) managed the habilitation program for the Norwegian patients. F.A. Chaudhry (Center for Molecular Biology and Neuroscience, University of Oslo, Norway) and J. Clayton-Smith (Department of Clinical Genetics, St Mary's Hospital, Manchester General, UK) contributed with valuable comments on this work. We thank B. Birkenes and K. Brandal for excellent technical assistance and R. Lyle (Department of Medical Genetics, Ullevål University Hospital, Oslo, Norway) for helpful suggestions. We are grateful to S. Mohammed (Clinical Genetics, Guy's Hospital, London, UK), W.B. Dobyns (Department of Human Genetics, University of Chicago, Chicago, USA), J. Rodriguez, and A. Srivastava (Greenwood Genetic Center, Greenwood, SC, USA) for providing DNA samples, and we thank Ban-Hock Toh (Monash University, Melbourne, Australia) and Jean Gruenberg (University of Geneva, Geneva, Switzerland) for gifts of antibodies. The study was financially supported by the Norwegian Eastern Regional Health Authorities, a National Institute of Neurological Diseases and Stroke (NINDS) grant (NS31564) (USA), a National Institute of Child Health and Human Development (NICHD) grant (HD2606) (USA), the Wellcome Trust (UK), and the South Carolina Department of Disabilities and Special Needs. None of the authors of this work has a financial interest related to this work.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Disease Gene Prediction, http://cgg.ebi.ac.uk/services/dgp/
Ensembl Genome Browser, http://www.ensembl.org
GeneSeeker, http://www.cmbi.ru.nl/GeneSeeker/
Greenwood Genetic Center XLMR website, http://ggc.org/XLMR.htm
International Genetics of Learning Disability (IGOLD) study, http://goldstudy.cimr.cam.ac.uk/
Online Mendelian Inheritance in man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
XLMR genes update website, http://xlmr.interfree.it/home.htm
References
- 1.Chiurazzi P., Hamel B.C., Neri G. XLMR genes: Update 2000. Eur. J. Hum. Genet. 2001;9:71–81. doi: 10.1038/sj.ejhg.5200603. [DOI] [PubMed] [Google Scholar]
- 2.Chiurazzi, P., Schwartz, C.E., Gecz, J., and Neri, G. XLMR genes: Update 2007. Eur. J. Hum. Genet. doi: 10.1038/sj.ejhg.5201994. [DOI] [PubMed]
- 3.Williams C.A., Beaudet A.L., Clayton-Smith J., Knoll J.H., Kyllerman M., Laan L.A., Magenis R.E., Moncla A., Schinzel A.A., Summers J.A. Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am. J. Med. Genet. A. 2006;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- 4.Williams C.A., Angelman H., Clayton-Smith J., Driscoll D.J., Hendrickson J.E., Knoll J.H., Magenis R.E., Schinzel A., Wagstaff J., Whidden E.M. Angelman syndrome: Consensus for diagnostic criteria. Angelman Syndrome Foundation. Am. J. Med. Genet. 1995;56:237–238. doi: 10.1002/ajmg.1320560224. [DOI] [PubMed] [Google Scholar]
- 5.Laan L.A., Vein A.A. Angelman syndrome: Is there a characteristic EEG? Brain Dev. 2005;27:80–87. doi: 10.1016/j.braindev.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Petersen M.B., Brondum-Nielsen K., Hansen L.K., Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: Estimated prevalence rate in a Danish county. Am. J. Med. Genet. 1995;60:261–262. doi: 10.1002/ajmg.1320600317. [DOI] [PubMed] [Google Scholar]
- 7.Stromme P., Hagberg G. Aetiology in severe and mild mental retardation: A population-based study of Norwegian children. Dev. Med. Child Neurol. 2000;42:76–86. doi: 10.1017/s0012162200000165. [DOI] [PubMed] [Google Scholar]
- 8.Thomson A.K., Glasson E.J., Bittles A.H. A long-term population-based clinical and morbidity profile of Angelman syndrome in Western Australia: 1953–2003. Disabil. Rehabil. 2006;28:299–305. doi: 10.1080/09638280500190631. [DOI] [PubMed] [Google Scholar]
- 9.Hou J.W., Wang T.R., Chuang S.M. An epidemiological and aetiological study of children with intellectual disability in Taiwan. J. Intellect. Disabil. Res. 1998;42:137–143. doi: 10.1046/j.1365-2788.1998.00104.x. [DOI] [PubMed] [Google Scholar]
- 10.Clayton-Smith J., Laan L. Angelman syndrome: A review of the clinical and genetic aspects. J. Med. Genet. 2003;40:87–95. doi: 10.1136/jmg.40.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams C.A., Lossie A., Driscoll D. Angelman syndrome: Mimicking conditions and phenotypes. Am. J. Med. Genet. 2001;101:59–64. doi: 10.1002/ajmg.1316. [DOI] [PubMed] [Google Scholar]
- 12.Jedele K.B. The overlapping spectrum of rett and angelman syndromes: A clinical review. Semin. Pediatr. Neurol. 2007;14:108–117. doi: 10.1016/j.spen.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Kong A., Cox N.J. Allele-sharing models: LOD scores and accurate linkage tests. Am. J. Hum. Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiffin N., Adie E., Turner F., Brunner H.G., van Driel M.A., Oti M., Lopez-Bigas N., Ouzounis C., Perez-Iratxeta C., Andrade-Navarro M.A. Computational disease gene identification: A concert of methods prioritizes type 2 diabetes and obesity candidate genes. Nucleic Acids Res. 2006;34:3067–3081. doi: 10.1093/nar/gkl381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Numata M., Petrecca K., Lake N., Orlowski J. Identification of a mitochondrial Na+/H+ exchanger. J. Biol. Chem. 1998;273:6951–6959. doi: 10.1074/jbc.273.12.6951. [DOI] [PubMed] [Google Scholar]
