

Abstract
We have identified and characterized two unrelated patients with prenatal onset of microcephaly, intrauterine growth retardation, feeding problems, developmental delay, and febrile seizures/epilepsy who both carry a de novo balanced translocation that truncates the DYRK1A gene at chromosome 21q22.2. DYRK1A belongs to the dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) family, which is highly conserved throughout evolution. Given its localization in both the Down syndrome critical region and in the minimal region for partial monosomy 21, the gene has been studied intensively in animals and in humans, and DYRK1A has been proposed to be involved in the neurodevelopmental alterations associated with these syndromes. In the present study, we show that truncating mutations of DYRK1A result in a clinical phenotype including microcephaly.
Main Text
DYRK1A [MIM 600855] is a member of the dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) family, which is evolutionarily conserved and participates in various cellular processes. DYRK1A has been investigated intensively, mainly because of the chromosomal localization of the gene, DYRK1A, within the Down syndrome critical region (DSCR [MIM 190685]) on chromosome 21.1 Several lines of evidence, both in human and in animal models, suggest that DYRK1A is involved in neurogenesis. The Drosophila ortholog of DYRK1A is the minibrain (mnb) gene, and its protein product is shown to play an essential role during postembryonic neurogenesis through regulation of the number of distinct neuronal cell types.2 The brain of the adult mutant mnb fly is smaller than that of the wild-type fly, and the reduction in the number of neuronal cells is mainly in the optic lobes and in parts of the central brain hemispheres, resulting in distinct behavioral abnormalities.2 Mice with one functional copy of Dyrk1a (Dyrk1a+/− mutants) display a brain size 30% smaller than of wild-type mice, and they show growth retardation, behavioral defects, and altered motor activity due to dopaminergic dysfunction.3–5 In humans, DYRK1A has been proposed to be associated with microcephaly and mental retardation, given its localization to the minimal overlapping region observed in patients with partial monosomy 21.6
Overexpression of DYRK1A in mice leads to increased brain weight and neuronal size, as well as learning and memory defects, neurodevelopmental delay, hyperactivity, and motor abnormalities.7–11 Interestingly, in Down syndrome patients the DYRK1A transcript and the protein product are overexpressed in the brain, which is microcephalic and characterized by defective cortical lamination, reduced cortical neurons, and abnormal synapses.12–15 The Down syndrome critical region of human chromosome 21 has been defined on the basis of the analysis of rare cases of partial trisomy 21, and the studies in human and animal models suggest that DYRK1A, which is one of the 20 genes located in this region, is a likely candidate for the neurodevelopmental alterations associated with Down syndrome.16 Interestingly, the pyramidal cells, which comprise 70% of the neurons in the cerebral cortex, have smaller dendritic arbors, shorter basal dendrites, and fewer dendritic spines than normal, both in Dyrk1a+/− mutants17 and in Ts65Dn mice triallelic for Dyrk1a.18 These studies with human and animal models, which are monoallelic or triallelic for DYRK1A/Dyrk1a/mnb, indicate that this gene is important in neural plasticity and necessary for the normal size and development of the brain in a dosage-sensitive way. In the present study we show, to our knowledge for the first time, that truncation of the human DYRK1A gene by translocation breakpoints results in a clinical phenotype including microcephaly.
