

Abstract
Autosomal-dominant arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) causes sudden cardiac death and is characterized by clinical and genetic heterogeneity. Fifteen unrelated ARVC families with a disease-associated haplotype on chromosome 3p (ARVD5) were ascertained from a genetically isolated population. Identification of key recombination events reduced the disease region to a 2.36 Mb interval containing 20 annotated genes. Bidirectional resequencing showed one rare variant in transmembrane protein 43 (TMEM43 1073C→T, S358L), was carried on all recombinant ARVD5 ancestral haplotypes from affected subjects and not found in population controls. The mutation occurs in a highly conserved transmembrane domain of TMEM43 and is predicted to be deleterious. Clinical outcomes in 257 affected and 151 unaffected subjects were compared, and penetrance was determined. We concluded that ARVC at locus ARVD5 is a lethal, fully penetrant, sex-influenced morbid disorder. Median life expectancy was 41 years in affected males compared to 71 years in affected females (relative risk 6.8, 95% CI 1.3–10.9). Heart failure was a late manifestation in survivors. Although little is known about the function of the TMEM43 gene, it contains a response element for PPARγ (an adipogenic transcription factor), which may explain the fibrofatty replacement of the myocardium, a characteristic pathological finding in ARVC.
Introduction
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D [MIM 107970]) is an inherited disorder, often involving both ventricles, and is characterized by ventricular tachycardia (VT), heart failure, sudden cardiac death (SCD), and fibrofatty replacement of cardiomyocytes.1 It is usually inherited as an autosomal-dominant disorder, although recessive forms exist.2 To date, eleven genetic loci have been mapped and seven genes have been identified.3–7 Five genes code for desmosomal proteins (desmoplakin [MIM 125647], plakophilin-2 [MIM 602861], desmoglein-2 [MIM 125671], desmocollin-2 [MIM 125645], and plakoglobin [MIM 173325]) that are predicted to succumb to mechanical stress.5,7 The remaining two genes are cardiac ryanadine receptor 2 (RYR2 [MIM 180902])8 and transforming growth factor beta-3 (TGFβ3 [MIM 190230]).9
Clinical diagnosis of ARVC is difficult because it relies on physiological and pathological testing for the presence of right ventricular structural and functional anomalies, fibrofatty replacement of the ventricular myocardium, premature ventricular contractions (PVCs) on Holter monitor, extended QRS, epsilon waves and T wave inversion on 12 lead ECG, and late potentials on signal-averaged ECG (SAECG).10 Phenotypic variation of ARVC, in both presentation and clinical course, has led to a modification of the McKenna diagnostic criteria10 to facilitate the identification of relatives at risk in ARVC families.11 ARVC can have both adverse (sudden death and heart failure)12 and favorable outcomes,13 which may be due to genetic heterogeneity, although clearly mutations at different loci do not explain intrafamilial variation14 or the sex influence toward greater clinical severity in males.15 There are no known studies to date that assess clinical features in mutation-negative subjects born at a priori 50% pedigree risk to determine mutation-specific cardiac features: Most studies assess small families to determine cardiac features and gene-specific (not mutation-specific) penetrance.16 Ultimately, the determination of the penetrance of mutation-specific clinical manifestations of ARVC will provide accurate, mutation-specific clinical information for appropriate genetic counseling and treatment options.
The ARVD5 locus (MIM 604400) on 3p was mapped in an extended eight generation family from the genetically isolated population of the island of Newfoundland, Canada.17 This family was first identified in the 1980s,18 and at least one family relative participated in right ventricular disconnection studies as a possible treatment for ARVC.19 We have identified 14 additional Newfoundland-ancestral families that share the ARVD5 haplotype and have previously shown that the use of prophylactic treatment with implantable cardioverter defibrillator (ICD) therapy in relatives at risk of ARVC greatly improved survival.20 We report here that we have identified the ARVD5 disease gene and determined the mutation-specific penetrance of its major clinical manifestations.
Subjects and Methods
Study Population
Fifteen of 150 unrelated families were referred either to the Newfoundland Provincial Medical Genetics Program or the Newfoundland Labrador genetics cardiomyopathy clinic because of a family history of cardiomyopathy and sudden death. These families were determined to have ARVC on the basis of clinical testing with established criteria,10 postmortem pathology, and an autosomal-dominant pattern of inheritance (Figures 1A and 1B). These families are characterized by deep genealogies, a Newfoundland ancestry, and a disease-associated haplotype originally identified in family AR1 used to map ARVD5 (Figure 1A).
Figure 1.
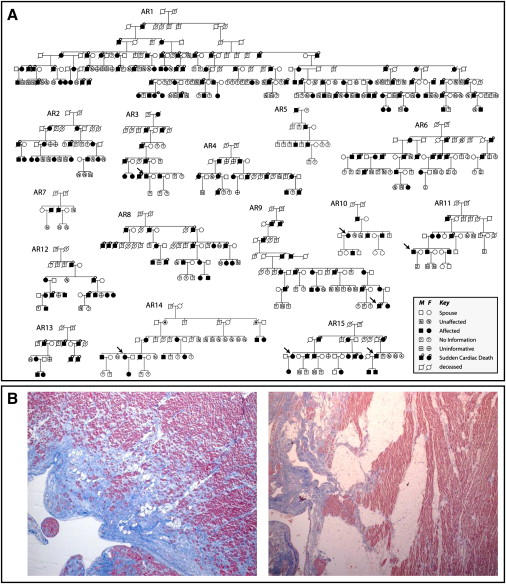
ARVC Families Linked to Chromosome 3p
(A) Pedigrees of 15 autosomal-dominant ARVC families linked to ARVD5. Affected subjects are shown as blackened squares (male) and circles (female). Subjects deceased because of SCD are noted by a circle above the symbol.
(B) Photomicrographs of paraffin-embedded postmortem right ventricular myocardium stained with masson trichrome showing fibrofatty replacement of myocytes from a male teenager after sudden cardiac death (left: 40× magnification) and a second-degree relative with sudden cardiac death in his eighth decade (right: 20× magnification). Pink represents normal myocardium, blue represents fiber, and white represents fat.
A total of 496 subjects born at a priori 50% risk (clinically affected and their first-degree relatives) were available for study across the 15 families. Subjects were determined to be clinically affected if they fulfilled the ARVC modified criteria11 (Figure 2). Blood samples from 295 subjects born at a priori 50% risk were collected, and informed consent was obtained in compliance with the Human Investigation Committee requirements of the Eastern Health Corporation of St. John's, Newfoundland, Canada (study number 00-176).
Figure 2.
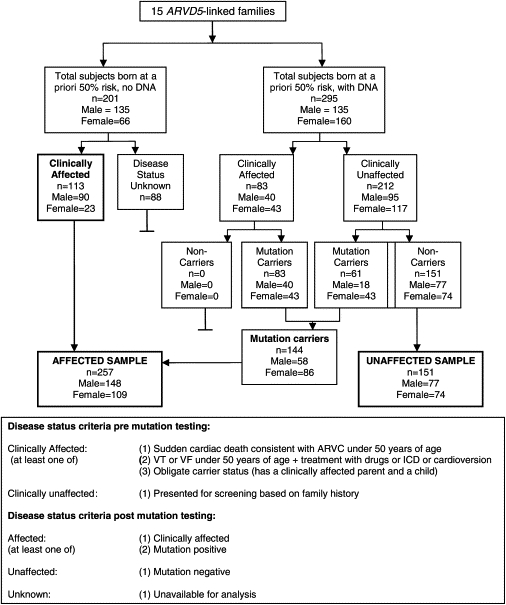
Workflow and Mutation Status of Subjects Born at a Priori 50% Risk of ARVC
Fine Mapping of the ARVD5 Locus
Mapping the ARVD5 locus in family AR1 (Figure 1A) previously defined a 9.3 cM disease region between markers D3S3610 and D3S3659.17 We genotyped 18 polymorphic microsatellite markers on clinically affected subjects across all families to identify a disease-associated haplotype at 3p25 that was shared among families from this genetic isolate and presumed to be ancestral. We identified key recombination events on the ARVD5 ancestral haplotype and used recombinations seen in two or more families to narrow the region.
