

Abstract
The genetic basis for association of the PARK11 region of chromosome 2 with familial Parkinson disease (PD) is unknown. This study examined the GIGYF2 (Grb10-Interacting GYF Protein-2) (TNRC15) gene, which contains the PARK11 microsatellite marker with the highest linkage score (D2S206, LOD 5.14). The 27 coding exons of the GIGYF2 gene were sequenced in 123 Italian and 126 French patients with familial PD, plus 131 Italian and 96 French controls. A total of seven different GIGYF2 missense mutations resulting in single amino acid substitutions were present in 12 unrelated PD index patients (4.8%) and not in controls. Three amino acid insertions or deletions were found in four other index patients and absent in controls. Specific exon sequencing showed that these ten sequence changes were absent from a further 91 controls. In four families with amino acid substitutions in which at least one other PD case was available, the GIGYF2 mutations (Asn56Ser, Thr112Ala, and Asp606Glu) segregated with PD. There were, however, two unaffected carriers in one family, suggesting age-dependent or incomplete penetrance. One index case (PD onset age 33) inherited a GIGYF2 mutation (Ile278Val) from her affected father (PD onset age 66) and a previously described PD-linked mutation in the LRRK2 gene (Ile1371Val) from her affected mother (PD onset age 61). The earlier onset and severe clinical course in the index patient suggest additive effects of the GIGYF2 and LRRK2 mutations. These data strongly support GIGYF2 as a PARK11 gene with a causal role in familial PD.
Introduction
Parkinson disease (PD [MIM 168600]) is a neurodegenerative disorder affecting 1%–2% of the population above age 60.1 The cause of dopaminergic neuronal loss in the nigro-striatal pathway and consequent bradykinesia, resting tremor, muscular rigidity, and postural instability that characterize PD is largely unknown. Although only 10%–30% of PD is familial,2 there is strong interest in identifying genes that contribute to familial PD, because these genes are expected to define mechanisms and new therapeutic approaches that apply to more common, sporadic forms of the disease.
Family-based whole-genome linkage scans have identified 13 chromosomal loci (PARK1 to PARK13) that show linkage to PD.3–14 Candidate-gene analysis at these genomic loci linked to PD has identified eight causative genes in PD (SNCA [MIM 163890], PARK2 [Parkin] [MIM 602544], UCHL1 [MIM 191342], PINK1 [MIM 608309], PARK7 [DJ1] [MIM 602533], LRRK2 [MIM 609007], ATP13A2 [MIM 610513], and HTRA2 [MIM 606441]).12,15–26 The mechanisms through which these genes contribute to the development of PD have not yet been clearly established. Autosomal-dominant transmission is associated with mutations in the SNCA, LRRK2, and, probably, UCHL1 and HTRA2 genes, whereas the four other genes are autosomal recessive. These genes only account for a small proportion of PD cases, except in specific clinical subgroups or populations. This is the case for the G2019S mutation in the LRRK2 gene found in up to 18% of Ashkenazi Jews and 39% of North African Arabs with PD but in only 1 to 3% of their corresponding controls.27,28
The PARK11 locus on chromosome 2q36-37 was initially identified by whole-genome linkage analysis in a population of PD patients with at least one first-degree affected relative.8,29,30 This linkage region was not confirmed in another PD population.31 However, an earlier association analysis,32 which represents a more powerful method for identifying risk-factor genes,33 also revealed significant association between markers in the PARK11 region and PD.
The PARK11 locus corresponds to an 18 cM interval between microsatellite markers D2S396 and D2S338, with LOD scores of 2.9 and 2.4, respectively,8,29,30 that contains 73 potential candidate genes (Figure 1). The PARK11 microsatellite marker D2S206, which has the highest LOD score of 5.14, is contained within intron 21 of a 27-exon gene encoding 1299 amino acids, which has been alternately designated GIGYF2 (Grb10-Interacting GYF Protein 2)34 or TNRC15 (Trinucleotide Repeat-Containing 15). We previously identified GIGYF2 by yeast two-hybrid screening as one of two homologous proteins, GIGYF1 (or PERQ1) and GIGYF2, interacting through their GYF domain with the Grb10 adaptor protein.34 Grb10 and the GIGYF proteins are of interest for their potential involvement in insulin-like growth factor (IGF) and insulin signaling.34–39 Because the IGFs and insulin have important effects in the central nervous system40,41 and are potentially associated with PD,42–45 we investigated the involvement of the GIGYF2 (TNRC15) gene in PD.
Figure 1.

Genetic Map of the PARK11 Locus, Extending from Markers D2S396 to D2S338
Microsatellite markers enclosed within boxes have been significantly linked to Parkinson disease.29 Dots indicate markers localized within the respective genes. Gene-mapping data are from the National Center for Biotechnology Information (NCBI) website.
Methods and Materials
Study Population
Index cases with familial Parkinson disease (PD) (n = 123), defined by the presence of at least one first-degree affected relative (parent, child, or sibling), and healthy controls (n = 131) were obtained from an Italian DNA bank assembled by the Parkinson Institute, Istituti Clinici di Perfezionamento (Milan, Italy) (Table 1). DNA from a second set of patients with familial PD compatible with dominant transmission (n = 126) and controls (n = 96) was obtained from the French Parkinson Disease Genetics Study Group (Paris, France) (Table 1). The clinical diagnosis of PD in both populations was established according to widely accepted criteria.46,47 This required the presence of bradykinesia and at least one of the following: resting tremor, rigidity, and postural instability; a positive response to dopaminergic therapy; the absence of atypical features or other causes of parkinsonism. Control DNAs were from unrelated individuals from the same populations. All controls were free of symptoms suggestive of PD, ≥ 45 years old, and with a negative family history for movement disorders at least in their first-degree relatives. The project was approved by the local ethical authorities, and written informed consent was obtained from all subjects.
Table 1.
Population Phenotype Descriptions
| Italian Population |
French Population |
|||
|---|---|---|---|---|
| PD n = 123 | Control n = 131 | PD n = 126 | Control n = 96 | |
| Parkinson age of onset, Control age of study entry (Mean ± SD) | 53 ± 12 | 69 ± 9 | 48 ± 12 | 64 ± 9 |
| Young onset, ≤ 21 years | 0 | 4 | ||
| Early onset, ≤ 45 years | 28 | 50 | ||
| Late onset, > 45 years | 95 | 72 | ||
| At least one first-degree relative affected (%) | 100 | 100 | ||
| Only father affected (%) | 25.6 | 33.9 | ||
| Only mother affected (%) | 24.0 | 32.2 | ||
| Both parents affected (%) | 0.8 | 0.9 | ||
| Only siblings affected (%) | 44.6 | 1.7 | ||
| Only progeny affected (%) | 0 | 6.0 | ||
| At least one parent and one sibling affected (%) | 5.0 | 18.3 | ||
| At least one parent and one progeny affected (%) | 0 | 4.4 | ||
| At least one parent, one sibling and one progeny affected (%) | 0 | 1.7 | ||
| At least one sibling and one progeny affected (%) | 0 | 0.9 | ||
| Consanguinity (%) | 8 | ND | 0 | ND |
| Sex ratio (F/M) | 54/69 | 75/56 | 63/63 | 61/35 |
| Resting tremor (%) | 80 | 0 | 78.3 | 0 |
| Bradykinesia (%) | 99 | 0 | 93.0 | 0 |
| Rigidity (%) | 78 | 0 | 88.9 | 0 |
| Dementia (%) | 5.9 | 0 | 11.7 | 0 |
| Disease duration (mean year ± SD) | 14.0 ± 6.4 | 0 | 10.2 ± 9.4 | 0 |
| Positive L-Dopa response (% of patients) | 98 | 0 | 94 | 0 |
| L-Dopa treatment duration (mean year ± SD) | 11.5 ± 6.6 | 0 | 8.25 ± 6.25 | 0 |
The L-dopa response was considered positive when at least a 30% improvement was observed on the basis of clinical assessment.