- 16.Wakabayashi S., Pang T., Su X., Shigekawa M. A novel topology model of the human Na(+)/H(+) exchanger isoform 1. J. Biol. Chem. 2000;275:7942–7949. doi: 10.1074/jbc.275.11.7942. [DOI] [PubMed] [Google Scholar]
- 17.Fafournoux P., Noel J., Pouyssegur J. Evidence that Na+/H+ exchanger isoforms NHE1 and NHE3 exist as stable dimers in membranes with a high degree of specificity for homodimers. J. Biol. Chem. 1994;269:2589–2596. [PubMed] [Google Scholar]
- 18.Murtazina R., Booth B.J., Bullis B.L., Singh D.N., Fliegel L. Functional analysis of polar amino-acid residues in membrane associated regions of the NHE1 isoform of the mammalian Na+/H+ exchanger. Eur. J. Biochem. 2001;268:4674–4685. doi: 10.1046/j.1432-1327.2001.02391.x. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura N., Tanaka S., Teko Y., Mitsui K., Kanazawa H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J. Biol. Chem. 2005;280:1561–1572. doi: 10.1074/jbc.M410041200. [DOI] [PubMed] [Google Scholar]
- 20.Nagy E., Maquat L.E. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 21.Christianson A.L., Stevenson R.E., van der Meyden C.H., Pelser J., Theron F.W., van Rensburg P.L., Chandler M., Schwartz C.E. X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J. Med. Genet. 1999;36:759–766. doi: 10.1136/jmg.36.10.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niedermeyer E., Lopes da Silva F.H. Lippincott Williams & Wilkins; Baltimore: 2005. Electroencephalography: Basic Principles, Clinical Applications, and Related Fields. [Google Scholar]
- 23.Brett C.L., Donowitz M., Rao R. Evolutionary origins of eukaryotic sodium/proton exchangers. Am. J. Physiol. Cell Physiol. 2005;288:C223–C239. doi: 10.1152/ajpcell.00360.2004. [DOI] [PubMed] [Google Scholar]
- 24.Miyazaki E., Sakaguchi M., Wakabayashi S., Shigekawa M., Mihara K. NHE6 protein possesses a signal peptide destined for endoplasmic reticulum membrane and localizes in secretory organelles of the cell. J. Biol. Chem. 2001;276:49221–49227. doi: 10.1074/jbc.M106267200. [DOI] [PubMed] [Google Scholar]
- 25.Brett C.L., Wei Y., Donowitz M., Rao R. Human Na(+)/H(+) exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. Am. J. Physiol. Cell Physiol. 2002;282:C1031–C1041. doi: 10.1152/ajpcell.00420.2001. [DOI] [PubMed] [Google Scholar]
- 26.Orlowski J., Grinstein S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 2004;447:549–565. doi: 10.1007/s00424-003-1110-3. [DOI] [PubMed] [Google Scholar]
- 27.Park M., Salgado J.M., Ostroff L., Helton T.D., Robinson C.G., Harris K.M., Ehlers M.D. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooke S.F., Bliss T.V. Plasticity in the human central nervous system. Brain. 2006;129:1659–1673. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- 29.Dindot S.V., Antalffy B.A., Bhattacharjee M.B., Beaudet A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 30.Simonsen A., Bremnes B., Ronning E., Aasland R., Stenmark H. Syntaxin-16, a putative Golgi t-SNARE. Eur. J. Cell Biol. 1998;75:223–231. doi: 10.1016/S0171-9335(98)80116-7. [DOI] [PubMed] [Google Scholar]
- 31.Bell S.M., Schreiner C.M., Schultheis P.J., Miller M.L., Evans R.L., Vorhees C.V., Shull G.E., Scott W.J. Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am. J. Physiol. 1999;276:C788–C795. doi: 10.1152/ajpcell.1999.276.4.C788. [DOI] [PubMed] [Google Scholar]
- 32.Laan L.A., den Boer A.T., Hennekam R.C., Renier W.O., Brouwer O.F. Angelman syndrome in adulthood. Am. J. Med. Genet. 1996;66:356–360. doi: 10.1002/(SICI)1096-8628(19961218)66:3<356::AID-AJMG21>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 33.Smith J.C. Angelman syndrome: Evolution of the phenotype in adolescents and adults. Dev. Med. Child Neurol. 2001;43:476–480. doi: 10.1017/s0012162201000871. [DOI] [PubMed] [Google Scholar]
- 34.Laan L.A., Renier W.O., Arts W.F., Buntinx I.M., vd Burgt I.J., Stroink H., Beuten J., Zwinderman K.H., van Dijk J.G., Brouwer O.F. Evolution of epilepsy and EEG findings in Angelman syndrome. Epilepsia. 1997;38:195–199. doi: 10.1111/j.1528-1157.1997.tb01097.x. [DOI] [PubMed] [Google Scholar]
- 35.Meldrum B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 36.Chaudhry F.A., Krizaj D., Larsson P., Reimer R.J., Wreden C., Storm-Mathisen J., Copenhagen D., Kavanaugh M., Edwards R.H. Coupled and uncoupled proton movement by amino acid transport system N. EMBO J. 2001;20:7041–7051. doi: 10.1093/emboj/20.24.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.