Screening Candidate ARVD5 Genes
A mutation-screening panel was established that comprised seven genomic DNA samples from four clinically affected subjects from three families (AR1, AR8, and AR15; Figure 1A) and three spouses (controls). All coding and noncoding exons and intron-exon boundaries of positional candidate genes for ARVD5 were sequenced. All sequences were amplified by polymerase-chain reaction (PCR) assay from genomic DNA in 25 μl reaction volume. Primer sequences are available from the authors on request. The PCR products were purified with 50% sephacryl (Amersham Biosciences) and MultiScreen HTS filter plates (Millipore Corporation). Purified PCR products were cycle sequenced in both forward and reverse directions with the use of BigDye Terminator V3.1 cycle sequencing kit on an automated ABI 3700 DNA analyzer (Applied Biosystems). Sequencing electropherograms were inspected manually and analyzed with Mutation Surveyor software (Transition Technologies). Sequencing variants found exclusively in clinically affected subjects on the mutation-screening panel were experimentally determined to reside on the ARVD5 haplotype by segregation analysis in family AR14 (Figure 1A). The allele frequencies of the ARVD5 sequencing variants were determined with Newfoundland-population-based controls obtained through random phone dialing, as part of a large colorectal cancer study.21 Newfoundland is a known genetic isolate where 98% of its residents are of English and Irish descent.22,23 We analyzed key recombinant families (AR2 and AR10) to determine which rare variants (<1% of the alleles screened) were retained on recombinant ancestral haplotypes in clinically affected subjects.
Expression of TMEM43 in Myocardium and Lymphocytes of Clinically Affected Subjects
Total RNA was extracted from Epstein Barr virus (EBV)-transformed B lymphocytes from two affected subjects and one unaffected control and also extracted from cardiac tissue from one affected subject and an unrelated control with Trizol (Invitrogen); this was followed by DNase1 treatment (Ambion). Complementary synthesis was performed and analyzed by both size fractionation and direct sequencing with overlapping primers designed to cover the complete coding sequence of TMEM43.
Bioinformatic Analysis
Conservation of the TMEM43 protein across species was determined with ClustalW and Weblogo.24–26 Potential protein localization, function, structure, and posttranslational modification sites were predicted with the online tools via the ExPASy web site.26 The effects of amino acid substitutions on protein function were predicted.27–31 Information on specific analyses is available upon request.
Clinical Assessment of Affected versus Unaffected Subjects
Clinical data were prospectively collected over 11 years. This involved annual visits by subjects to the genetics cardiomyopathy clinic in which 12 lead ECGs, Holter monitors, MRIs, signal-averaged ECGs, and echocardiograms were done. All cardiac anomalies were noted after clinical testing. Clinical data were also obtained retrospectively from medical records including “at-risk” relatives not seen in clinic and autopsy results. Subjects were categorized as affected, unaffected, or unknown (Figure 2). Only subjects from well-ascertained sibships (in which disease status was known in ≥50% of siblings) were included in this study.
Anomalies on 12 lead ECGs were determined by two physicians blind to disease status. Left ventricular enlargement (LVE) was defined as ≥2 standard deviations (SDs) above a predicted mean: left ventricular end diastolic diameter (LVEDD) >112% (>2 SD).32 Late potentials on SAECG were classified on the basis of recognized criteria: QRS (filtered QRS duration) >114 ms, LAS (low-amplitude signals) >38 ms, and RMS (root-mean-square voltage of the terminal 40 ms of filtered QRS) <20 ms.33 The presence of arrhythmias in the form of PVCs was determined from Holter monitor analysis. Heart failure was classified according to the New York Heart Association (NYHA) functional classification.34 Subjects were identified with heart failure (NYHA categories 1–4) if they presented prospectively in the study with heart failure or it was documented in a medical record. Date of death was confirmed via autopsy records and archival records.
Penetrance is defined as the proportion of subjects who have a specified genotype known to cause a disease and who have any signs or symptoms of the disease.35 To determine the disease penetrance of ARVC at ARVD5, we assessed a subset of disease features based on the ARVC modified diagnostic criteria11 from the first available clinical test result to show an anomaly. We could then assess which clinical features truly segregated with affection status in this population. A Kaplan Meier analysis was then done with a subset of subjects who were alive at the start of the prospective study (1996), who had an available medical record, and who were either mutation positive or an obligate carrier (60 males and 77 females). On the basis of the analysis of prevalent disease features, a male subject was considered “penetrant” at the age when any one of the following clinical events occurred: (1) SCD, (2) VT on clinical testing, (3) heart failure, (4) >200 PVCs in a 24 hr period, (5) LVE >2 SD, (6) QRS > 110 ms on 12 lead ECG, or (7) any late potential on SAECG.11 Female subjects were considered penetrant for all these features apart from QRS >110 ms on 12 lead ECG and RMS or LAS on SAECG. We also determined the penetrance of two major morbid outcomes of the disease, death and heart failure, using both the prospective and retrospective data set (affected subjects n = 257, 148 males, 109 females; unaffected subjects, n = 151, 77 males, 74 females). For the heart-failure analysis, only those from this group with an available medical record were used.
Cardiac tests were not available in all subjects at a priori 50% risk, mainly because of SCD as a presenting feature for many family members. For example, of 114 affected subjects who did not have an available echocardiogram, 86 were deceased (75%), whereas only two were dead in 50 unaffected subjects with no echocardiogram. Testing is in progress in this latter group.
Statistical Analysis
Comparisons between affected versus unaffected subjects were calculated by the Kaplan Meier product limit method with censoring occurring at the time of ICD therapy, heart transplantation, or last follow-up (defined as the age at the last clinic visit). Relative risk was calculated with Cox's Regression model. A p value of <0.05 was considered significant (SPSS software, version 14, Chicago, IL).
Results
Fine Mapping and Candidate Gene Screening
Comparison of the ARVD5 ancestral haplotypes at 3p25 across clinically affected subjects identified key recombinations and reduced the disease region to a 2.36 Mb interval containing 20 annotated genes (Figures 3A and 3B; Table 1). Bidirectional resequencing of the 20 physical candidate ARVD5 genes revealed 240 variants (Table S1 available online). Nineteen variants were found exclusively in clinically affected subjects on the mutation-screening panel, and 11 were determined to reside on the ARVD5 ancestral haplotype through segregation analysis in AR14 (Table 2 and Figure 4). Screening of population controls showed that five of these variants were rare (<1% of the alleles screened; Table 2). Only one of the five, TMEM43 1073C→T (S358L) (Figure 5D), was shared by all clinically affected subjects across the 15 families and was retained on key recombinant ARVD5 haplotypes identified in clinically affected subjects from families AR10 (Figure 6) and AR2 (data not shown). This suggested that TMEM43 is ARVD5. Additional supportive evidence included clinically unaffected adult subjects who shared distal sections of the ARVD5 haplotype and lacked the TMEM43 mutation (e.g., Figure 4). All available spouses (n = 47) and population controls (n = 161) were negative for the TMEM43 mutation (416 mutation-negative chromosomes).