The study population was analyzed for the LRRK2 G2019S mutation. Six of the 123 Italian patients with familial PD (4.8%) carried the G2019S mutation, which was absent from the 131 Italian controls.48 In the French population, only two of the 126 familial-PD patients (1.6%) and one of the 96 controls (1.0%) carried the G2019S mutation.27,49
GIGYF2 Gene Sequencing and Mutation Analysis
Genomic-primer design for all exons was performed with Vector NTI-Suite 6.0 (InforMax, Invitrogen). Each exon and at least 50 bp of flanking intronic sequence was PCR amplified from genomic DNA with the primer pairs listed in Table 2. PCR amplification was carried out with Taq DNA polymerase (FastStart Taq, 5U/μl, Roche) and 25 pmol of forward and reverse primers. PCR was performed in 50 μl volumes with conditions of 95°C for 2 min followed by 35 cycles of 95°C for 30 s, annealing temperature for 30 s (58°C for exons 1,3,4,6,8–10,13–15,17,18,21,23–27; 53°C for exons 2,22; 47°C for exons 5,19; 59°C for exon 7; 56°C for exons 11,12,16; and 61°C for exon 20), and 68°C for 1 min, with a final 68°C hold for 5 min. Exons 9 and 10 plus the intervening intron were small enough to be amplified in a single PCR reaction. The PCR products were purified via Montage PCR96 Plate vacuum filtration (Millipore) and sequenced with either the forward or reverse primer designed for the PCR amplification, except for several with a sequencing primer indicated by “seq” after the primer name in Table 2. Sequencing products were analyzed on an ABI 3730xl or ABI 3700 sequencer with Sequence Analysis 3.3 software (Applied Biosystems). All 27 coding exons of the GIGYF2 gene were sequenced in the 249 familial-PD cases and 227 controls. All missense mutations, deletions, and insertions were subcloned for confirmation. Purified PCR-amplification products were vector ligated with the TOPO-TA cloning sequencing kit (Invitrogen). At least six recombinant colonies were selected at random for each variation and sequenced.
Table 2.
GIGYF2 Gene Primers Used for PCR and Sequencing Analysis
| Primer Code | Sequence | PCR | Sequencing | Primer Size (bp) | PCR Size (bp) |
|---|---|---|---|---|---|
| GG2-E1F | GTCTGTTCCTACCTCTCTCACT | PCR | 22 | 485 (E1F-E1R) | |
| GG2-E1F seq | ACAGGTTTCTTCACATA | SEQ | 17 | ||
| GG2-E1R | GTTTTCAGCGAGGCACATAA | PCR | SEQ | 20 | |
| GG2-E2F | CTGATACTTTGAGATAG | PCR | SEQ | 17 | 643 (E2F-E2R) |
| GG2-E2R | GCTCCTCCTGTTGGCTCTTT | PCR | SEQ | 20 | |
| GG2-E3F | TGCTAAGGACACATACAGTTGC | PCR | SEQ | 22 | 735(E3F-E3R) |
| GG2-E3R | TGATTTTCTCCCCTCCCCAA | PCR | SEQ | 20 | |
| GG2-E4F | GTGTTATTTAGATGGTTGCT | PCR | SEQ | 20 | 728 (E4F-E4R) |
| GG2-E4R | AAGTTCTCTATCCTCCTCCT | PCR | SEQ | 20 | |
| GG2-E5F | ATTTCACTATTCAACTGT | PCR | 18 | 910 (E5F-E5R) | |
| GG2-E5F seq | TTTATGGTGGTGAATGTGTGC | SEQ | 21 | ||
| GG2-E5R | CAAAAACACCTCTAATGCTCA | PCR | 21 | ||
| GG2-E5R seq | GATGTTCAATACTTA | SEQ | 17 | ||
| GG2-E6F | TAGAAAAATGGGAAAGTGAGTGTC | PCR | SEQ | 24 | 807(E6F-E6R) |
| GG2-E6R | TGGCATTATTTTCTCCTCAG | PCR | 20 | ||
| GG2-E6R seq | CTGATACAAAAGCAAAGAT | SEQ | 19 | ||
| GG2-E7F | GGCGTTTGTGTGTGTGGATGAGG | PCR | SEQ | 23 | 625 (E7F-E7R) |
| GG2-E7R | CTTTCCCTTATTTCAGTCC | PCR | SEQ | 19 | |
| GG2-E8F | CTTTAGAGTATGTTGCCTGG | PCR | SEQ | 20 | 662 (E8F-E8R) |
| GG2-E8R | TCTTATCGGGTTCTTTGGC | PCR | SEQ | 19 | |
| GG2-E9-10F | TTAGAGGATGCTGAAGAAG | PCR | SEQ | 19 | 678 (E9-10F - E9-10R) |
| GG2-E9-10R | GCATAAGCAGAAGCCTAACT | PCR | SEQ | 20 | |
| GG2-E11F | GTAGTTAGCCTTCACAGA | PCR | SEQ | 18 | 296 (E11F-E11R) |
| GG2-E11R | TAAGAGGAGACAAAAAGAAT | PCR | SEQ | 20 | |
| GG2-E12F | ATACCATTTAGAGTTGACCA | PCR | 20 | 873 (E12F-E12R) | |
| GG2-E12F seq | TGGCATTGTTGGGAGAAGTTAC | SEQ | 22 | ||
| GG2-E12R | GGGATGCTCAACTGGTATGT | PCR | SEQ | 20 | |
| GG2-E13F | TTGACAGATGCCACCTCG | PCR | SEQ | 18 | 321 (E13F-E13R) |
| GG2-E13R | GTGTTTCCGTGACTAATA | PCR | SEQ | 18 | |
| GG2-E14F | GATGTCTGAGTTTTTATTC | PCR | SEQ | 19 | 325 (E14F-E14R) |
| GG2-E14R | AGGTGAGACTTGAGAGCAGA | PCR | SEQ | 20 | |
| GG2-E15F | CTGAATAGACCACTTTGC | PCR | 18 | 557 (E15F-E15R) | |
| GG2-E15F seq | TTTCTAATAATGTCTCCTGGG | SEQ | 21 | ||
| GG2-E15R | CGCTTATCTCCACCTGAAAT | PCR | SEQ | 20 | |
| GG2-E16F | CAAATAACCTGGAGAACCTGC | PCR | 21 | 443 (E16F-E16R) | |
| GG2-E16F seq | GCAAATGACAAGAACAAG | SEQ | 18 | ||
| GG2-E16R | ATAAAAGAGATGCTGACT | PCR | SEQ | 18 | |
| GG2-E17F | GCTCATTTAGGCACAGGGA | PCR | SEQ | 19 | 634 (E17F-E17R) |
| GG2-E17R | CGGGCTTTTTGGCTGCTACG | PCR | 20 | ||