Patient 1 (Figures 1A–1E) was the first child of healthy nonconsanguineous Danish parents. The family history was unremarkable. The mother was hospitalized during pregnancy from week 25 onward due to cervical dilatation (4 cm). The boy was born by vaginal delivery at 37 weeks. Birth weight was 2286 g (−3 SD), birth length was 45 cm (−3 SD), and head circumference was 29.5 cm (−3 SD). Besides microcephaly, minor dysmorphic features including large low-set ears, long philtrum, and micrognathia were noticed at birth. The neonatal period was complicated by poor feeding due to diminished sucking reflexes. A systolic murmur was noted, but echocardiography did not reveal any abnormality. Several incidences of a downward gaze (“setting-sun” sign) indicating hydrocephalus were noticed, but cranial ultrasound was normal. He was released from hospital at 16 days of age. He was hospitalized on several occasions due to feeding problems, recurrent otitis, and a single septic episode. At the two months of age, bilateral inguinal hernia was observed. The repeated downward gaze persisted and was followed by episodes of hyperextension of the neck. Repeated cranial-ultrasound examinations did not reveal hydrocephalus or any other abnormality. Brain MRI performed at three months of age showed, in addition to microcephaly, hypogenesis of the corpus callosum (Figure 1F). Some of his developmental milestones were slightly delayed. His first eye contact and first smile occurred at three months of age. He started to crawl at 8.5 months of age, and he walked at 15 months. His head circumference corresponded to < −3 SD at 8.5 months. At 20 months, he was happy and content. He was able to play with toys and spoke his first few words during this time. His spatial skills were still limited. His weight was 10 kg (−2 SD), height was 77.5 cm (−3 SD), and head circumference was 43.2 cm (< −3 SD). He was diagnosed with bilateral hypermetropia (+6 dpt). He had a total of seven episodes of febrile convulsions, of a duration of up to 2 min each, induced by bacterial infections and fever. Interictal EEG was normal. Hand stereotypes (hand-washing) were observed to occur when he felt insecure. The patient is at present 24 months of age.
Figure 1.
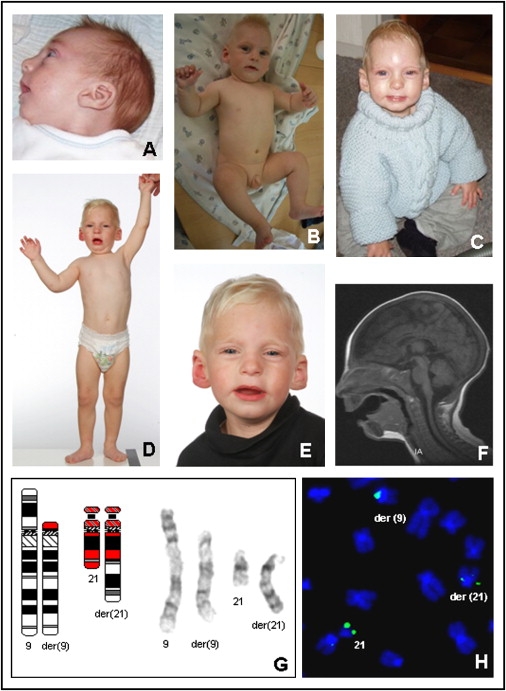
Clinical Pictures and Laboratory Investigations of Patient 1
The pictures were taken when the patient was 3 months (A), 9 months (B), 14 months (C), and 24 months (D and E) of age. Magnetic-resonance imaging (F) at three months of age showed microcephaly and hypogenesis of corpus callosum. Cytogenetic analysis revealed a translocation with the karyotype 46,XY,t(9;21)(p12;q22) (G). For mapping of the breakpoints of the translocation, fluorescence in situ hybridization (FISH) was carried out with the use of BAC clones, according to standard procedures. The BAC clone RP11-315B15, which contains DYRK1A, spans the chromosome 21 breakpoint, as shown by signals on the normal and the derivative chromosomes (H).
Cytogenetic analysis revealed a de novo balanced translocation t(9;21)(p12;q22) (Figure 1G). The chromosome 9 breakpoint was mapped to 9p11.2 by fluorescence in situ hybridization (FISH) within the BAC clone RP11-268E1 covering a gene-empty region with several segmental duplications (data not shown). The chromosome 21 breakpoint was mapped to 21q22.2 within the overlapping region of the BAC clones RP11-315B15 (Figure 1H), RP11-777J19, and RP11-119A10 (chromosome position: 37,669,622–37,704,000, UCSC March 2006 freeze) (data not shown). The FISH results revealed that the breakpoint was within the second intron of the DYRK1A gene14 (Figure 2). Whole-genome array CGH was carried out with the use of a submegabase-resolution tiling-path BAC array consisting of the human genome high-resolution 32 k re-arrayed clone set, the 1 Mb Sanger set, and a set of 390 subtelomeric clones as described previously19 and deletions or duplications around the translocation breakpoints or elsewhere in the genome were excluded.