Figure 3.
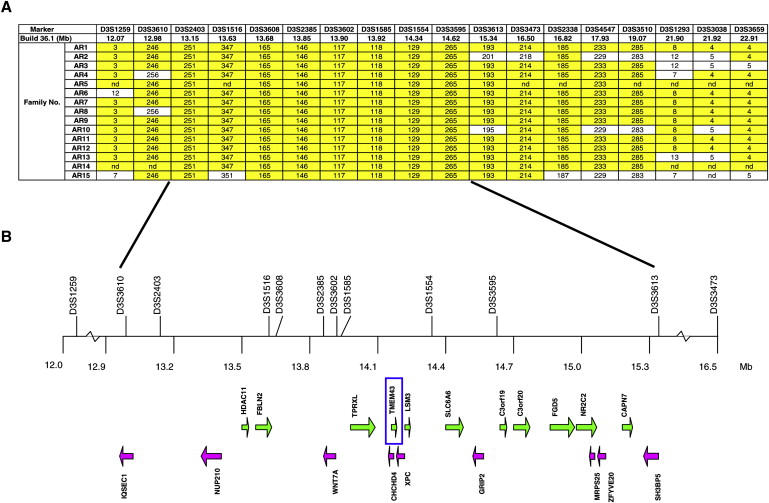
Physical Map of the ARVD5 Critical Region
(A) Summary recombinant ARVD5 haplotypes identified across the 15 ARVC families with microsatellite markers (alleles are either numbered [1–9] or given in base pairs).
(B) The physical map of the ARVD5 critical region. Physical distances were captured from the March 2006 freeze of the UCSC Genome Browser. Arrows show the direction of transcription of each annotated gene.
Table 1.
The 20 Physical Candidate Genes for ARVD5
| Genes | Accession Number | MIM Number | Strand | Genomic Position |
Exons | |
|---|---|---|---|---|---|---|
| Start | End | |||||
| IQSEC1 | NM_014869 | 610166 | − | 13003536 | 12917079 | 13 |
| NUP210 | NM_024923 | 607703 | − | 13436809 | 13332737 | 40 |
| HDAC11 | NM_024827 | 607226 | + | 13496824 | 13521834 | 10 |
| FBLN2 | NM_001004019 | 135821 | + | 13565625 | 13654922 | 18 |
| WNT7A | NM_004625 | 601570 | − | 13896619 | 13835083 | 4 |
| TPRXL | AK092426 | 611167 | + | 13953902 | 14082480 | 3 |
| CHCHD4 | NM_144636 | 611077 | − | 14141323 | 14128584 | 4 |
| TMEM43 | NM_024334 | na | + | 14141546 | 14160180 | 12 |
| XPC | NM_004628 | 278720 | − | 14195143 | 14161651 | 16 |
| LSM3 | NM_014463 | 607283 | + | 14195341 | 14214840 | 4 |
| SLC6A6 | NM_003043 | 186854 | + | 14419110 | 14503973 | 15 |
| GRIP2 | NM_001080423 | na | − | 14558592 | 14510177 | 25 |
| C3orf19 | NM_016474 | na | + | 14668278 | 14689167 | 11 |
| C3orf20 | NM_032137 | na | + | 14691658 | 14789544 | 17 |
| FGD5 | NM_152536 | na | + | 14835810 | 14950899 | 20 |
| NR2C2 | NM_003298 | 601426 | + | 14964240 | 15065782 | 15 |
| MRPS25 | NM_022497 | na | − | 15081820 | 15065024 | 4 |
| ZFYVE20 | NM_022340 | 609511 | − | 15115659 | 15086584 | 14 |
| CAPN7 | NM_014296 | 606400 | + | 15222737 | 15269426 | 21 |
| SH3BP5 | NM_004844 | 605612 | − | 15349108 | 15271250 | 9 |
| Total | 275 | |||||
Table 2.
Sequencing Variants Identified Exclusively in Clinically Affected Subjects
| Gene Name | Accession number | Variant Nomenclature | Classification | Allele Frequency(# of chromosomes) | |
|---|---|---|---|---|---|
| HDAC11 | NM_024827 | c.369+18_369+19insG | Noncoding | nd | nd |
| TMEM43 | NM_024334 | c.1073C→T | Missense (S > L) | 0/322 | 0.00%∗ |
| TMEM43 | NM_024334 | c.1203+115T→C | Noncoding | nd | nd |
| XPC | NM_004628 | c.2823+684G→C | Noncoding | nd | nd |
| SLC6A6 | NM_003043 | c.1-27420G→A | Noncoding | nd | nd |
| SLC6A6 | NM_003043 | c.599+370A→G | Noncoding | dbSNP | 46.00% |
| SLC6A6 | NM_003043 | c.733-1226A→G | Noncoding | 50/90 | 55.60% |
| FGD5 | NM_152536 | c.934G→A | Missense (V > M) | 2/318 | 0.60%∗ |
| FGD5 | NM_152536 | c.2186+22G→A | Noncoding | nd | nd |
| FGD5 | NM_152536 | c.2187-82G→A | Noncoding | nd | nd |
| FGD5 | NM_152536 | c.2220G→T | Synonymous (L > L) | nd | nd |
| FGD5 | NM_152536 | c.2613+50C→T | Noncoding | nd | nd |
| FGD5 | NM_152536 | c.3085-74G→A | Noncoding | 15/166 | 9.00% |
| NR2C2 | NM_003298 | c.855+70G→A | Noncoding | 8/88 | 9.10% |
| NR2C2 | NM_003298 | c.1848+365T→A | Noncoding | 16/90 | 17.80% |
| NR2C2 | NM_003298 | c.1848+2965_1848+2966insGATA | Noncoding | 23/126 | 18.30% |
| MRPS25 | NM_022497 | c.522+1059G→A | Noncoding | 0/138 | 0.00%∗ |
| CAPN7 | NM_014296 | c.1289+68C→T | Noncoding | 1/162 | 0.01%∗ |
| CAPN7 | NM_014296 | c.1430-28T→C | Noncoding | 0/160 | 0.00%∗ |
Eleven of the 19 variants were determined to be in phase on the ARVD5 ancestral haplotype and were subsequently sequenced on population controls (italics). The allele frequency of variants previously reported in the NCBI dbSNP was used. Rare variants (<1% of the alleles screened) are marked with an asterisk.
Figure 4.