| GG2-E17R seq | AGCAATGTTTATGTCCTG | SEQ | 18 | ||
| GG2-E18F | GAGTGTTCCTGTGCTTGGTT | PCR | SEQ | 20 | 239 (E18F-E18R) |
| GG2-E18R | GGTCAGAGTTCCCAGATACA | PCR | SEQ | 20 | |
| GG2-E19F | ATACTTTTTACTCCAGAT | PCR | SEQ | 18 | 538 (E19F-E19R) |
| GG2-E19R | TGAGCAAAAACCAGAAAGAT | PCR | SEQ | 20 | |
| GG2-E20F | GCTTTCTCTGTCATCTCTAT | PCR | SEQ | 20 | 516 (E20F-E20R) |
| GG2-E20R | CTGGGTGATGGGAGTGAGAC | PCR | SEQ | 20 | |
| GG2-E21F | TGGAGTTGAGAGAAATGC | PCR | SEQ | 18 | 308 (E21F-E21R) |
| GG2-E21R | GTAACCACTAACCCATAAT | PCR | SEQ | 19 | |
| GG2-E22F | TATCTAAAAACAGGAATC | PCR | SEQ | 18 | 329 (E22F-E22R) |
| GG2-E22R | TAAAAAGATAAGAAGACAGT | PCR | SEQ | 20 | |
| GG2-E23F | CGCCAGCAGAGGGAGTTGAT | PCR | 20 | 550 (E23F-E23R) | |
| GG2-E23F seq | AGTATTATCTTTTCTGAC | SEQ | 18 | ||
| GG2-E23R | CATAGGTGTGGTCATTTTG | PCR | SEQ | 19 | |
| GG2-E24F | CATCCATCCAGTATTGCCA | PCR | SEQ | 19 | 305 (E24F-E24R) |
| GG2-E24R | GTCCTGCTCTGTCATTCCAC | PCR | SEQ | 20 | |
| GG2-E25F | ATGTGGTATGGATGTTGTGATTAT | PCR | 24 | 616 (E25F-E25R) | |
| GG2-E25F seq | TTACCAAGTGTCTGTCATTA | SEQ | 20 | ||
| GG2-E25R | TGGGAGATACACAAAGGT | PCR | 18 | ||
| GG2-E25R seq | CATTTCTTGGGTTACTCA | SEQ | 18 | ||
| GG2-E26F | CTGAGTGGGATGTCTTGGTCTA | PCR | 22 | 395 (E26F-E26R) | |
| GG2-E26F seq | AGGGTGAGCCAAGTCTGAATCTGC | SEQ | 24 | ||
| GG2-E26R | GCACTCCTCCAACACTCA | PCR | SEQ | 18 | |
| GG2-E27F | ACAGACTTACATTCCAGC | PCR | SEQ | 18 | 246 (E27F-E27R) |
| GG2-E27R | AAGAGTAAATGGTGAGTAAGTGT | PCR | SEQ | 23 |
After the completion of these analyses, the eight exons found to contain mutations (exons 2, 4, 8, 9, 11, 14, 25, and 26) were PCR amplified and sequenced in 91 additional DNA samples obtained from the French Parkinson Disease Genetics Study Group.
Statistical Analysis
Data are presented as mean ± standard deviation. A database including genetic and phenotypic data for each PD patient and control was constructed in Statview 5.1 (SAS Institute, Cary, NC), and all statistical analyses were performed with this program. Allelic frequency (q) of GIGYF2 gene variation was expressed as relative frequency in 2n chromosomes (with “n” as the number of subjects). Statistical significance was determined by Chi square testing (p < 0.05). Hardy-Weinberg equilibrium was considered for a Hardy-Weinberg Chi square test < 3.84.
Results
GIGYF2 was screened for mutations as a candidate gene at the PARK11 locus by complete sequencing of all 27 coding exons in the index cases of families with PD from Caucasian populations (n = 249). This included 123 unrelated Italian PD patients and 126 unrelated French PD patients with at least one affected first-degree relative. For comparison, sequencing of the 27 coding exons of GIGYF2 was also performed in 131 Italian and 96 French unrelated controls. The general characteristics of PD index cases and controls are shown in Table 1. Although most patients developed PD after age 45 (n = 167), there were also cases with early (n = 78) and juvenile onset (n = 4). Most patients had only one affected first-degree relative.
We identified seven different heterozygous amino acid changes in the GIGYF2 gene in 12 unrelated index cases with PD (PmutX) (Table 3). None of these seven GIGYF2 mutations found in PD patients were observed in 227 controls. They consist of seven single-nucleotide changes resulting in amino acid substitutions in exon 2 (Asn56Ser), exon 4 (Thr112Ala), exon 8 (Ile278Val), exon 9 (Ser335Thr), exon 11 (Asn457Thr), exon 14 (Asp606Glu), and exon 26 (Val1242Ile) (Table 3, Figure 2). The Asn56Ser substitution was found in four unrelated PD patients (one Italian and three French), and the Asn457Thr substitution was present in three unrelated PD patients (two Italian and one French). All other amino acid substitutions were observed only in a single PD patient. In contrast, sequencing of all coding exons in 227 unrelated control individuals detected a single 71-year-old healthy Italian woman without a family history of PD, with an amino acid substitution in exon 25 (His1171Arg, Ctrlmut-1) (Table 3, Figure 2), which was not found in the PD population or other control subjects. The eight exons shown to contain mutations in the PD patients and the single control (exons 2, 4, 8, 9, 11, 14, 25, 26) then were sequenced in a further 91 control DNA samples from the French Parkinson Disease Genetics Study Group. In this set of additional controls, none of the identified mutations and no new variations were found.
Table 3.