Figure 2.
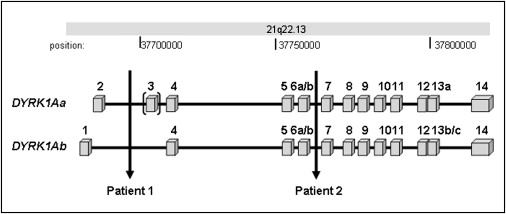
The Genomic Region Covered by the DYRK1A Gene
The locations of the chromosomal breakpoints of the translocation patients are indicated by vertical arrows. The DYRK1A gene is organized in 17 exons (boxes) and has two transcription start sites, with exon 1 and exon 2 as alternative first exons. The use of these alternative start sites yields two major transcripts, DYRK1Aa (5182 nt) and DYRK1Ab (5287 nt), which are both truncated by the translocation breakpoints in patients 1 and 2. The bracket indicates that exon 3 is alternatively spliced. The chromosome band and base-pair positions are shown above the figure.
Patient 2 (Figures 3A–3E) was the second child of healthy nonconsanguineous German parents with uneventful family history. The older sister is phenotypically normal. The pregnancy was complicated by polyhydramnios. Microcephaly and growth retardation were noticed at the 28th week of gestation. The patient was born at term, and her birth weight was 2400 g (−3 SD), birth length was 47 cm (−2 SD), and head circumference was 30 cm (−3 SD). Difficulties in feeding required enteral feeding, and feeding problems persisted during infancy. Gross motor development was delayed: She sat alone at 15 months of age and walked at two years of age. At one year of age, the first febrile convulsion was noted during a severe bronchiopulmonary infection. Antiepileptic treatment with primidone was started after the third generalized tonic-clonic seizure at 19 months of age. Additional focal seizures and further EEG abnormalities at three years of age led to therapy with valproate and ethosuccimide. Reduction of antiepileptic medication was not tolerated. At 21 months of age, height was 85 cm (mean for age), weight was 9270 g (−3 SD), and head circumference was 41 cm (< −3 SD). Echocardiography at 18 months of age showed a small ventricular septal defect and mild aortic-valve insufficiency. At 10.5 years of age, height was 130 cm (−1.5 SD), weight was 21 kg (−3 SD), and head circumference was 45.9 cm (< −3 SD). On clinical reexamination at 13 years of age, the patient was severely mentally retarded and did not have any speech. Her height was 142 cm (−1.5 SD), her weight was 26 kg (−3 SD), and her head circumference was 46.5 cm (< −3 SD). She had mild dysmorphic facial features, including asymmetrical head, flat philtrum, small upper lip, large ears, and abnormal whorls of hair. She had pectus excavatum, kyphosis, and slight scoliosis, and her skin was very thin. Terminal phalanges of hands and feet were shortened and broad. Feeding of the patient continued to be extremely difficult. A CT scan of her brain at seven years of age showed head asymmetry with left occipital flattening and mild enlargement of the ventricles (Figure 3F). MRI of the brain at 12 years of age did not reveal further structural changes (Figures 3G and 3H).
Figure 3.
