

Abstract
Unobstructed vision requires a particular refractive index of the lens, a measure based on the organization of the structural proteins within the differentiated lens cells. To ensure an intact lens structure, homeostasis within the lens cells is indispensable. Alterations of the lens structure result in opacity and lead to cataract. Renal glucosuria is defined by elevated glucose level in the urine without hyperglycemia and without evidence of morphological renal anomalies. In a Swiss family with autosomal dominant juvenile cataract, microcornea, and renal glucosuria, we have identified a nonsense mutation in a member of the carboxylic acid transporter family SLC16. The underlying gene defect in SLC16A12 resides within a 3 cM region on chromosome 10q23.13 defined by linkage mapping of this phenotype. We found tissue-specific variability of SLC16A12 transcript levels in control samples, with high expression in the eye and kidney, the two organs affected by this syndrome. This report demonstrates biological relevance of this solute carrier. We hypothesize that SLC16A12 is important for lens and kidney homeostasis and discuss its potential role in age-related cataract.
Main Text
Lens transparency, a requirement of unobstructed vision, is achieved by ordered events of cell differentiation accompanied by controlled arrangement of proteins, mainly crystallins. Differentiation of the lens cells follows a precise sequence of events.1 Mitotic activity of a small number of lens epithelial cells (LEC) provides a continuous supply of new cells that, upon signal-induced differentiation, will begin with a cellular elongation process, followed by the breakdown of the nucleus and organelles. Concomitantly, some but not all metabolic activity ceases. Tightly packed, highly elongated cells comprise the several millimeter-thick lens structure. Changes in this structure, composition, or the assembly of the structural proteins, of which crystallines make about 90%, will result in alteration of the refractive index. This increasing opacity of the lens is termed cataract. Defined by age of onset, one distinguishes between congenital (infantile), juvenile, and age-related cataract. The first two, also referred to as childhood cataract, show wide heterogeneity with respect to the genetic and phenotypic aspects.2 Frequently, mutations that disturb the development of the lens occur in structural lens proteins and will lead to childhood cataract. Among the genetic factors that influence age-related cataract, no genes with mutations have yet been identified. Genes involved in recessively or dominantly inherited cataract encode structural components of the lens cells but also components of the cytoskeleton, of the cell membrane, transcription factors, metabolic proteins, chromatin-modifying protein −4B, and the gene TMEM114, encoding a protein with four predicted transmembrane domains but of unknown function.3–6
Occasionally, cataract is accompanied by additional symptoms, among them microcornea.4,5 Of particular interest to this work is a Swiss family with juvenile cataract, associated with microcornea and renal glucosuria.7 Although renal glucosuria is not considered a disease, affected individuals show characteristic elevation of glucose concentration in the urine, without evidence of other renal tubular defects. The pattern of inheritance has been described as codominant with variable penetrance.8 In the family described by Vandekerckhove and colleagues,7 9 of 12 cataract patients also showed elevated levels of glucose in their urine, in the absence of any other renal or metabolic abnormalities (Figures 1 and 2; Table 1).
Figure 1.
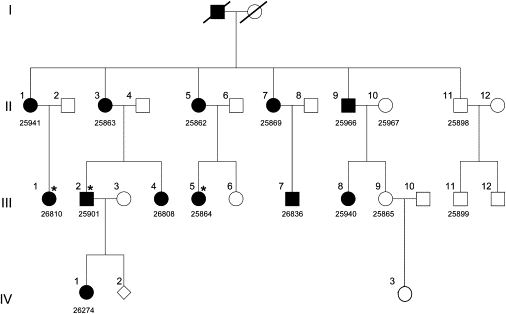
Pedigree of Swiss Family Segregating Juvenile Cataract with Microcornea and Glucosuria
Modified after Vandekerckhove et al.7 Filled symbols represent affected status for all three phenotypes, with three exceptions indicated by star; III-1 and III-2 are negative and III-5 is borderline for glucosuria (Table 1). Five-digit laboratory identification numbers (given below pedigree symbols) were assigned prior to DNA extraction. Family members IV-2 and IV-3 were not tested for any of the three phenotypes.