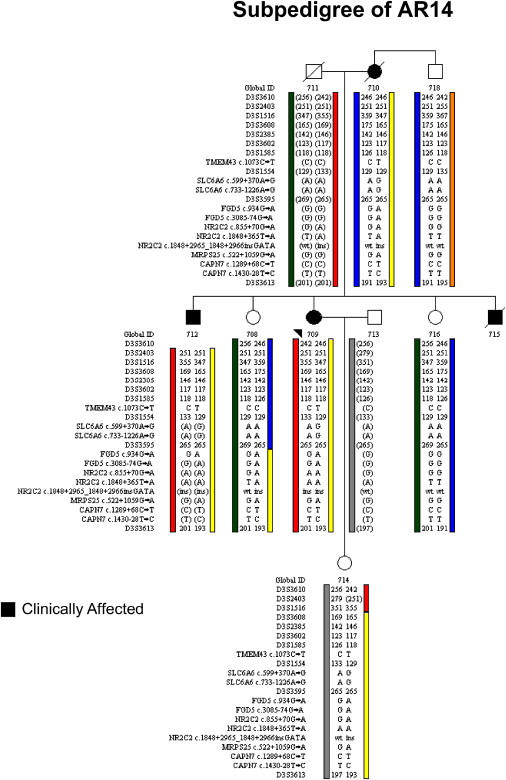
Segregation Analysis in Subpedigree AR14
Of the 19 variants found exclusively in clinically affected subjects on the mutation-screening panel, only 11 were found to reside on the ARVD5 ancestral haplotype (yellow) through segregation analysis of clinically affected subjects (Global IDs 709, 710, and 712). Note that a clinically unaffected subject (Global ID 708) inherited a recombinant ARVD5 haplotype from her clinically affected mother that lacks TMEM43. Alleles in brackets have been inferred.
Figure 5.
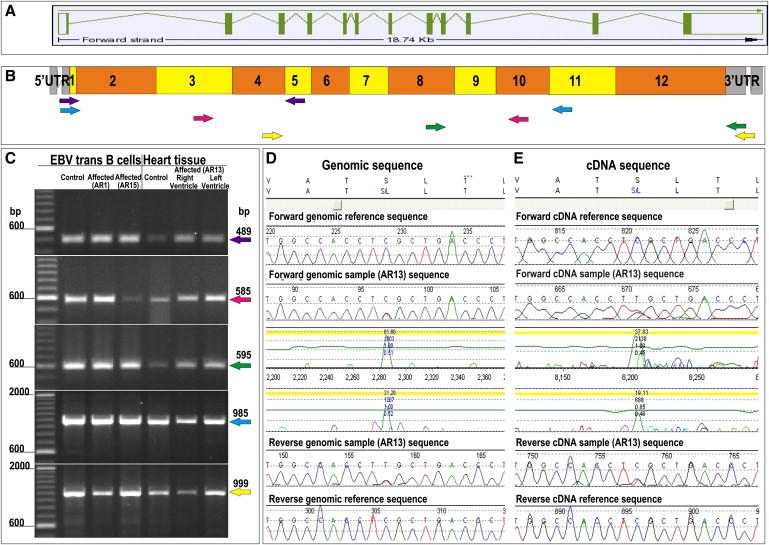
Gene Structure and Mutation Analysis of TMEM43
(A) Gene structure of TMEM43. Exons are represented by boxes. Translated exons are solid green, and untranslated exons are clear. Introns are represented by green lines.
(B) Coverage of primers designed to amplify cDNA showing position of PCR primer pairs: exons 1–4 (purple), exons 4–9 (red), exons 9–12 (green), exons 1–10 (blue), and 5–12 (yellow).
(C) PCR products amplified from cDNA of EBV-transfected B cells of affected subjects from the mutation-screening panel (affected AR1 and affected AR15) and unaffected (control) subjects. cDNA of heart tissue from both the left and right ventricle of an affected subject (AR13) and a heart biopsy from a control subject were size fractionated by electrophoresis.
(D and E) Forward and reverse sequencing traces showing the TMEM43 1073C→T mutation of an affected subject's (AR13) genomic and cDNA. The amino acid translations (top) shows the S358L amino acid substitution.
Figure 6.
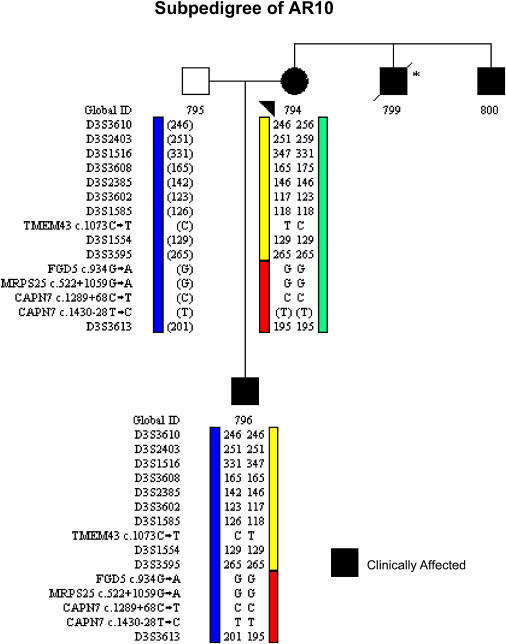
Segregation Analysis in Subpedigree AR10
Clinically affected subjects (Global IDs 794 and 796) only have one of the five rare variants because of a historical recombination event on the ARVD5 haplotype (yellow). Alleles in brackets have been inferred.
ARVC at Locus ARVD5 Is Caused by a Missense Mutation in TMEM43
The longest isoform of transmembrane protein 43 (TMEM43, GenBank accession number NM_024334) has 12 exons (Figure 5A) predicting a 400 amino acid protein that is 98% similar to the mouse protein (Figure 7). This conserved gene is found across all eukaryotic and prokaryotic species (Figure 7). We were able to extract full-length TMEM43 cDNA from white blood cells and cardiac tissue from both patients and controls, demonstrating that TMEM43 is expressed in both blood and cardiac tissue and that the TMEM43 1073C→T (S358L) mutation does not appear to affect splicing (Figures 5B, 5C, and 5E).
Figure 7.
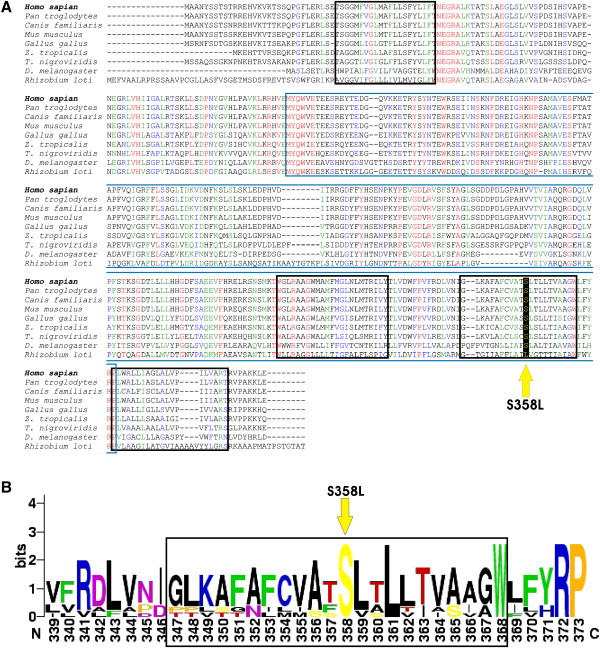
Multiple Alignment of the TMEM43 Gene across Eight Eukaryotic and Prokaryotic Species
(A) Clustal W align was used to align orthologs from Homo sapiens (NP_077310), Pan troglodytes (XP_516299), Canis familiaris (XP_541751), Mus musculus (NP_083042), Gallus gallus (XP_414378), Zenopus tropicalis (UP10004D5297), Tetraodon nigroviridis (Q4RXL8), Drosophila melanogaster (NP_64162), and Rhizobium loti (Q98HF3).25 The blue box outlines the DUF1625 domain, and the black boxes outline predicted transmembrane domains. Completely conserved residues are red, strongly similar residues are green, and weakly similar residues are blue. The S358L mutation is marked by a yellow arrow.