GIGYF2 Mutations and Gene-Sequence Variation Identified in PD and Control Populations
| Allele Frequency (q) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Exon | Variation Reference | Nucleotide Change | Amino Acid Change | Distance (bp) | Major Alleles | PD 498 Chr. | Control 454 Chr. | p Value |
| GIGYF2 Mutations | ||||||||
| Ex2 | PDmut-1 | c.167A→G | Asn56Ser (AAC-AGC) | 16776 | A | 0.008 | 0.000 | 0.056 |
| Ex4 | PDmut-2 | c.334A→G | Thr112Ala (ACA-GCA) | 25326 | A | 0.002 | 0.000 | 0.339 |
| Ex8 | PDmut-3 | c.832A→G | Ile278Val (ATA-GTA) | 59866 | A | 0.002 | 0.000 | 0.339 |
| Ex9 | PDmut-4 | c.1003T→A | Ser335Thr (TCT-ACT) | 60129 | T | 0.002 | 0.000 | 0.339 |
| Ex11 | PDmut-5 | c.1370A→C | Asn457Thr (AAT-ACT) | 63886 | A | 0.006 | 0.000 | 0.098 |
| Ex14 | PDmut-6 | c.1818C→G | Asp606Glu (GAC-GAG) | 78790 | C | 0.002 | 0.000 | 0.339 |
| Ex25 | PDmut-7 | c.3625_3648del24 | Del LPQQQQQQ 1209-1216 | 116588 | 0.004 | 0.000 | 0.184 | |
| Ex25 | PDmut-8 | c.3648_3649insCAACAG | Ins QQ 1217 | 116611 | 0.002 | 0.000 | 0.357 | |
| Ex25 | PDmut-9 | c.3661_3672del12 | Del PPQQ 1221-1224 | 116624 | 0.002 | 0.000 | 0.357 | |
| Ex26 | PDmut-10 | c.3724G→A | Val1242Ile (GTA-ATA) | 119378 | G | 0.002 | 0.000 | 0.339 |
| Ex25 | Ctrlmut-1 | C.3512A→G | His1171Arg (CAT-CGT) | 116475 | A | 0.000 | 0.002 | 0.286 |
| GIGYF2 Variants | ||||||||
| Ex11 | Variant-rs2289912 | c.1378C→A | Pro469Thr (CCT-ACT) | 63894 | C | 0.018 | 0.020 | 0.843 |
| Ex25 | Variant-1 | c.3613_3633del21 | Del QQQQLPQ 1205-1211 | 116576 | 0.056 | 0.057 | 0.924 | |
| Ex25 | Variant-rs10555297 | c.3630_3632del3 | Del Q 1210 | 116593 | DelACA | 0.464 | 0.457 | 0.843 |
| Ex25 | Variant-2 | c.3633_3634insCAG | Ins Q 1212 | 116596 | 0.016 | 0.021 | 0.555 | |
| Ex25 | Variant-3 | c.3673_3684del12 | Del PQQQ 1225-1228 | 116636 | 0.018 | 0.007 | 0.144 | |
| GIGYF2 SNPs | ||||||||
| Ex2 | SNP-1 | c.69T→C | Ser23 (AGT-AGC) | 16678 | T | 0.002 | 0.002 | 0.948 |
| Ex8 | SNP-2 | c.828G→C | Gly276 (GGG-GGC) | 59862 | G | 0.002 | 0.002 | 0.948 |
| Ex9 | SNP-3 | c.1047T→C | Asp349 (GAT-GAC) | 60173 | T | 0.002 | 0.000 | 0.407 |
| Ex12 | SNP-rs2305138 | c.1554G→A | Glu518 (GAG-GAA) | 65187 | G | 0.054 | 0.035 | 0.159 |
| Ex13 | SNP-4 | c.1716G→T | Ala572 (GCG-GCT) | 75626 | G | 0.006 | 0.000 | 0.150 |
| Ex21 | SNP-5 | c.2835G→T | Ser945 (TCG-TCT) | 108989 | G | 0.004 | 0.000 | 0.241 |
| Ex22 | SNP-rs3816334 | c.2940G→A | Gln980 (CAG-CAA) | 113169 | G | 0.281 | 0.319 | 0.198 |
| Ex25 | SNP-rs7563724 | c.3630A→G | Pro1210 (CCA-CCG) | 116593 | A | 0.014 | 0.021 | 0.445 |
| Ex25 | SNP-rs12328151 | c.3651G→A | Pro1217 (CCG-CCA) | 116614 | G | 0.227 | 0.225 | 0.948 |
| Ex27 | SNP-6 | c.3855G→A | Ser1285 (TCG-TCA) | 125899 | G | 0.006 | 0.009 | 0.615 |
Figure 2.

Chromatograms Illustrating GIGYF2 Gene Mutations
All show analysis by forward sequencing, except for the exon 4 (Thr112Ala) mutation, which was sequenced in reverse, and insertion (Ins QQ 1217) and deletions (Del LPQQQQQQ 1209–1216 and Del PPQQ 1221–1224), for which individual alleles were sequenced after subcloning.
Most of the seven single amino acid substitutions identified in GIGYF2 in patients represent conservative amino acid sequence changes, but alignment of the human GIGYF2 protein sequence with 12 other species demonstrates that they occur within highly conserved amino acid blocks and involve residues that are highly, although not absolutely, conserved across species (Figure 3). In addition, these amino acid substitutions are significantly more frequent in PD patients (12 of 249) than in controls (1 of 227) (p < 0.003).
Figure 3.

Alignment of GIGYF2-Mutation Protein-Sequence Regions in Multiple Species
Overall identity of available amino acid sequences of various species with Homo sapiens (gi 42476299) is: Pan troglodytes (gi 114583902) 99%, Macaca mulatta (gi 109101490) 99%, Canis familaris (gi 73993973) 97%, Bos taurus (gi 156120403) 95%, Equus caballus (gi 149711637) 95%, Rattus norvegicus (gi 149016387) 94%, Mus musculus (gi 34365797) 93%, Gallus gallus (gi 118094814) 86%, Monodelphis domestica (gi 126314299) 86%, Danio rerio (gi 71834468) 56%, Xenopus laevis (gi 148223517) 56%, and Ornithorhynchus anatinus (gi 149599305) 39%. The dotted lines correspond to sequence regions not yet available or sequence regions of known sequence but nonhomology.
Among the 12 PD probands with GIGYF2 mutations, eight had at least one parent affected, one had both parents affected, and three had a single affected sibling (Figure 4). PD was present by medical history across three successive generations in three of the families and across two generations in five of the families (Figure 4). Among the probands of families with GIGYF2 missense mutations, there were a total of six females and six males with the diagnosis of PD, and the occurrence of PD in two or three successive generations in most families is consistent with autosomal-dominant inheritance. DNA samples were available from at least one affected relative in four families. In family 068-004, PD was present in three successive generations and the affected mother of the male proband was shown to have the same Asp606Glu mutation (Figure 4). The proband was diagnosed with PD at age 42, his mother at age 83. In family 057-009, the male index case inherited the Asn56Ser mutation from his affected mother. The age of PD onset was 41 in the proband and 73 in the mother. Among eight siblings of the proband, three were genotyped. Two of these, a dizygotic female twin and a brother, bear the same mutation but do not have clinically apparent PD. Their ages at the time of most-recent medical history were 61 and 73 years. Follow-up will be required to determine whether they ultimately develop PD. Another unaffected brother, age 67, does not carry the Asn56Ser mutation. In family 28-V-0056, the male index case had an affected brother, and both carried the Thr112Ala mutation. Although both brothers had onset at age 43, neither their mother, who died at age 72, nor their father, who died at age 52, had known PD (Figure 4).
Figure 4.