Figure 2.
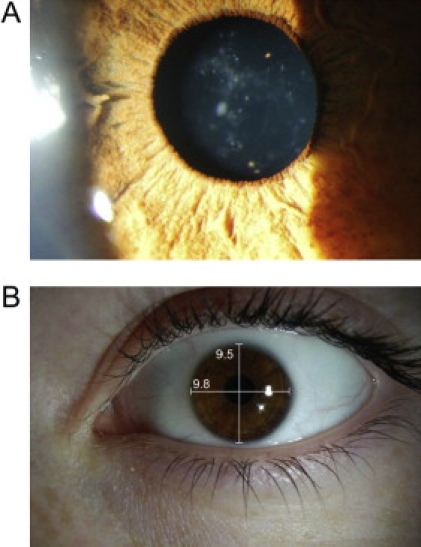
Cataract and Microcornea Phenotype of Patient III-5
Preoperative cortico-nuclear cataract in right eye is shown in (A) and microcornea (9.8 × 9.5 mm) in (B).
Table 1.
Summary of Clinical Data
| Pedigree ID | Cataract | Microcornea | Glucosuria |
|---|---|---|---|
| II-1 | + | + | + |
| II-3 | + | + | + |
| II-5 | + | + | + |
| II-7 | + | + | +a |
| II-9 | + | + | + |
| II-11 | - | - | - |
| III-1 | + | + | -a |
| III-2 | + | + | - |
| III-4 | + | + | + |
| III-5 | + | + | +/−b |
| III-7 | + | + | + |
| III-8 | + | + | + |
| III-9 | - | - | +c |
| III-11 | - | - | - |
Pedigree identification numbers are taken from Figure 1. Presence/absence of cataract, microcornea, and renal glucosuria is given as +/−. Assignment of microcornea was given if values were below 11.0 mm. Glucosuria was evaluated as positive (+) if glucose concentration was above 0.8 mmol/L.
Glucose values were generally obtained from postprandial samples except for patients II-7 and III-1.
Test performed during pregnancy.
Borderline value.
Determination of the underlying genetic defect and whether cataract and glucosuria are caused by the same pathogenic alteration was subject of this study. We began with linkage analysis with the Affymetrix GeneChip Human Mapping 10K Array, version 2.0 (Affymetrix, Santa Clara, CA). Nonparametric linkage analysis with all genotypes of a chromosome simultaneously was carried out with MERLIN, and parametric linkage analysis was performed by the program ALLEGRO9 assuming a disease allele frequency of 0.0001 and autosomal dominant inheritance with full penetrance for cataract. Haplotypes were reconstructed with ALLEGRO and presented graphically with HaploPainter.10 Results predicted that the disease-causing mutation for the juvenile cataract and microcornea maps to an interval on chromosome 10q23.31 (Figure 3) of 3 cM, spanning between the SNP markers rs701826 and rs2254391 (Figure 4). For the glucosuria phenotype, no significant LOD score was obtained (data not shown), probably resulting from incomplete penetrance. Calculations of 50% penetrance for affected patients revealed a LOD score slightly above 2 on a region of chromosome 10, which overlaps with the 3 cM interval for cataract. These findings suggest that more affected family members are required to obtain a significant LOD score for glucosuria.
Figure 3.
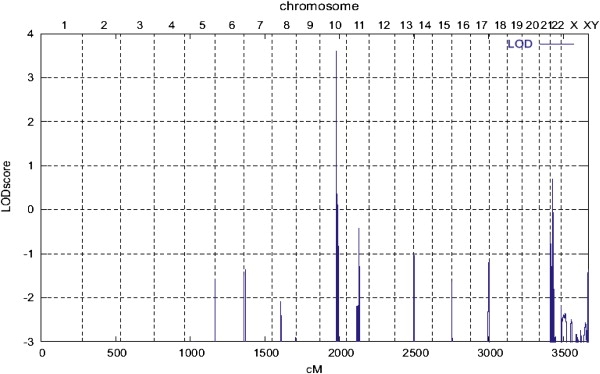
LOD Scores across the Genome for the Phenotype of Cataract with Microcornea
Chromosome number and genetic distances in cM (centi Morgan) is horizontally displayed; LOD score is given along the vertical axis.