(B) The web logo format was used for aligning eukaryotic species. The third transmembrane domain is outlined (black box). The S358L mutation is marked by a yellow arrow.
Bioinformatic analysis of TMEM43 predicts it to be a cytoplasmic membrane protein with several potential posttranslation modification sites (Figure 8). Unlike the transmembrane proteins of the desmosome (desmocollin and desmoglein), TMEM43 does not have a cadherin domain. Furthermore, protein sequence alignments with desmocollin and desmoglein show less than 10% identity and less than 12% similarity.26 The mutation, S358L, occurs within the third predicted transmembrane domain and is highly conserved in mammalian, avian, amphibian, and insect orthologs (Figures 7A and 7B and Figure 8). Interestingly, a leucine at this position is found in the bacterium Rhizobium loti, but it is not found in any multicellular organisms (Figures 7A and 7B). The S358L mutation is predicted to be deleterious (Table 3), but little is known about the function of TMEM43 protein.
Figure 8.
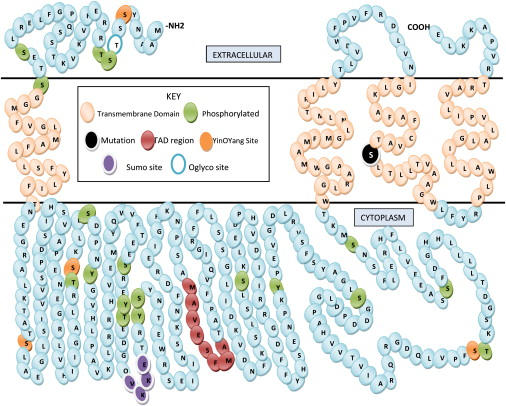
Predicted Topography of the TMEM43 Protein
Indicated are transmembrane domains (beige), phosphorylation sites (green), a transactivation domain (red), YingOYang sites (orange), a SUMO attachment site (purple), and an O-glycosylation site (blue open). The extracellular and cytoplasmic regions may be reversed: There is evidence supporting either orientation.
Table 3.
Prediction of the TMEM43 1073C→T, S358L, Mutation Effect
| Program | SIFT | Panther | PolyPhen | SNPs3D | PMut |
|---|---|---|---|---|---|
| S358L mutation | deleterious | deleterious | Benign | deleterious | deleterious |
Five different bioinformatic programs were used for predicting the effect of the S358L mutation. Note that sequence homology used by the PolyPhen analyses was calculated with alignments of orthologs from Eukaryota and bacteria.
TMEM43 Mutation Screening in ARVD5 Linked Families
We sequenced genomic DNA from all available subjects born at a priori 50% risk (n = 295) across the 15 ARVC families for the presence of the 1073C→T TMEM43 mutation. All clinically affected subjects (n = 83/83; 40 males, 43 females) and 28.8% of clinically unaffected subjects (n = 61/212; 18 males, 43 females) were mutation carriers (Figure 2). Interestingly, 57% (35/61) of unaffected mutation carriers were found on subsequent testing to have clinical signs of ARVC. These included ectopy on Holter monitor (≥200 PVCs over 24 hr11), extended QRS on 12 lead ECG or SAECG,10 or an enlarged left ventricle (>2 SD above the mean). The remaining 26 clinically unaffected mutation carriers, 3 males and 23 females, were at a median age of 22 years and 33 years respectively. The 151 subjects who had no clinical signs and who did not have the TMEM43 variant were considered unaffected (Figure 2).
Clinical Assessment of Penetrance in ARVD5
12 Lead ECG
Data on a total of 297 subjects (167 affected, 130 unaffected) that had at least one 12 lead surface ECG was available. Extended QRS >110 ms was significantly different between affected versus unaffected males (Table 4). Epsilon waves and T wave inversion commonly seen in ARVC10 was seen in less than 3% of affected subjects in this cohort.
Table 4.
Prevalent Cardiac Features on First Clinical Test
| 12 Lead ECG | Affected |
Unaffected |
Affected Males versus Unaffected Males, χ2 | Affected Females versus Unaffected Females, χ2 | ||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | Total | Male | Female | Total | |||
| Subjects Tested (N) | 78 | 89 | 167 | 64 | 66 | 130 | ||
| Mean Age (SD) | 30.0 (12.1) | 37.7 (15.8) | 33.1 (16.0) | 38.2 (13.2) | ||||
| QRS >110 ms n (%) | 25 (32) | 8 (9) | 33 (19.8) | 4 (6) | 2 (3) | 6 (4.6) | p ≤ 0.001 | p ≤ 0.2 ns |
| Holter Monitor | ||||||||
| Subjects Tested (N) | 67 | 79 | 146 | 49 | 44 | 93 | ||
| Mean Age (SD) | 31.1 (13.3) | 37.7 (15.5) | 33.1 (15.9) | 38.2 (12.8) | ||||
| PVCs ≥200/24 hr n (%) | 47 (70) | 47 (59) | 94 (64.4) | 0 (0) | 1 (2) | 1 (1.1) | p < 0.001 | p < 0.001 |
| ≥1 run ns VT n (%) | 13 (19) | 18 (23) | 31 (21.2) | 0 (0) | 0 (0) | 0 (0.0) | p < 0.01 | p < 0.001 |
| Echocardiograph | ||||||||
| Subjects Tested (N) | 67 | 76 | 143 | 50 | 51 | 101 | ||
| Mean Age (SD) | 31.6 (13.2) | 39.6 (15.1) | 32.5 (14.9) | 38.3 (13.3) | ||||
| LVE >2 SD n (%) | 35 (52) | 27 (20) | 62 (43.3) | 10 (20) | 4 (8) | 14 (13.9) | p < 0.001 | p < 0.001 |
| SAECG | ||||||||
| Subjects Tested (N) | 32 | 65 | 97 | 50 | 51 | 101 | ||
| Mean Age (SD) | 30.9 (14.5) | 39.8 (14.5) | 36.7 (16.3) | 38.9 (11.0) | ||||
| QRS >114 ms n (%) | 19 (59) | 19 (29) | 38 (39.2) | 10 (20) | 2 (4) | 12 (11.9) | p ≤ 0.001 | p ≤ 0.001 ns |
| RMS <20 ms n (%) | 15 (47) | 18 (28) | 33 (34.0) | 12 (24) | 9 (18) | 21 (20.8) | p ≤ 0.05 | p ≤ 0.3 ns |
| LAS >38 ms n (%) | 17 (53) | 21 (32) | 38 (39.2) | 14 (28) | 13 (25) | 27 (26.7) | p ≤ 0.025 | p ≤ 0.5 ns |
This table shows 12 lead ECG, Holter, echocardiographic, and SAECG manifestations used in penetrance analysis in subjects from 15 families with ARVC due to a mutation in TMEM43. Abbreviations are used as follows: PVC, premature ventricular complex; VT, ventricular tachycardia; LVE, left ventricular enlargement indexed to height and weight; RMS, root mean square voltage of the terminal 40 ms of filtered QRS; and LAS, low-amplitude signals.
Holter Monitor
Of the 239 subjects (146 affected, 93 unaffected) with at least one Holter monitor report, the most prevalent feature was PVCs ≥200 and the presence of at least one run of nonsustained VT (Table 4), both significantly more common in affected than unaffected subjects.