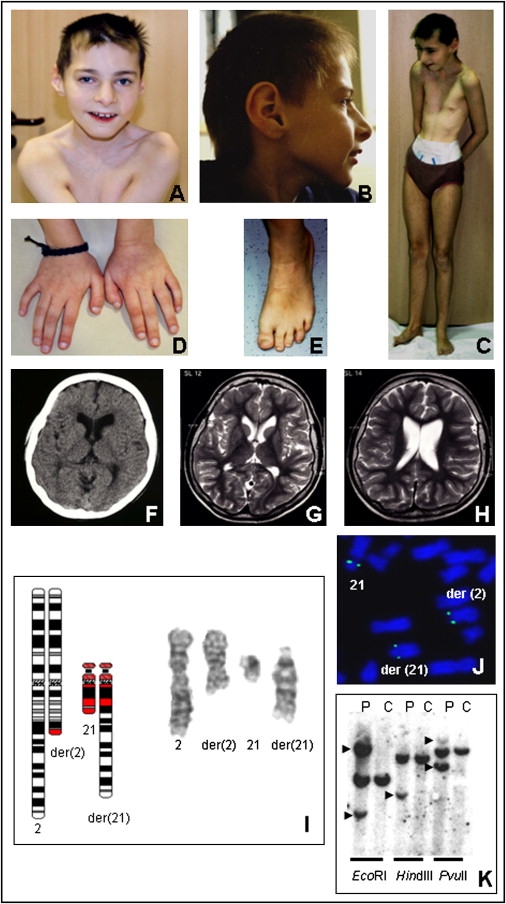
Clinical Pictures and Laboratory Investigations of Patient 2
The pictures were taken when the patient was 12 years of age. She has asymmetry of the head, flat philtrum, small upper lip, abnormal whorls of hair (A); microcephaly and large ears (B); pectus excavatum (C); and shortened and broad terminal phalanges of the hands and feet (D and E). Computerized tomography (F) at seven years of age and magnetic-resonance imaging (G and H) at 12 years of age showed microcephaly with head asymmetry and mild enlargement of the ventricles. Cytogenetic analysis revealed a translocation with the karyotype 46,XX,t(2;21)(q22;q22) (I). For mapping of the breakpoints of the translocation, FISH was carried out with the use of YAC, BAC, PAC, or cosmid clones according to standard procedures. The YAC clone 662A10, which contains DYRK1A, spans the chromosome 21 breakpoint, as shown by signals on the normal and the derivative chromosomes (J). Chromosome 21 breakpoint was fine mapped with Southern-blot hybridization (K). Genomic DNA from the patient (“P”) and the control (“C”) was digested with three restriction enzymes and hybridized with the breakpoint-spanning probe (chromosome position: 37,778,809–37,780,325). Aberrant bands representing the junction fragments are indicated by arrowheads.
Cytogenetic analysis indicated a de novo balanced translocation t(2;21)(q22;q22) (Figure 3I). Using FISH analysis, we mapped the chromosome 21 breakpoint within the YAC clone 662A10 (Figure 3J), which contained the DYRK1A gene, and the chromosome 2 breakpoint was mapped within the YAC clone 670G1 (data not shown). Cloning and sequence analysis of the junction fragments indicated that the chromosome 2 breakpoint was within intron 39 of the putative tumor-suppressor gene, LRP1B (low density lipoprotein-related protein 1B [MIM 608766]) (after nucleotide 141,257,981 of chromosome 2, UCSC March 2006 freeze) (data not shown). Mapping of the chromosome 21 breakpoint by Southern-blot analysis (Figure 3K) followed by cloning and sequencing of the junction fragment indicated that DYRK1A was disrupted in intron 6 following exon 6a/6b (after nucleotide 37,779,572 of chromosome 21, UCSC March 2006 freeze) (Figure 3). Whole-genome BAC-array CGH and 244 K oligonucleotide CGH array (Agilent) showed a 590 kb duplication at Xp22 (chromosome position: 13,895,000–14,489,000, UCSC March 2006 freeze) comprising the GEMIN8 gene (GenBank; see Web Resources section) and the first exon of the glycine receptor gene (GLRA2, MIM 305990) (data not shown). Parental origin of this duplication could not be investigated because DNA samples from the parents were not available.
In two unrelated translocation carriers, the DYRK1A gene was truncated by the chromosome 21 breakpoints. The features of microcephaly, intrauterine growth retardation, postnatal feeding problems, and large ears observed in these patients resemble the phenotypes of the patients with partial monosomy 21.6,20–22 In partial monosomy 21 patients, the common deleted region for microcephaly and mental retardation has been narrowed down to a 1.2 Mb region flanked by the markers D21S334 and D21S55 (chromosome position: 36,737,455-38,012,252).6 This region covers ten genes, including DYRK1A, which has been suggested as the candidate gene for the common neurological phenotypes of these patients. Our findings therefore strongly suggest that haploinsufficiency of DYRK1A is causative for the common features, including microcephaly, observed in partial monosomy 21 patients. These findings, together with the brain phenotypes of fly (mnb) and mouse (Dyrk1a+/−) mutants with one functional copy of Dyrk1a, render this evolutionarily conserved gene as a microcephaly gene in humans.