Figure 4.
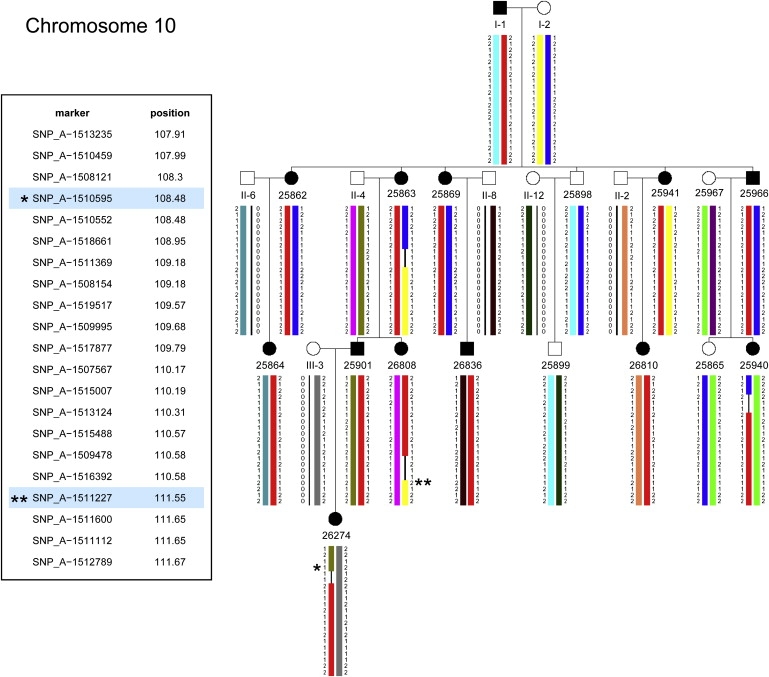
Haplotypes for the Cataract-Linked Region on Chromosome 10
Recombinations in patients 26808 (III-4) and 26274 (IV-1) define a critical interval of 2.6 Mega bases (Mb) delimited by markers SNP_A_1510595 (rs701826) (∗) and SNP_A_1511227 (rs2254391) (∗∗) at positions 108.48 and 111.55 cM, respectively. Disease chromosome in red.
The NCBI map viewer (Build 36.2, August 2007) displayed 31 genes and 3 phenotypes (selection shown in Table 2) within the linkage interval on chromosome 10q23.31. Among the phenotypes, none seemed obviously related to cataract. Distal to this linkage interval maps the homeobox gene PITX3 (MIM 602669). Mutations in this gene are known to cause the dominant form of congenital cataract and anterior segment mesenchymal dysgenesis (ASMD).11,12 We performed sequence analysis in the DNA of one affected patient (II-1) without finding a mutation (data not shown; primer sequences are available upon request).
Table 2.