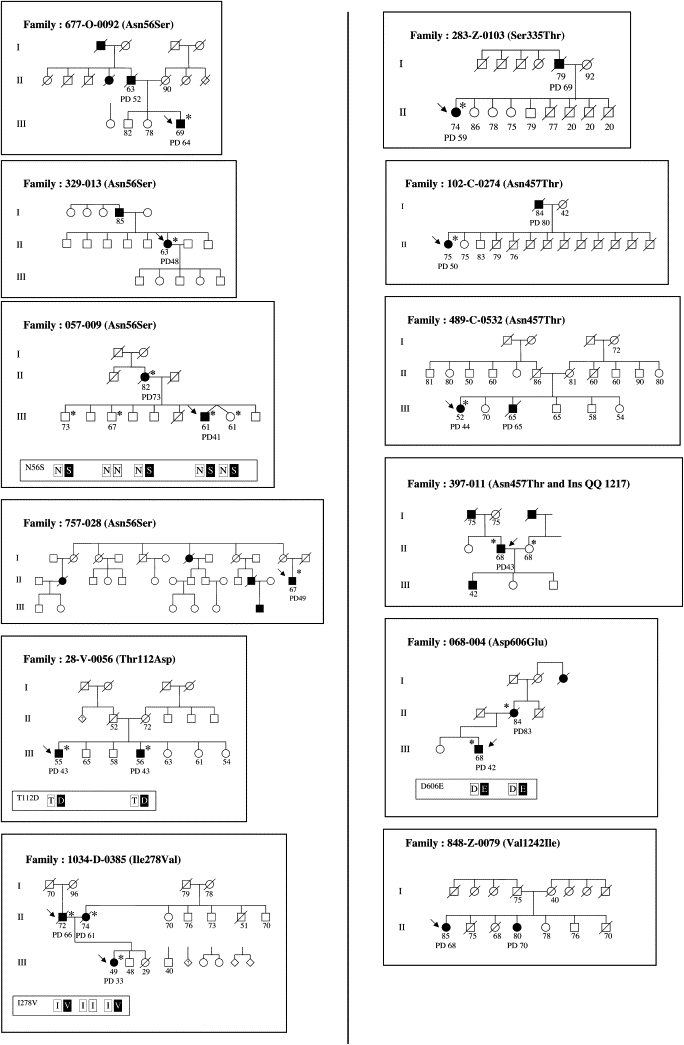
Pedigrees of Patients with GIGYF2 Missense Mutations
Arrows indicate the index cases. Patients with PD are in black, with current age or age at death indicated if known. Age of onset is the number adjacent to PD. Numbers under other family members correspond to the best-available estimate of current age or the age at death based on information provided by the index cases. Asterisks indicate individuals from whom DNA was obtained. When information on mutation segregation was available, the genotypes are indicated in the box below the pedigree.
In the Italian family 1034-D-0385, the female index case inherited the Ile278Val candidate mutation from her father. Sequence analysis of a second PD-associated gene, LRRK2, in this family revealed the previously described LRRK2 Ile1371Val mutation in the index case and her mother.50 The father had PD onset at age 66, the mother at age 61, and the index case bearing the two different gene mutations had PD onset at the much earlier age of 31 years. She has had progressive, severe PD with marked fluctuations in clinical course in spite of initial response to L-dopa therapy. At 50 years of age, she currently is partially compensated on intraduodenal L-dopa infusions.
Three other nucleotide variations were found in four index cases with PD and none of the controls. They consisted of a deletion of eight (Del LPQQQQQQ 1209–1216) and four codons (Del PPQQ 1221–1224) and an insertion of two codons (Ins QQ 1217) in exon 25 (Table 3). The Del LPQQQQQQ 1209–1216 deletion was detected in two unrelated French PD patients. The InsQQ1217 insertion was associated with the Asn457Thr missense mutation in a patient with onset at age 43. These deletion and insertion sequences are in a region of Gln (Q) and Pro (P) repeats in exon 25 that is not conserved across species. Furthermore, four other insertions and deletions have been observed in this region, with similar frequencies in PD patients and controls. One was previously described in dbSNP (rs10555297, Table 3), and three are new (Variant-1, -2, and -3, Table 3). We therefore speculate that the rare deletion and insertion sequences identified in PD patients could represent less-frequent variants without functional significance.
In addition, we found one amino acid substitution already described in dbSNP (rs2289912) (Table 3), as well as ten nonsynonymous SNPs, including four previously described in dbSNP (SNP-rsxxxx) (Table 3), and six new SNPs (SNP-X) (Table 3). All these variants appear to be polymorphisms on the basis of their similar frequencies in PD cases and controls.
Discussion
Our study has examined GIGYF2 (TNRC15) as a candidate gene for PARK11. This was accomplished by complete sequencing of all 27 coding exons of the GIGYF2 gene in two different populations of Caucasian patients with familial PD and in population-matched controls (123 Italian PD patients and 131 controls, and 126 French PD patients and 96 controls). We found seven different missense mutations in the GIGYF2 gene in 12 index patients with PD from Italy (n = 7) and France (n = 5). The following evidence supports the pathogenic role of these amino acid changes: i) they are not found in geographically matched controls (a single amino acid change was detected in only one of 127 controls) (p < 0.003); ii) the seven PD missense mutations (Asn56Ser, Thr112Ala, Ile278Val, Ser335Thr, Asn457Thr, Asp606Glu, and Val1242Ile) and the control mutation (His171Arg) were absent from an additional 91 controls; iii) they involve amino acids that are relatively conserved across 12 nonhuman species; iv) when available affected relatives were sampled, they were also found to carry the same missense mutation (except for one of the parents in the case with bilineal transmission of PD). These data provide strong support for a role of mutations in the GIGYF2 gene as a frequent cause of familial PD, at least in Italy and France, and as a major contributing factor at the PARK11 locus. The other sequence variants found in patients are probably not causative, because they affect a nonconserved region containing highly polymorphic repeats rich in Q and P residues. They are more likely rare polymorphisms.
The 12 index cases with PD and GIGYF2 missense mutations include six women and six men with a mean age at onset of 48.7 ± 10.2 years (range 33–68). The clinical features of these patients did not differ from those of patients with typical idiopathic PD (Table 4). Unilateral tremor and bradykinesia appeared at onset in most of the mutation carriers, all of whom responded to L-dopa and developed dyskinesia and/or motor fluctuations as the disease progressed.
Table 4.