Echocardiography
Of the 244 subjects (143 affected, 101 unaffected) that had at least one 2D echocardiogram available for analysis, the most prevalent feature was LVE based on LVEDD (Table 4),32,36 and LVE was significantly different between affected and unaffected subjects.
SAECG
Of the 198 subjects (97 affected, 101 unaffected) who had at least one SAECG available for analysis, all late potentials were significantly more common in affected versus unaffected males; only QRS was significant in females (Table 4).
ARVC Penetrance
We used 137 subjects (60 affected males, 77 affected females) to determine penetrance. Median age to develop an ARVD5 associated phenotype was 32 years (95% CI 28–35) for males and 44 years (95% CI 39-48) for females, with 100% of males and females penetrant by 63 and 76 years, respectively (Figure 9 and Table 5). Males were twice as likely to reveal the disease phenotype than females (RR 2, 99% CI 1.2–3.3) (p ≤ 0.0001). The commonest clinical features for which subjects were initially penetrant were ectopy (44%) and LVE (27%), and those were followed by VT (9%), QRS >110 ms (9%), late potentials on SAECG (7%), SCD (2%), and heart failure (2%).
Figure 9.
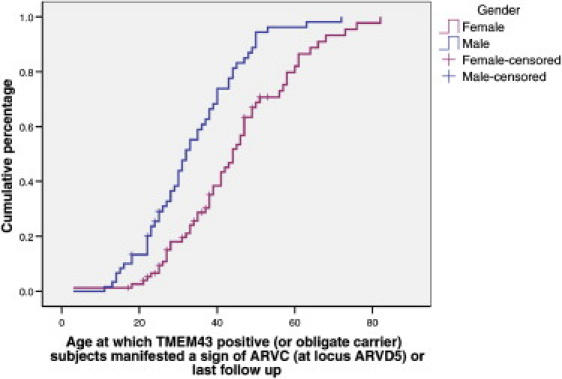
ARVC Penetrance Caused by the TMEM43 1073C→T Mutation
Cumulative proportion by age at which male (n = 60) and female (n = 77) mutation positive affected subjects manifested a first clinical sign of ARVC. Subjects were censored at last follow-up.
Table 5.
Percentage of Subject's Penetrant by Each Age
| Age (years) | 11–20 | 21–30 | 31–40 | 41–50 | 51–60 | 61–70 | 71–80 |
|---|---|---|---|---|---|---|---|
| Males | 13 | 39 | 68 | 89 | 96 | 100 | 100 |
| Females | 3 | 18 | 38 | 67 | 80 | 97 | 100 |
This table shows the cumulative percentage of subjects penetrant by decade.
Heart Failure
Fourteen of 89 affected males developed heart failure at a median age of 63 years (95% CI 41–84 years) compared with no unaffected males (p ≤ 0.0001, log rank) (Figure 10Ai). One unaffected female at age 79 developed heart failure compared to seven affected females (median age 73 years, 95% CI 69–77 years) (p ≤ 0.001, log rank) (Figure 10Aii). Affected males were three times more likely to develop heart failure than females (RR 3.4, 95% CI 1.36–8.57, p ≤ 0.009) (Figure 10Aiii).
Figure 10.
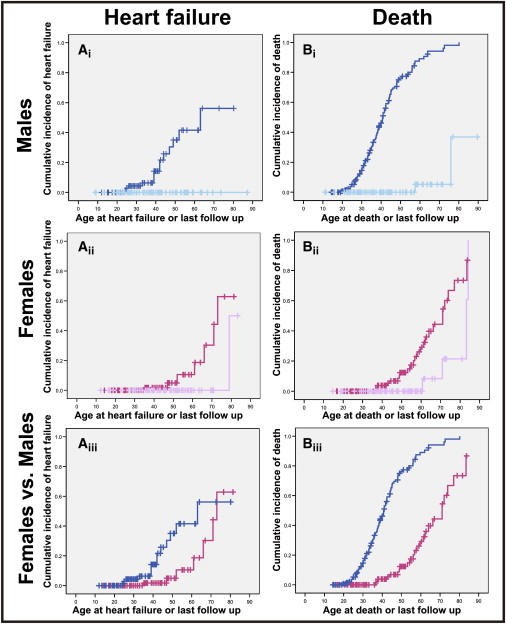
Time to Event Analysis of Heart Failure and Death in Affected Subjects
(Ai–Aiii) Cumulative incidence of heart failure; panels describe the following: (Ai) affected males (n = 89, dark blue) compared to unaffected males (n = 71, light blue) (p ≤ 0.0001: log rank); (Aii) affected females (n = 87, dark pink) compared to unaffected females (n = 68, light pink) (p ≤ 0.001, log rank); and (Aiii) affected males (dark blue) versus affected females (dark pink).
(Bi–Biii) Life expectancy; panels describe the following: (Bi) time to death in affected males (n = 148, dark blue) and unaffected males (n = 77, light blue); (Bii) time to death in affected females (n = 109, dark pink) and unaffected (n = 74, light pink); and (Biii) affected males (dark blue) versus affected females (dark pink).
Life Expectancy
In affected subjects (n = 257), there were 123 (48%) deaths (99 males and 24 females). Sudden cardiac death occurred in 86% (86/99) of the males and 42% (10/24) of the females. Survival was significantly reduced in affected versus unaffected subjects. Median survival was 41 compared to 83 years (p ≤ 0.0001, log rank) in affected versus unaffected males (Figure 10Bi) and 71 compared to 83 years in affected versus unaffected females (p ≤ 0.002, log rank) (Figure 10Bii). The relative risk of dying was 6.8 times greater in affected males versus affected females (95% C.I: 4.3–10.9, p ≤ 0.0001) (Figure 10Biii).
Discussion
Mutations in desmosomal genes underlie several genetic subtypes of ARVC, suggesting that ARVC is primarily a disease of the desmosome.5,7 We used a positional mapping approach and 15 ARVC families that shared a disease-associated haplotype on 3p from a genetic isolate to identify ARVD5. All clinically affected subjects that were screened for the TMEM43 1073C→T mutation (n = 83) were mutation carriers. This mutation was not detected in spouses or population controls. The TMEM43 gene is expressed in cardiac tissue and is evolutionarily conserved, and the amino acid substitution is predicted to be deleterious.
Recently, signaling pathways have been implicated in ARVC pathogenesis.7,37 For example, plakoglobin, when freed from desmosomal complexes, translocates to the nucleus where it competes and opposes the action of β-catenin and downregulates the canonical Wnt/β-catenin signaling pathway.37 Suppression of the canonical Wnt/β-catenin signaling upregulates two adipogenic transcription factors, C/EBP-α (MIM 116897) and PPARγ (MIM 601487).37 A genome-wide scan for peroxisome proliferator response elements (PPREs) identified 1085 potential target genes of PPARγ, including TMEM43.38 If TMEM43 is a part of an adipogenic pathway regulated by PPARγ, then perhaps dysregulation of this pathway may explain the fibrofatty replacement of the myocardium in ARVC patients.
Studies that assess the ARVC phenotype in patients with mutations in plakophilin-2,39 desmoglein-2,16 and plakoglobin2 typically evaluate clinical features in mutation carriers only. The assumption is that all cardiac signs present in affected subjects are due to the underlying genetic defect. However, it may be that some signs are due to the genetic background of either the family or the source population. The size of the ARVC cohort in this study provides an opportunity to compare and define ARVC-specific clinical features in affected and unaffected subjects.