Phenotype and Loci that Map to the Linkage Interval on Chromosome 10
| Symbol |
Description |
MIM |
Position |
|---|---|---|---|
| Phenotypes | |||
| TNFRSF6 | tumor necrosis factor receptor superfamily, member 6 | 134637 | 10q24.1 |
| LIPA | Wolman disease, liposomal acid lipase deficiency | 278000 | 10q24-q25 |
| SCZD11 | schizophrenia susceptibility locus | 608078 | 10q22.3 |
| Loci | MIM or Ensembl | GeneID | |
| LIPF | lipase, gastric | 601980 | 8513 |
| LIPK | lipase, family member K | ENSG00000204021 | 643414 |
| LIPN | member N | ENSG00000204020 | 643418 |
| LIPM | member M | ENSG00000173239 | 340654 |
| ANKRD22 | ankyrin repeat domain 22 | ENSG00000152766 | 118932 |
| STAMBPL1 | STAM binding protein-like 1 | ENSG00000138134 | 57559 |
| ACTA2 | actin, alpha 2, smooth muscle, | ENSG00000107796 | 59 |
| FAS | Fas (TNF receptor superfamily, member 6) | 134637 | 355 |
| CH25H | cholesterol 25-hydroxylase | 604551 | 9023 |
| LIPA | lipase A, (Wolman disease) | 278000 | 3988 |
| IFIT2 | interferon-induced protein with tetratricopeptide repeats 2 | 147040 | 3433 |
| IFIT3 | repeats 3 | 604650 | 3437 |
| IFIT1L | repeats 1-like | ENSG00000204010 | 439996 |
| IFIT1 | repeats 1 | 147690 | 3434 |
| IFIT5 | repeats 5 | ENSG00000152778 | 24138 |
| SLC16A12 | solute carrier family 16, member 12 (monocarboxylic acid transporter 12) | ENSG00000152779 | 387700 |
| MIRN107 | microRNA 107 | 406901 | |
| PANK1 | pantothenate kinase 1 | 606160 | 53354 |
| MPHOSPH1 | M-phase phosphoprotein 1 | 605498 | 9585 |
| HTR7 | 5-hydroxytryptamine (serotonin) receptor 7 (adenylate cyclase-coupled) | 182137 | 3363 |
| RPP30 | ribonuclease P/MRP 30kDa subunit | 606115 | 10556 |
| ANKRD1 | ankyrin repeat domain 1 (cardiac muscle) | 609599 | 27063 |
| NUDT9P1 | nudix (nucleoside diphosphate linked moiety X)-type motif 9 pseudogene 1 | 119369 | |
| PCGF5 | polycomb group ring finger 5 | ENSG00000180628 | 84333 |
| HECTD2 | HECT domain containing 2 | ENSG00000165338 | 143279 |
| PPP1R3C | protein phosphatase 1, regulatory (inhibitor) subunit 3C | 602999 | 5507 |
| TNKS2 | tankyrase, TRF1-interacting ankyrin-related ADP-ribose polymerase 2 | 607128 | 80351 |
From the NCBI map viewer (Build 36.2, August 2007), we selected 27 annotated genes, and all phenotypes located to the affected region on chromosome 10q23.31 are shown. For identification, MIM code, Ensembl code, and/or GeneID (NCBI) is given.
Considering the function of each of the 31 genes within the linkage interval, we reasoned that FAS and SLC16A12 were potential candidate genes. FAS (MIM 134637), encoding the tumor necrosis factor receptor super family member 6, could play a role during differentiation of lens cells.13 DNA sequence analysis did not reveal a coding region mutation, but upstream of the transcription unit we detected a deletion of six thymidin residues. This alteration was also found in unaffected family members, so it was excluded as disease causing. In addition, a deletion of seven thymidin residues at the same site has been reported as SNP rs3074157.
Metabolic requirements of the lens cells can be accommodated by establishing a transport system for small molecules. The gene encoding the solute carrier SLC16A12 (ENSG00000152779, NCBI GeneID 387700) belongs to a family of 14 monocarboxylate transporters.14 All members display an average size of 40–50 kDa and are characterized by 12 transmembrane domains (TMDs). Besides DNA sequence and gene annotation for SLC16A12, no information on its genetic and biochemical properties was available. We sequenced the five coding exons (3 to 8) including approximately 50–100 base pairs of their respective flanking introns and UTR regions (primer information in Table 3) and found a heterozygous mutation in exon 6: c.643C→T, which is predicted to lead to a premature termination codon p.Q215X (Figure 5). This mutation was found in all 12 cataract patients of the Swiss family whereas the three unaffected individuals (II-11, III-9, and III-11) did not carry this alteration. It was also absent in 370 normal alleles, two of which were from an unrelated spouse of the family (II-10) (Figure 1). Thus, we considered SLC16A12 as the gene associated with the development of this cataract.
Table 3.