Phenotype of Patients with GIGYF2 Mutations
| Patient Number |
677-O-0092 |
057-009 |
329-013 |
757-028 |
28-V-0056 |
1034-D-0385 |
283-Z-0103 |
102-C-0274 |
489-C-0532 |
397-011 |
068-004 |
848-Z-0079 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GIGYF2 Mutation |
Asn56Ser |
Thr112Asp |
Ile278Val |
Ser335Thr |
Asn457Thr |
Asp606Glu |
Val1242Ile |
|||||
| Sex (M/F) |
M |
M |
F |
M |
M |
F |
F |
F |
F |
M |
M |
F |
| Origin (ITA/FR) | ITA | FR | FR | FR | ITA | ITA | ITA | ITA | ITA | FR | FR | ITA |
| Age of Onset (yr) | 64 | 41 | 48 | 49 | 43 | 33 | 59 | 50 | 44 | 43 | 42 | 68 |
| Disease Duration at Examination (yr) | 7 | 9 | 10 | 15 | 14 | 19 | 15 | 25 | 8 | 25 | 15 | 17 |
| Resting Tremor | − | ND | + | − | + | + | + | + | + | + | + | + |
| Bradykinesia | + | ND | + | + | + | + | + | + | + | + | + | + |
| Rigidity | + | ND | + | + | + | + | + | + | + | + | + | + |
| Dementia | − | ND | − | − | − | − | − | − | − | − | − | − |
| L-Dopa Response | + | ND | + | + | + | + | + | + | + | + | + | + |
| L-Dopa Duration (yr) | 7 | ND | 8 | 10 | 10 | 15 | 13 | 21 | 3 | 25 | 7 | 17 |
| UPDRS (ON) | 14 | ND | ND | 9 | 19 | 24 | 22 | 26 | 13 | 35 | 9 | 18 |
| UPDRS (OFF) | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | 16 | 37 |
| H & Y (ON) | 2.5 | ND | ND | 1.5 | 2.5 | 3 | 3 | 3 | 2 | 2.5 | 2.5 | 2.5 |
| H & Y (OFF) | ND | ND | ND | ND | ND | 4 | 3 | ND | ND | ND | ND | 3 |
The GIGYF2 missense mutations are all found in the heterozygous state compatible with autosomal-dominant transmission. This inheritance pattern is also supported by the observation of parent-child transmission in 11 of the 12 pedigrees. However, age-dependent penetrance or reduced penetrance is likely. In family 20-V-0056, the parents of the two affected siblings died without being diagnosed with PD, and in family 757-028, two siblings with the mutation are still healthy even though they are older than their affected brother. It will be important to precisely evaluate the penetrance of GIGYF2 mutations and to determine their frequency in idiopathic PD.
It has been suggested that interactions between multiple genetic factors could have a role in the development of PD.52 In the first report on the PARK11 locus, the LOD score of 5.14 for the entire study population was noted to decrease to 4.12 when 11 of 65 families with known Parkin mutations were excluded.30 In the present study, we provide further evidence for gene interaction involving the PARK11 locus in an individual with co-occurrence of Ile278Val GIGYF2 and Ile1371Val LRRK2 mutations, who had a much earlier onset of PD than did either parent with single gene mutations. Given that GIGYF2 and LRRK2 mutations have each been found in approximately 5% and 6% of familial PD patients, respectively, it should be possible to identify additional patients with both mutations to further test their potential interaction. Although the functional role of the LRRK2 protein is not known, it contains a mixed-function kinase-like sequence, and the most common LRRK2 mutations (G2019S and Arg1441Cys) enhance the kinase activity.53 Since GIGYF2 was initially identified through its binding to the Grb10 adaptor protein34 and Grb10 is thought to function by linking proteins to tyrosine kinases,36 it will be important in future studies to determine whether GIGYF2 interacts with the LRRK2 kinase.
The function of the GIGYF2 protein is presently unknown. Consistent with the development of a neurodegenerative disease in humans with GIGYF2 mutations, GIGYF2 mRNA is strongly expressed in multiple regions of the central nervous system (Novartis Gene Expression Atlas). Together with the homologous GIGYF1 protein, GIGYF2 was cloned from a mouse-cDNA-expression library by yeast 2-hybrid screening with an N-terminal fragment of the Grb10 adaptor protein.34 The Grb10 N-terminus contains a proline-rich region, and both GIGYF proteins contain a GYF motif, which had previously been shown to bind proline-rich sequences.54 With the yeast 2-hybrid method, the GYF domains of GIGYF1 and GIGYF2 were confirmed to interact with the Grb10 proline-rich region.34 The GYF domain is named for a Gly-Tyr-Phe triad and forms a bulge-helix-bulge structure important for binding to proline sequences.54 On the basis of its flanking amino acid residues, the GYF domain of GIGYF2 can be classified as a member of the SMY2 subtype.55 There is evidence that this class of GYF domains has a preferred pro-rich binding motif that is present in several proteins with roles in mRNA splicing. GYF-domain-containing proteins in general are hypothesized to function in mRNA splicing and/or other aspects of mRNA processing, such as nuclear export.
The interaction between the GIGYF proteins and the Grb10 adaptor protein evident in the yeast 2-hybrid system has been confirmed in mammalian cells expressing endogenous levels of Grb10 and a myc-tagged GYF-domain-containing fragment of the GIGYF1 protein.34 Further studies demonstrated that this fragment of GIGYF1 was recruited to activated IGF-I receptors, presumably via the Grb10 adaptor. Of interest, overexpression of the GIGYF1 fragment resulted in augmented IGF-I receptor signaling. Similar studies with full-length GIGYF1 or GIGYF2 have not been possible, because the full-length proteins form toxic inclusion bodies in cells when overexpressed, presumably as a consequence of protein self-aggregation.34 In future experiments at GIGYF2 cellular concentrations below the threshold for inclusion-body formation, it will be important to determine whether the association of GIGYF2 with PD involves its interaction with Grb10 and the IGF and/or insulin signaling pathways. It will also be important to determine whether the mutations that we have identified lead to the development of PD by altering GIGYF2 interactions with Grb10 and the IGF and/or insulin pathways or other functions of the GIGYF2 protein.
Web Resources
The URLs for data presented herein are as follows:
Novartis Gene Expression Atlas, http://expression.gnf.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Parkinson Institute DNA Bank, http://www.parkinson.it/dnabank.html
Acknowledgments
We thank the patients and their relatives for their contributions. The Italian population DNA samples were from the “Human Genetic Bank of Patients Affected by Parkinson Disease and Parkinsonisms” of the Parkinson Institute, Istituti Clinici di Perfezionamento (Italy). This DNA bank is supported by the Italian Telethon Foundation (GTF04007) and by the Fondazione Grigioni per il Morbo di Parkinson. We also thank the French Parkinson Disease Genetics Study Group (Y. Agid, A.-M. Bonnet, M. Borg, A. Brice, E. Broussolle, P. Damier, A. Destée, A. Dürr, F. Durif, S. Lesage, E. Lohmann, P. Pollak, O. Rascol, F. Tison, C. Tranchant, M. Vérin, F. Viallet, and M. Vidailhet) and the DNA and Cell Bank of the Institut Fédératif des Neurosciences (IFR 070) (CHU Pitié-Salpêtrière, AP-HP, Paris, France) for sample preparation. We express gratitude to V. Bonifati and A. Di Fonzo for sharing their LRRK2 data on family 1034-D-0385, and to S. de la Monte and J. Klysik for their contributions to the study-project evolution. This work was supported by grants from the Fondation Fértilité et Stérilité, France (C.L.), the Association Française des Femmes Dîplomées des Universités (Paris, France) (C.L.), the Association France Parkinson (Paris, France) (A.B., C.L.), the Agence Nationale de la Recherche (ANR) (ANR-05-NEUR-019 to A.B.), the Hallett Center for Diabetes and Endocrinology, (RI, USA) (R.J.S.), and the National Institutes of Health (NIH) (MD, USA) (DK43038 to R.J.S.).