The two patients presented here have overlapping clinical phenotypes including microcephaly, large ears, intrauterine growth retardation, postnatal feeding problems, and febrile seizures, but they have different degrees of developmental delay, and patient 2 is more severely affected than patient 1. A plausible explanation might be disruption of LRP1B by the chromosome 2 breakpoint in patient 2, but given that Lrp1b−/− mice appear normal,23 it is unlikely that this aberration has a major impact on the clinical phenotype. Patient 2 also has a duplication of unknown origin at Xp22. The duplication included the GEMIN8 gene, which has an essential role in organization and function of the survival-motor-neuron complex.24 Even though it is unlikely that the duplication of X chromosome sequences in a female would cause an abnormal phenotype, the possibility that a hypothetically increased dosage of GEMIN8 in the critical tissues might affect the disease severity cannot be excluded.
In summary, our finding of two unrelated patients with microcephaly and truncation of DYRK1A demonstrates that this gene plays a critical role in human brain development and that haploinsufficiency of DYRK1A causes microcephaly in humans, probably through mechanisms that control neural differentiation. In addition, our results, together with previous findings of DYRK1A overexpression in brains of Down syndrome patients, strongly suggest that in humans the dosage of the associated kinase needs to be very tightly regulated and that both an increase and a decrease in the dosage could result in a disease phenotype.
Acknowledgments
We gratefully acknowledge the patients and their parents for giving their consent to publish clinical data and photos. This study has been approved by the Local Scientific Committee (Reg. Nr. 2004-2-08). We thank Elisabeth Larsen, Ingrid Kjær, Minna Becher, and Anita Niebuhr for their expert technical assistance; Jutta Wirth and Andreas Schröer for their help during the initial phase of patient 2 breakpoint mapping, and Marie-Laure Yaspo for chromosome 21 cosmid clones. This work was supported by the National Genome Research Network (NGFN, project numbers 01GR0105 and 01GR0414). Wilhelm Johannsen Centre for Functional Genome Research is established by the Danish Research Foundation.
Web Resources
The URLs for data presented herein are as follows:
UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Nikolaienko O., Nguyen C., Crinc L.S., Cios K.J., Gardiner K. Human chromosome 21/Down syndrome gene function and pathway database. Gene. 2005;364:90–98. doi: 10.1016/j.gene.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 2.Tejedor F., Zhu X.R., Kaltenbach E., Ackermann A., Baumann A., Canal I., Heisenberg M., Fischbach K.F., Pongs O. Minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- 3.Fotaki V., Dierssen M., Alcántara S., Martínez S., Martí E., Casas C., Visa J., Soriano E., Estivill X., Arbonés M.L. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002;22:6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fotaki V., Martínez De Lagrán M., Estivill X., Arbonés M., Dierssen M. Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav. Neurosci. 2004;118:815–821. doi: 10.1037/0735-7044.118.4.815. [DOI] [PubMed] [Google Scholar]
- 5.Martinez de Lagran M., Bortolozzi A., Millan O., Gispert J.D., Gonzalez J.R., Arbones M.L., Artigas F., Dierssen M. Dopaminergic deficiency in mice with reduced levels of the dual-specificity tyrosine-phosphorylated and regulated kinase 1A, Dyrk1A(+/−) Genes Brain Behav. 2007;6:569–578. doi: 10.1111/j.1601-183X.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto N., Ohashi H., Tsukahara M., Kim K.C., Soeda E., Niikawa N. Possible Narrowed Assignment of the Loci of Monosomy 21- Associated Microcephaly and Intrauterine Growth Retardation to a 1.2-Mb Segment at 21q22.2. Am. J. Hum. Genet. 1997;60:997–999. [PMC free article] [PubMed] [Google Scholar]