Primer Information
| Gene - Exon - Direction | Sequence | Purpose |
|---|---|---|
| SLC16A12 ex3f | gtctgccccagtctagtattca | genomic sequencing |
| SLC16A12 ex3r | cggaaatacacacacaccaca | genomic sequencing |
| SLC16A12 ex4f | ccctgtggtggttgaacact | genomic sequencing |
| SLC16A12 ex4r | tggctttggctgaagatagg | genomic sequencing |
| SLC16A12 ex5f | tctattccaaccctgctgct | genomic sequencing |
| SLC16A12 ex5r | ccagctctgtttaactgctagg | genomic sequencing |
| SLC16A12 ex6af | gaatgactggtgaggggaga | genomic sequencing |
| SLC16A12 ex6ar | aacagaacggagacggctaa | genomic sequencing |
| SLC16A12 ex6bf | cggggagccttactcattct | genomic sequencing |
| SLC16A12 ex6br | agtaccagcaagggagatgc | genomic sequencing |
| SLC16A12 ex7f | cacaatgggaaagccatctc | genomic sequencing |
| SLC16A12 ex7r | atggttttgggggctcttat | genomic sequencing |
| SLC16A12 ex8f | caaagttacaattggtggtgct | genomic sequencing |
| SLC16A12 ex8r | agttatgagcacaaatcccaaa | genomic sequencing |
| SLC16A12exon3f | caggaagtcactggacagca | RT-PCR |
| SLC16A12exon5r | caggaagtcactggacagca | RT-PCR |
| SLC16A12exon4_5f | gtgtgaccatgctctgtgct | RT-PCR |
| SLC16A12exon6r | aagacaaagcccccaagaat | RT-PCR |
| RPII_cDNA_N20_F | tgtggagatcttcacggtgct | RT-PCR |
| RPII_cDNA_N234_R | cataagcacgtccaccgtttc | RT-PCR |
The name of primers contains information about the gene (SLC16A12 or POLR2A, exon, and direction, forward [f,F] or reverse [r,R]). The sequence of primers points 5′ to 3′.
Figure 5.
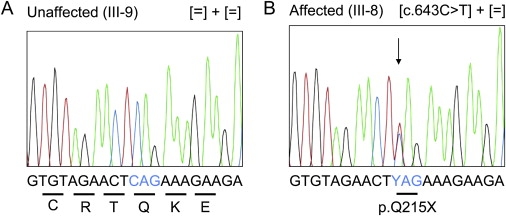
Mutation Screening in SCL16A12
Electropherogram shows the mutation in exon 6 within the context of 21 nucleotides. Shown are both the DNA sequence of the unaffected individual III-9 (A) and the heterozygous change of C→T (Y) in the affected individual III- 8 (B). The genotypes are given in brackets. Translation codons are underlined and amino acid identity is written below with single letter code.
Knowledge of the expression pattern of SLC16A12 would aid in understanding its effect on cataract with microcornea and glucosuria. For that purpose, we performed RT-PCR experiments based on established procedures15 with RNA from several organs, including the two affected structures, lens and kidney (Figure 6). In general, the solute carrier was detected in retina, brain, lung, kidney, liver, and testis, although at remarkably different levels. We compared the amount of SLC16A12 transcripts with that of the endogenous control transcripts from RNA polymerase II gene, and we estimated that the solute carrier seemed most highly expressed in kidney, followed by retina, lung, and testis and very weakly in brain and liver (Figure 6B). The expected RT-PCR fragment was not detected in blood cells (data not shown). In addition, we assayed SLC16A12 transcripts isolated from human retina, retinal pigment epithelial cells (RPE), and lens of a 47-year-old eye donor lacking any signs of cataract and confirmed the expression pattern we had seen from purchased RNA (Figures 6B and 6C). Importantly, SLC16A12 transcripts were also detected in the human lens (Figure 6C). Because only a very small portion of the lens, namely the lens epithelial cells (LECs), may be transcriptionally active, we concluded that expression of SLC16A12 in the LECs must be relatively high. Our RT-PCR data show that SLC16A12 expression is regulated in a cell/tissue-specific manner. These observations concur with the reported expression pattern of other SLC16 family members, which can be either ubiquitous or tissue specific.14,16 Tissue-specific regulation of SLC16A12 expression is further supported by the lack of additional manifesting symptoms in the Swiss family.