By using the benefit of a control group, we evaluated modified clinical criteria11 in affected versus unaffected subjects. We included LVE, a clinical feature that is noted in other ARVC studies40–44 and that is prevalent in this cohort. For all clinical features examined, a significant difference was found between affected versus unaffected males. Females were significantly different for all features with the exception of RMS and LAS on SAECG and a QRS >110 ms on 12 lead ECG.
For the penetrance study, we followed subjects who were alive at the start of the study (1996), who had a medical record, and who had a genetic diagnosis (mutation carriers or obligate carriers). The results showed that ARVC was fully penetrant in males by the age of 63 and in females by the age of 76; ectopy and LVE were often the first presenting features, and few presented with death or heart failure. Therefore, in order to assess the penetrance of heart failure and death, we used all affected subjects. Heart failure and death were morbid outcomes at early ages in both males and females, with far more serious early events in males. In both sexes, heart failure occurs as a later manifestation in subjects who did not succumb to SCD. These major manifestations define ARVC, because of the TMEM43 mutation, as a lethal, fully penetrant, sex-influenced, autosomal-dominant disorder.
Biases exist in studies that use both retrospective and prospective data; the use of well-ascertained sibships may mitigate some of these. Although formal assessment of right ventricular structure is not available for the majority of subjects in this cohort, the cardiomyopathy fulfills diagnostic criteria for ARVC,11 it has been historically diagnosed as ARVC,17,19 and fibrofatty replacement of the myocardium is present. The involvement of the left ventricle in this cohort supports the recent call to place ARVC within a group called “arrhythmic cardiomyopathies,” which would take into account various presentations of disease.45 These findings may not be applicable to other forms of inherited ARVC. However, it is likely that mutations in TMEM43 are present in other populations. Issues of classification in the cardiomyopathies in general will be solved by their underlying genetic etiology; thus, disease-specific genotype/phenotype information will become the most useful and relevant.
This study has the advantage of having access to past medical records and extensive genealogical data, and a single cardiac-care team followed all subjects. Clinical signs due to the genetic background of either the family or the source population were controlled for by the use of unaffected subjects from well-ascertained sibships. This genetic subtype does not show the typical ARVC ECG findings of T wave inversion or epsilon waves.10 A direct benefit of this process therefore has been the provision of accurate mutation-specific clinical data for precise genetic counseling and follow-up of those at-risk.20
We have identified the gene that causes ARVC at locus ARVD5. A missense mutation in TMEM43 causes a fully penetrant, sex-influenced lethal form of ARVC. Although little is known about the function of this gene, it encodes a transmembrane protein that could be a target of PPARγ, and this may explain the fibrofatty replacement of the myocardium in ARVC patients. Future directions include functional studies of the TMEM43 protein.
Supplemental Data
One table is available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI dbSNP, http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp&cmd=search&term=
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UCSC Genome Browser, http://genome.ucsc.edu/
Acknowledgments
The authors are supported by a career award from the Canadian Institutes of Health Research-Regional Partnerships Program (to T.-L.Y.). K.H. and L.T. were supported by a grant from the Ernst and Berta Grimmke Foundation, Düsseldorf, Germany. This project was funded by the Canadian Institutes of Health Research, Genome Canada (Atlantic Medical Genetics and Genomics Initiative), Newfoundland and Labrador Centre for Applied Health Research, Janeway Children's Hospital Foundation, Memorial University Opportunities Fund, the General Hospital Foundation and St. Judes Medical Research Grant, Canada. We dedicate this paper to the ARVC families in Newfoundland, who live daily with the reality of this disease. Without their continued support and participation, this research could not occur. We thank Barbara Peddle, Pauline Ryan, Karen Williams, Carol Tilley, Liz Kernaghan, Sara MacKay, Fiona Curtis, and Elizabeth Dicks for clinical assistance, Dr. Roger Green for control samples, and Dante Galutria, Sigrid Milan, Ingrid Pardoe, Nancy Whalen, Yoella Teplisky, Susan Stuckless, and Fahad Chowdhury for technical assistance. Special thanks to Dr. Laura Gillespie and Dr. Gary Paterno for help with protein prediction, Dr. Christina Templeton and Dr. Suryakant Shah for pediatric cardiology consultation, and Dr. Adrian Lear, Dr. William Marshall, and Dr. Elizabeth Ives for their early interest in this disease.
References
- 1.Fontaine G., Fontaliran F., Frank R. Arrhythmogenic right ventricular cardiomyopathies: Clinical forms and main differential diagnoses. Circulation. 1998;97:1532–1535. doi: 10.1161/01.cir.97.16.1532. [DOI] [PubMed] [Google Scholar]
- 2.Protonotarios N., Tsatsopoulou A., Anastasakis A., Sevdalis E., McKoy G., Stratos K., Gatzoulis K., Tentolouris K., Spiliopoulou C., Panagiotakos D. Genotype-phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J. Am. Coll. Cardiol. 2001;38:1477–1484. doi: 10.1016/s0735-1097(01)01568-6. [DOI] [PubMed] [Google Scholar]
- 3.Heuser A., Plovie E.R., Ellinor P.T., Grossmann K.S., Shin J.T., Wichter T., Basson C.T., Lerman B.B., Sasse-Klaassen S., Thierfelder L. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006;79:1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pilichou K., Nava A., Basso C., Beffagna G., Bauce B., Lorenzon A., Frigo G., Vettori A., Valente M., Towbin J. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricularcardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 5.Sen-Chowdhry S., Syrris P., McKenna W. Genetics of right ventricular cardiomyopathy. J. Cardiovasc. Electrophysiol. 2005;16:927–935. doi: 10.1111/j.1540-8167.2005.40842.x. [DOI] [PubMed] [Google Scholar]
- 6.Dokuparti M.V., Pamuru P.R., Thakkar B., Tanjore R.R., Nallari P. Etiopathogenesis of arrhythmogenic right ventricular cardiomyopathy. J. Hum. Genet. 2005;50:375–381. doi: 10.1007/s10038-005-0273-5. [DOI] [PubMed] [Google Scholar]
- 7.MacRae C.A., Birchmeier W., Thierfelder L. Arrhythmogenic right ventricular cardiomyopathy: moving toward mechanism. J. Clin. Invest. 2006;116:1825–1828. doi: 10.1172/JCI29174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiso N., Stephan D.A., Nava A., Bagattin A., Devaney J.M., Stanchi F., Larderet G., Brahmbhatt B., Brown K., Bauce B. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum. Mol. Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 9.Beffagna G., Occhi G., Nava A., Vitiello L., Ditadi A., Basso C., Bauce B., Carraro G., Thiene G., Towbin J.A. Regulatory mutations in transforming growth factor-[beta]3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 10.McKenna W.J., Thiene G., Nava A., Fontaliran F., Blomstrom-Lundquist C., Fontaine G., Camererini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamid M.S., Norman M., Quraishi A., Firoozi S., Thaman R., Gimeno J.R., Sachdev B., Rowland E., Elliott P.M., McKenna W.J. Prospective evaluation of relatives for familal arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J. Am. Coll. Cardiol. 2002;40:1445–1450. doi: 10.1016/s0735-1097(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 12.Corrado D., Basso C., Thiene G. CARDIOMYOPATHY: Arrhythmogenic right ventricular cardiomyopathy: Diagnosis, prognosis, and treatment. Heart. 2000;83:588–595. doi: 10.1136/heart.83.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nava A., Bauce B., Basso C., Muriago M., Rampazzo A., Villanova C., Daliento L., Buja G., Corrado D., Danieli G.A. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2000;36:2226–2233. doi: 10.1016/s0735-1097(00)00997-9. [DOI] [PubMed] [Google Scholar]