References
- 1.Lang A.E., Lozano A.M. Parkinson's disease. First of two parts. N. Engl. J. Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 2.Payami H., Zareparsi S. Genetic epidemiology of Parkinson's disease. J. Geriatr. Psychiatry Neurol. 1998;11:98–106. doi: 10.1177/089198879801100207. [DOI] [PubMed] [Google Scholar]
- 3.Funayama M., Hasegawa K., Kowa H., Saito M., Tsuji S., Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 2002;51:296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- 4.Gasser T., Muller-Myhsok B., Wszolek Z.K., Oehlmann R., Calne D.B., Bonifati V., Bereznai B., Fabrizio E., Vieregge P., Horstmann R.D. A susceptibility locus for Parkinson's disease maps to chromosome 2p13. Nat. Genet. 1998;18:262–265. doi: 10.1038/ng0398-262. [DOI] [PubMed] [Google Scholar]
- 5.Hicks A.A., Petursson H., Jonsson T., Stefansson H., Johannsdottir H.S., Sainz J., Frigge M.L., Kong A., Gulcher J.R., Stefansson K., Sveinbjornsdottir S. A susceptibility gene for late-onset idiopathic Parkinson's disease. Ann. Neurol. 2002;52:549–555. doi: 10.1002/ana.10324. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y., Fallon L., Lashuel H.A., Liu Z., Lansbury P.T.J. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 7.Matsumine H., Saito M., Shimoda-Matsubayashi S., Tanaka H., Ishikawa A., Nakagawa-Hattori Y., Yokochi M., Kobayashi T., Igarashi S., Takano H. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2–27. Am. J. Hum. Genet. 1997;60:588–596. [PMC free article] [PubMed] [Google Scholar]
- 8.Pankratz N., Nichols W.C., Uniacke S.K., Halter C., Rudolph A., Shults C., Conneally P.M., Foroud T. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am. J. Hum. Genet. 2002;71:124–135. doi: 10.1086/341282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polymeropoulos M.H., Higgins J.J., Golbe L.I., Johnson W.G., Ide S.E., Di Iorio G., Sanges G., Stenroos E.S., Pho L.T., Schaffer A.A. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 1996;274:1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- 10.Schultheis P.J., Hagen T.T., O'Toole K.K., Tachibana A., Burke C.R., McGill D.L., Okunade G.W., Shull G.E. Characterization of the P5 subfamily of P-type transport ATPases in mice. Biochem. Biophys. Res. Commun. 2004;323:731–738. doi: 10.1016/j.bbrc.2004.08.156. [DOI] [PubMed] [Google Scholar]
- 11.Scott W.K., Nance M.A., Watts R.L., Hubble J.P., Koller W.C., Lyons K., Pahwa R., Stern M.B., Kolcher A., Hiner B.C. Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA. 2001;286:2239–2244. doi: 10.1001/jama.286.18.2239. [DOI] [PubMed] [Google Scholar]
- 12.Strauss K.M., Martins L.M., Plun-Favreau H., Marx F.P., Kautzmann S., Berg D., Gasser T., Wszolek Z., Muller T., Bornemann A. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 13.Valente E.M., Bentivoglio A.R., Dixon P.H., Ferraris A., Ialongo T., Frontali M., Albanese A., Wood N.W. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 2001;68:895–900. doi: 10.1086/319522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Duijn C.M., Dekker M.C., Bonifati V., Galjaard R.J., Houwing-Duistermaat J.J., Snijders P.J., Testers L., Breedveld G.J., Horstink M., Sandkuijl L.A. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 2001;69:629–634. doi: 10.1086/322996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonifati V., Rizzu P., van Baren M.J., Schaap O., Breedveld G.J., Krieger E., Dekker M.C., Squitieri F., Ibanez P., Joosse M. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 16.Brice A. Genetics of Parkinson's disease: LRRK2 on the rise. Brain. 2005;128:2760–2762. doi: 10.1093/brain/awh676. [DOI] [PubMed] [Google Scholar]
- 17.Hedrich K., Kann M., Lanthaler A.J., Dalski A., Eskelson C., Landt O., Schwinger E., Vieregge P., Lang A.E., Breakefield X.O. The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset parkinsonism. Hum. Mol. Genet. 2001;10:1649–1656. doi: 10.1093/hmg/10.16.1649. [DOI] [PubMed] [Google Scholar]
- 18.Ibanez P., Bonnet A.M., Debarges B., Lohmann E., Tison F., Pollak P., Agid Y., Durr A., Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 19.Ibanez P., De Michele G., Bonifati V., Lohmann E., Thobois S., Pollak P., Agid Y., Heutink P., Durr A., Brice A. Screening for DJ-1 mutations in early onset autosomal recessive parkinsonism. Neurology. 2003;61:1429–1431. doi: 10.1212/01.wnl.0000094121.48373.fd. [DOI] [PubMed] [Google Scholar]
- 20.Ibanez P., Lesage S., Lohmann E., Thobois S., De Michele G., Borg M., Agid Y., Durr A., Brice A. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain. 2006;129:686–694. doi: 10.1093/brain/awl005. [DOI] [PubMed] [Google Scholar]
- 21.Lesage S., Lohmann E., Tison F., Durif F., Durr A., Brice A. Rare heterozygous parkin variants in French early-onset Parkinson's disease patients and controls. J. Med. Genet. 2007;45:43–46. doi: 10.1136/jmg.2007.051854. [DOI] [PubMed] [Google Scholar]
- 22.Lincoln S., Vaughan J., Wood N., Baker M., Adamson J., Gwinn-Hardy K., Lynch T., Hardy J., Farrer M. Low frequency of pathogenic mutations in the ubiquitin carboxy-terminal hydrolase gene in familial Parkinson's disease. Neuroreport. 1999;10:427–429. doi: 10.1097/00001756-199902050-00040. [DOI] [PubMed] [Google Scholar]
- 23.Paisan-Ruiz C., Lang A.E., Kawarai T., Sato C., Salehi-Rad S., Fisman G.K., Al-Khairallah T., St George-Hyslop P., Singleton A., Rogaeva E. LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology. 2005;65:696–700. doi: 10.1212/01.wnl.0000167552.79769.b3. [DOI] [PubMed] [Google Scholar]
- 24.Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 25.Ramirez A., Heimbach A., Grundemann J., Stiller B., Hampshire D., Cid L.P., Goebel I., Mubaidin A.F., Wriekat A.L., Roeper J. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 26.Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 27.Lesage S., Durr A., Tazir M., Lohmann E., Leutenegger A.L., Janin S., Pollak P., Brice A. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N. Engl. J. Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- 28.Ozelius L.J., Senthil G., Saunders-Pullman R., Ohmann E., Deligtisch A., Tagliati M., Hunt A.L., Klein C., Henick B., Hailpern S.M. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 29.Pankratz N., Nichols W.C., Uniacke S.K., Halter C., Murrell J., Rudolph A., Shults C.W., Conneally P.M., Foroud T. Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinson disease families. Hum. Mol. Genet. 2003;12:2599–2608. doi: 10.1093/hmg/ddg270. [DOI] [PubMed] [Google Scholar]