Figure 6.
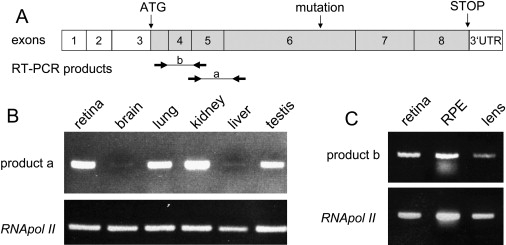
Expression Studies of SLC16A12
(A) Schematic representation of exons. Protein coding regions are displayed in darker shade. Translation initiates within exon 3 (vertical arrow, ATG) and terminates within exon 8 (vertical arrow, STOP). Mutation, c.643C→T, in exon 6 (vertical arrow) is predicted to lead to a premature termination. Positions of primers are indicated by forward and reverse horizontal arrows, yielding RT-PCR product a (exon spanning 4_5 to exon 6) and product b (exon 3 to exon 5).
(B) RT-PCR analyses from human tissues with commercially available mRNA. Primers to yield product a were used to amplify SLC16A12 transcripts. RNA Polymerase II (POLR2A) transcripts served as endogenous control.
(C) RT-PCR analysis from tissues isolated from a single human donor eye. Primers to yield product b of SLC16A12 and of POLR2A (control) were used for amplification. Lens RNA was 3-fold concentrated compared to the other samples.
This report provides the first evidence (to our knowledge) for the physiological function of SLC16A12. In combination with the knowledge of the transport function of other SLC16 isoforms, a prediction of molecular activity is possible. These transporters are highly conserved throughout evolution and can be found in prokaryotes as well as eukaryotes, from yeast to mammals. They can transport lactate, aromatic amino acids, short-chain fatty acids, butyrate, ketones, or thyroid hormone in a proton-dependent or -independent fashion.14 Subcellular localization of some family members in the eye and also kidney points to highly specific tasks of molecular transport.17,18 Although neither the localization in the lens or kidney nor substrate specificity of this transporter is known, we speculate that its reduction would interfere with homeostasis. In the lens, solutes need to move from the cortical lens epithelial cells to the inner fiber cells. In the kidney, solutes also need to move between tubular cells and blood.
Prediction of membrane topology19 revealed a 536 amino acid protein containing 12 transmembrane domains (TMDs) with both termini located intracellularly. Whereas the large intracellular loop and both termini show high variability in their amino acid sequence among the different SLC16 family members, highest conservation is found in the first and fifth TMD.14 The p.Q215X mutation in SLC16A12 is located within the large cytoplasmic loop near the fourth TMD (Figure 7), predicted to result in a truncated protein with severely impaired or completely absent transport function. The premature termination codon might cause mRNA decay of the mutant allele, possibly by the mechanism of nonsense-mediated decay,20 but a dominant-negative effect or gain of function of the truncated protein can not be excluded. Consequently, reduced amounts of the normal allele in the patients could account for a gradual, progressive nature of the cataract. The resulting deficiency in transportation of metabolites in the lens could lead to alteration of structural components of the fiber cells and the refractive index, contributing to the development of cataract. Similarly, defective transport in the kidney could lead to excessive accumulation of glucose in the urine, making SLC16A12, directly or indirectly, involved in glucose transport. Because the causes of glucosuria can be heterogeneous, other factors, singly or in combination with deficient SLC transporter activity, could result in the highly variable phene of glucosuria.
Figure 7.
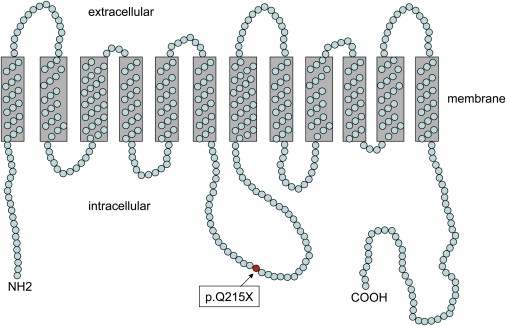
Schematic Representation of the Predicted Secondary Structure of SLC16A12
Prediction of membrane topology revealed a 536 amino acid protein with 12 transmembrane domains separated by intra- and extracellular domains of varying lengths with both termini (NH2 and COOH) located intracellularly. Amino acid glutamin (Gln, Q) at position 215 is mutated to a stop in the patients described herein (red circle).