- 14.Wlodarska E.K., Konka M., Zaleska T., Ploski R., Cedro K., Pucilowska B., Bekiesinska-Figatowska M., Rydlewska-Sadowska W., Ruzyllo W., Hoffman P. Arrhythmogenic right ventricular cardiomyopathy in two pairs of monozygotic twins. Int. J. Cardiol. 2005;105:126–133. doi: 10.1016/j.ijcard.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 15.Blomstrom-Lundqvist C., Sabel K., Olsson S. A long term follow-up of 15 patients with arrhythmogenic right ventricular dysplasia. Br. Heart J. 1987;58:477–488. doi: 10.1136/hrt.58.5.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Syrris P., Ward D., Asimaki A., Evans A., Sen-Chowdhry S., Hughes S.E., McKenna W.J. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: A genotype-phenotype characterization of familial disease. Eur. Heart J. 2007;28:581–588. doi: 10.1093/eurheartj/ehl380. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad F., Li D., Karibe A., Gonzalez O., Tapscott T., Hill R., Weilbaecher D., Blackie P., Furey M., Gardner M. Localisation of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p23. Circulation. 1998;98:2791–2795. doi: 10.1161/01.cir.98.25.2791. [DOI] [PubMed] [Google Scholar]
- 18.Marshall W., Furey M., Larsen B., Rose J., Sharratt G., Sussex B., Virmani S., Ko P., LeBlanc P., Nolan R. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988;319:174–175. doi: 10.1056/NEJM198807213190312. [DOI] [PubMed] [Google Scholar]
- 19.Guiraudon G., Klein G., Gulaumshein S., Paiinvin G., DelCompo C., Gonzales J., Ko P. Total disconnection of right ventricular free wall: Surgical treatment of right ventricular tachycardia associated with right ventricular dysplasia. Circulation. 1983;67:463–470. doi: 10.1161/01.cir.67.2.463. [DOI] [PubMed] [Google Scholar]
- 20.Hodgkinson K., Parfrey P., Bassett A., Kupprion C., Drenckhahn J., Norman M., Thierfelder L., Stuckless S., Dicks E., McKenna W. The impact of implantable cardioverter defibrillator therapy on survival in autosomal dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5) J. Am. Coll. Cardiol. 2005;45:400–408. doi: 10.1016/j.jacc.2004.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green R.C., Green J.S., Buehler S.K., Robb J.D., Daftary D., Gallinger S., McLaughlin J.R., Parfrey P.S., Younghusband H.B. Very high incidence of familial colorectal cancer in Newfoundland: A comparison with Ontario and 13 other population-based studies. Fam. Cancer. 2007;6:53–62. doi: 10.1007/s10689-006-9104-x. [DOI] [PubMed] [Google Scholar]
- 22.Mannion J. Memorial University; St. John's: 1977. The Peopling of Newfoundland: Essays in Historical Geography. Institute of Social and Economic Research. [Google Scholar]
- 23.Rahman P., Jones A., Curtis J., Bartlett S., Peddle L., Fernandez B., Freimer N. The Newfoundland population: A unique resource for genetic investigation of complex diseases. Hum. Mol. Genet. 2003;12:R167–R172. doi: 10.1093/hmg/ddg257. [DOI] [PubMed] [Google Scholar]
- 24.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Combet C., Blanchet C., Geourjon C., Deleage G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 26.Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R.D., Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng P.C., Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas P.D., Kejariwal A., Campbell M.J., Mi H., Diemer K., Guo N., Ladunga I., Ulitsky-Lazareva B., Muruganujan A., Rabkin S. PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue P., Melamud E., Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrer-Costa C., Gelpi J.L., Zamakola L., Parraga I., de la Cruz X., Orozco M. PMUT: A web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 32.Henry W.L., Gardin J.M., Ware J.H. Echocardiographic measurements in normal subjects from infancy to old age. Circulation. 1980;62:1054–1061. doi: 10.1161/01.cir.62.5.1054. [DOI] [PubMed] [Google Scholar]
- 33.Breithardt G., Cain M., El-Sherif N. Standards for analysis of ventricular late potentials using high resolution or signal-averaged electrocardiography. A statement by a task force committee of the european society of cardiology, the American Heart Association, and the American College of Cardiology. J. Am. Coll. Cardiol. 1991;17:999–1006. doi: 10.1016/0735-1097(91)90822-q. [DOI] [PubMed] [Google Scholar]
- 34.NYHA CC . Little, Brown & Co; Boston: 1964. Nomenclature and Criteria for Diagnosis: Diseases of the Heart and Blood Vessels. pp. 114. [Google Scholar]
- 35.Nussbaum R., McInnes R., Willard H. W.B Saunders; Philadelphia: 2001. Thompson and Thompson: Genetics in Medicine. [Google Scholar]
- 36.Henry W.L., Ware J., Gardin J.M., Hepner S.I., McKay J., Weiner M. Echocardiographic measurements in normal subjects. Growth related changes that occur between infancy and early adulthood. Circulation. 1978;57:278–285. doi: 10.1161/01.cir.57.2.278. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Gras E., Lombardi R., Giocondo M.J., Willerson J.T., Schneider M.D., Khoury D.S., Marian A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemay D.G., Hwang D.H. Genome-wide identification of peroxisome proliferator response elements using integrated computational genomics. J. Lipid Res. 2006;47:1583–1587. doi: 10.1194/jlr.M500504-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Syrris P., Ward D., Asimaki A., Sen-Chowdhry S., Ebrahim H.Y., Evans A., Hitomi N., Norman M., Pantazis A., Shaw A.L. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:356–364. doi: 10.1161/CIRCULATIONAHA.105.561654. [DOI] [PubMed] [Google Scholar]
- 40.Lindstrom L., Nylander E., Larsson H., Wranne B. Left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy - a scintigraphic and echocardiographic study. Clin. Physiol. Funct. Imaging. 2005;25:171–177. doi: 10.1111/j.1475-097X.2005.00607.x. [DOI] [PubMed] [Google Scholar]
- 41.Lobo F.V., Silver M.D., Butany J., Heggtveit H.A. Left ventricular involvement in right ventricular dysplasia/cardiomyopathy. Can. J. Cardiol. 1999;15:1239–1247. [PubMed] [Google Scholar]
- 42.Matsuo S., Sato Y., Nakae I., Masuda D., Yomota M., Ashihara T., Horie M. Left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy demonstrated by multidetector-row computed tomography. Int. J. Cardiol. 2007;115:e129–e131. doi: 10.1016/j.ijcard.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 43.Pinamonti B., Pagnan L., Bussani R., Ricci C., Silvestri F., Camerini F. Right ventricular dysplasia with biventricular involvement. Circulation. 1998;98:1943–1945. doi: 10.1161/01.cir.98.18.1943. [DOI] [PubMed] [Google Scholar]
- 44.Norman M., Simpson M., Mogensen J., Shaw A., Hughes S., Syrris P., Sen-Chowdhry S., Rowland E., Crosby A., McKenna W.J. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 2005;112:636–642. doi: 10.1161/CIRCULATIONAHA.104.532234. [DOI] [PubMed] [Google Scholar]
- 45.Sen-Chowdhry S., Syrris P., Ward D., Asimaki A., Sevdalis E., McKenna W. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.