- 30.Pankratz N., Nichols W.C., Uniacke S.K., Halter C., Rudolph A., Shults C., Conneally P.M., Foroud T. Significant linkage of Parkinson disease to chromosome 2q36–37. Am. J. Hum. Genet. 2003;72:1053–1057. doi: 10.1086/374383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prestel J., Sharma M., Leitner P., Zimprich A., Vaughan J.R., Durr A., Bonifati V., De Michele G., Hanagasi H.A., Farrer M. PARK11 is not linked with Parkinson's disease in European families. Eur. J. Hum. Genet. 2005;13:193–197. doi: 10.1038/sj.ejhg.5201317. [DOI] [PubMed] [Google Scholar]
- 32.Maraganore D.M., de Andrade M., Lesnick T.G., Strain K.J., Farrer M.J., Rocca W.A., Pant P.V., Frazer K.A., Cox D.R., Ballinger D.G. High-resolution whole-genome association study of Parkinson disease. Am. J. Hum. Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 34.Giovannone B., Lee E., Laviola L., Giorgino F., Cleveland K.A., Smith R.J. Two novel proteins that are linked to insulin-like growth factor (IGF-I) receptors by the Grb10 adapter and modulate IGF-I signaling. J. Biol. Chem. 2003;278:31564–31573. doi: 10.1074/jbc.M211572200. [DOI] [PubMed] [Google Scholar]
- 35.Dufresne A.M., Smith R.J. The adapter protein GRB10 is an endogenous negative regulator of insulin-like growth factor signaling. Endocrinology. 2005;146:4399–4409. doi: 10.1210/en.2005-0150. [DOI] [PubMed] [Google Scholar]
- 36.Holt L.J., Siddle K. Grb10 and Grb14: enigmatic regulators of insulin action–and more? Biochem. J. 2005;388:393–406. doi: 10.1042/BJ20050216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langlais P., Dong L.Q., Ramos F.J., Hu D., Li Y., Quon M.J., Liu F. Negative regulation of insulin-stimulated mitogen-activated protein kinase signaling by Grb10. Mol. Endocrinol. 2004;18:350–358. doi: 10.1210/me.2003-0117. [DOI] [PubMed] [Google Scholar]
- 38.Laviola L., Giorgino F., Chow J.C., Baquero J.A., Hansen H., Ooi J., Zhu J., Riedel H., Smith R.J. The adapter protein Grb10 associates preferentially with the insulin receptor as compared with the IGF-I receptor in mouse fibroblasts. J. Clin. Invest. 1997;99:830–837. doi: 10.1172/JCI119246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mori K., Giovannone B., Smith R.J. Distinct Grb10 domain requirements for effects on glucose uptake and insulin signaling. Mol. Cell. Endocrinol. 2005;230:39–50. doi: 10.1016/j.mce.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Russo V.C., Gluckman P.D., Feldman E.L., Werther G.A. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 2005;26:916–943. doi: 10.1210/er.2004-0024. [DOI] [PubMed] [Google Scholar]
- 41.Schulingkamp R.J., Pagano T.C., Hung D., Raffa R.B. Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci. Biobehav. Rev. 2000;24:855–872. doi: 10.1016/s0149-7634(00)00040-3. [DOI] [PubMed] [Google Scholar]
- 42.Craft S., Watson G.S. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–178. doi: 10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 43.Hu G., Jousilahti P., Bidel S., Antikainen R., Tuomilehto J. Type 2 diabetes and the risk of Parkinson's disease. Diabetes Care. 2007;30:842–847. doi: 10.2337/dc06-2011. [DOI] [PubMed] [Google Scholar]
- 44.Offen D., Shtaif B., Hadad D., Weizman A., Melamed E., Gil-Ad I. Protective effect of insulin-like-growth-factor-1 against dopamine-induced neurotoxicity in human and rodent neuronal cultures: possible implications for Parkinson's disease. Neurosci. Lett. 2001;316:129–132. doi: 10.1016/s0304-3940(01)02344-8. [DOI] [PubMed] [Google Scholar]
- 45.Takahashi M., Yamada T., Tooyama I., Moroo I., Kimura H., Yamamoto T., Okada H. Insulin receptor mRNA in the substantia nigra in Parkinson's disease. Neurosci. Lett. 1996;204:201–204. doi: 10.1016/0304-3940(96)12357-0. [DOI] [PubMed] [Google Scholar]
- 46.Hughes A.J., Daniel S.E., Kilford L., Lees A.J. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes A.J., Daniel S.E., Lees A.J. Improved accuracy of clinical diagnosis of Lewy body Parkinson's disease. Neurology. 2001;57:1497–1499. doi: 10.1212/wnl.57.8.1497. [DOI] [PubMed] [Google Scholar]
- 48.Goldwurm S., Di Fonzo A., Simons E.J., Rohe C.F., Zini M., Canesi M., Tesei S., Zecchinelli A., Antonini A., Mariani C. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson's disease and originates from a common ancestor. J. Med. Genet. 2005;42:e65. doi: 10.1136/jmg.2005.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lesage S., Ibanez P., Lohmann E., Pollak P., Tison F., Tazir M., Leutenegger A.L., Guimaraes J., Bonnet A.M., Agid Y. G2019S LRRK2 mutation in French and North African families with Parkinson's disease. Ann. Neurol. 2005;58:784–787. doi: 10.1002/ana.20636. [DOI] [PubMed] [Google Scholar]
- 50.Di Fonzo A., Tassorelli C., De Mari M., Chien H.F., Ferreira J., Rohe C.F., Riboldazzi G., Antonini A., Albani G., Mauro A. Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson's disease. Eur. J. Hum. Genet. 2006;14:322–331. doi: 10.1038/sj.ejhg.5201539. [DOI] [PubMed] [Google Scholar]
- 51.(1987). Fahn S, Elton, RL, and the UPDRS Development Committee (Unified Parkinson's). Disease Rating Scale: Recent Developments in Parkinson's Disease, Volume 2, S. Fahn, C.D. Marsden, D. Calne, and M. Goldstein, eds. (Macmillan Healthcare Information, Florham Park, NJ, USA), 153–163.
- 52.Gasser T. Genetics of Parkinson's disease. Curr. Opin. Neurol. 2005;18:363–369. doi: 10.1097/01.wco.0000170951.08924.3d. [DOI] [PubMed] [Google Scholar]
- 53.West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freund C., Dotsch V., Nishizawa K., Reinherz E.L., Wagner G. The GYF domain is a novel structural fold that is involved in lymphoid signaling through proline-rich sequences. Nat. Struct. Biol. 1999;6:656–660. doi: 10.1038/10712. [DOI] [PubMed] [Google Scholar]
- 55.Kofler M.M., Freund C. The GYF domain. FEBS J. 2006;273:245–256. doi: 10.1111/j.1742-4658.2005.05078.x. [DOI] [PubMed] [Google Scholar]