Although several arguments have been presented that support a model in which cataract and glucosuria are caused by the same mutation, we can not rule out that the two diseases may segregate independently. In case of an unlinked locus for glucosuria, examples of potential candidates include the chaperone protein CD147, which facilitates subcellular sorting of SLC16 members17 or proteins involved in glucose transportation, i.e., GLUT proteins21 or SLC5A2.8
Age-related cataract, which is the most common cause for avoidable blindness worldwide, is known to be dependent on both environmental risk factors and genetic factors. A twin study on the cortical type of age-related cataract implies the action of dominant genes to account for genetic heritability of about 50%.22,23 The progressive course of juvenile cataract described here, resulting most likely from defective transport of small molecules, suggests the potential role of the SLC16A12 transporter in age-related cataracts as well. Depending on the type and location of mutations within the SLC16A12 transporter, more or less severe forms of cataract would be expected, which may also vary in the time of onset. We propose that mutations in a solute carrier such as SLC16A12 could lead to the age-related form of cataract. Knowledge of the substrate may open new venues for nonsurgical treatment.
Taken together, we show for the first time (to our knowledge) the biological relevance of the solute carrier SLC16A12 and suggest a function in the establishment and/or maintenance of homeostasis in the lens and probably also in the kidney.
Acknowledgments
We would like to thank the family for participation in this study; Jaya Balakrishnan, Esther Glaus, and Philippe Reuge (Berger laboratory) for DNA preparations; C. Becker (Nürnberg laboratory) for expert technical assistance in providing the SNP genotype data from Affymetrix microarrays; Gabor Matyas (Berger laboratory) for providing the RNA II Polymerase primers for RT-PCR experiments and for invaluable support with DNA sequencing; and Adrian Knoepfel (Berger laboratory) for sequencing of the FAS promoter. We are also grateful for the donation of the human eyes from the eye bank at the University of Zurich. This work was funded in part by the German Federal Ministry of Sciences and Education through the National Genome Research Network (grant 01GR0416 to P.N.) and by a scientific grant from the eye clinic of the Kanton Hospital Luzern, Switzerland.
Web Resources
The URLs for data presented herein are as follows:
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PredictProtein, http://www.predictprotein.org/
References
- 1.Augusteyn R.C. Growth of the human eye lens. Mol. Vis. 2007;13:252–257. [PMC free article] [PubMed] [Google Scholar]
- 2.Francis P.J., Berry V., Bhattacharya S.S., Moore A.T. The genetics of childhood cataract. J. Med. Genet. 2000;37:481–488. doi: 10.1136/jmg.37.7.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jamieson R.V., Farrar N., Stewart K., Perveen R., Mihelec M., Carette M., Grigg J.R., McAvoy J.W., Lovicu F.J., Tam P.P. Characterization of a familial t(16;22) balanced translocation associated with congenital cataract leads to identification of a novel gene, TMEM114, expressed in the lens and disrupted by the translocation. Hum. Mutat. 2007;28:968–977. doi: 10.1002/humu.20545. [DOI] [PubMed] [Google Scholar]
- 4.Lorenz B. Genetic examination in cases of congenital cataract. Ophthalmologe. 2007;104:559–565. doi: 10.1007/s00347-007-1557-2. [DOI] [PubMed] [Google Scholar]
- 5.Shiels A., Hejtmancik J.F. Genetic origins of cataract. Arch. Ophthalmol. 2007;125:165–173. doi: 10.1001/archopht.125.2.165. [DOI] [PubMed] [Google Scholar]
- 6.Shiels A., Bennett T.M., Knopf H.L., Yamada K., Yoshiura K., Niikawa N., Shim S., Hanson P.I. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am. J. Hum. Genet. 2007;81:596–606. doi: 10.1086/519980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandekerckhove K., Lange A.P., Herzog D., Schipper I. Juvenile cataract associated with microcornea and glucosuria: a new syndrome. Klin. Monatsbl. Augenheilkd. 2007;224:344–346. doi: 10.1055/s-2007-962943. [DOI] [PubMed] [Google Scholar]
- 8.Santer R., Kinner M., Lassen C.L., Schneppenheim R., Eggert P., Bald M., Brodehl J., Daschner M., Ehrich J.H., Kemper M. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J. Am. Soc. Nephrol. 2003;14:2873–2882. doi: 10.1097/01.asn.0000092790.89332.d2. [DOI] [PubMed] [Google Scholar]
- 9.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 10.Thiele H., Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 11.Burdon K.P., McKay J.D., Wirth M.G., Russell-Eggit I.M., Bhatti S., Ruddle J.B., Dimasi D., Mackey D.A., Craig J.E. The PITX3 gene in posterior polar congenital cataract in Australia. Mol. Vis. 2006;12:367–371. [PubMed] [Google Scholar]
- 12.Semina E.V., Ferrell R.E., Mintz-Hittner H.A., Bitoun P., Alward W.L., Reiter R.S., Funkhauser C., Daack-Hirsch S., Murray J.C. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat. Genet. 1998;19:167–170. doi: 10.1038/527. [DOI] [PubMed] [Google Scholar]
- 13.Futter C.E., Crowston J.G., Allan B.D. Interaction with collagen IV protects lens epithelial cells from Fas-dependent apoptosis by stimulating the production of soluble survival factors. Invest. Ophthalmol. Vis. Sci. 2005;46:3256–3262. doi: 10.1167/iovs.05-0086. [DOI] [PubMed] [Google Scholar]
- 14.Halestrap A.P., Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- 15.Neidhardt J., Glaus E., Barthelmes D., Zeitz C., Fleischhauer J., Berger W. Identification and characterization of a novel RPGR isoform in human retina. Hum. Mutat. 2007;28:797–807. doi: 10.1002/humu.20521. [DOI] [PubMed] [Google Scholar]
- 16.Halestrap A.P., Price N.T. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J. 1999;343:281–299. [PMC free article] [PubMed] [Google Scholar]
- 17.Deora A.A., Philp N., Hu J., Bok D., Rodriguez-Boulan E. Mechanisms regulating tissue-specific polarity of monocarboxylate transporters and their chaperone CD147 in kidney and retinal epithelia. Proc. Natl. Acad. Sci. USA. 2005;102:16245–16250. doi: 10.1073/pnas.0504419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Philp N.J., Wang D., Yoon H., Hjelmeland L.M. Polarized expression of monocarboxylate transporters in human retinal pigment epithelium and ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 2003;44:1716–1721. doi: 10.1167/iovs.02-0287. [DOI] [PubMed] [Google Scholar]
- 19.Rost B., Yachdav G., Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holbrook J.A., Neu-Yilik G., Hentze M.W., Kulozik A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 21.Wallner E.I., Wada J., Tramonti G., Lin S., Kanwar Y.S. Status of glucose transporters in the mammalian kidney and renal development. Ren. Fail. 2001;23:301–310. doi: 10.1081/jdi-100104714. [DOI] [PubMed] [Google Scholar]
- 22.Hammond C.J., Snieder H., Spector T.D., Gilbert C.E. Genetic and environmental factors in age-related nuclear cataracts in monozygotic and dizygotic twins. N. Engl. J. Med. 2000;342:1786–1790. doi: 10.1056/NEJM200006153422404. [DOI] [PubMed] [Google Scholar]
- 23.Hammond C.J., Duncan D.D., Snieder H., de Lange M., West S.K., Spector T.D., Gilbert C.E. The heritability of age-related cortical cataract: the twin eye study. Invest. Ophthalmol. Vis. Sci. 2001;42:601–605. [PubMed] [Google Scholar]