- 7.Branchi I., Bichler Z., Minghetti L., Delabar J.M., Malchiodi-Albedi F., Gonzalez M.C., Chettouh Z., Nicolini A., Chabert C., Smith D.J. Transgenic mouse in vivo library of human Down syndrome critical region 1: association between DYRK1A overexpression, brain development abnormalities, and cell cycle protein alteration. J. Neuropathol. Exp. Neurol. 2004;63:429–440. doi: 10.1093/jnen/63.5.429. [DOI] [PubMed] [Google Scholar]
- 8.Smith D.J., Stevens M.E., Sudanagunta S.P., Bronson R.T., Makhinson M., Watabe A.M., O'Dell T.J., Fung J., Weier H.U., Cheng J.F. Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nat. Genet. 1997;16:28–36. doi: 10.1038/ng0597-28. [DOI] [PubMed] [Google Scholar]
- 9.Altafaj X., Dierssen M., Baamonde C., Martí E., Visa J., Guimerà J., Oset M., González J.R., Flórez J., Fillat C. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum. Mol. Genet. 2001;10:1915–1923. doi: 10.1093/hmg/10.18.1915. [DOI] [PubMed] [Google Scholar]
- 10.Martínez de Lagrán M., Altafaj X., Gallego X., Martí E., Estivill X., Sahún I., Fillat C., Dierssen M. Motor phenotypic alterations in TgDyrk1a transgenic mice implicate DYRK1A in Down syndrome motor dysfunction. Neurobiol. Dis. 2004;15:132–142. doi: 10.1016/j.nbd.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Ahn K.J., Jeong H.K., Choi H.S., Ryoo S.R., Kim Y.J., Goo J.S., Choi S.Y., Han J.S., Ha I., Song W.J. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol. Dis. 2006;22:463–472. doi: 10.1016/j.nbd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Wisniewski K.E. Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. Am. J. Med. Genet. Suppl. 1990;7:274–281. doi: 10.1002/ajmg.1320370755. [DOI] [PubMed] [Google Scholar]
- 13.Golden J.A., Hyman B.T. Development of the superior temporal neocortex is anomalous in trisomy 21. J. Neuropathol. Exp. Neurol. 1994;53:513–520. doi: 10.1097/00005072-199409000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Guimera J., Casas C., Estivill X., Pritchard M. Human minibrain homologue (MNBH/DYRK1): characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics. 1999;57:407–418. doi: 10.1006/geno.1999.5775. [DOI] [PubMed] [Google Scholar]
- 15.Dowjat W.K., Adayev T., Kuchna I., Nowicki K., Palminiello S., Hwang Y.W., Wegiel J. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci. Lett. 2007;413:77–81. doi: 10.1016/j.neulet.2006.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ronan A., Fagan K., Christie L., Conroy J., Nowak N.J., Turner G. Familial 4.3 Mb duplication of 21q22 sheds new light on the Down syndrome critical region. J. Med. Genet. 2007;44:448–451. doi: 10.1136/jmg.2006.047373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benavides-Piccione R., Dierssen M., Ballesteros-Yánez I., Martínez de Lagrán M., Arbonés M.L., Fotaki V., DeFelipe J., Elston G.N. Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/− mouse. Neurobiol. Dis. 2005;20:115–122. doi: 10.1016/j.nbd.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Dierssen M., Benavides-Piccione R., Martínez-Cué C., Estivill X., Flórez J., Elston G.N., DeFelipe J. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cereb. Cortex. 2003;13:758–764. doi: 10.1093/cercor/13.7.758. [DOI] [PubMed] [Google Scholar]
- 19.Erdogan F., Chen W., Kirchhoff M., Kalscheuer V.M., Hultschig C., Muller I., Schulz R., Menzel C., Bryndorf T., Ropers H.H. Impact of low copy repeats on the generation of balanced and unbalanced chromosomal aberrations in mental retardation. Cytogenet. Genome Res. 2006;115:247–253. doi: 10.1159/000095921. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto Y., Ogasawara N., Gotoh A., Komiya H., Nakai H., Kuroki Y. A case of 21q-syndrome with normal SOD-1 activity. Hum. Genet. 1979;48:321–327. doi: 10.1007/BF00272832. [DOI] [PubMed] [Google Scholar]
- 21.Chettouh Z., Croquette M.F., Delobel B., Gilgenkrants S., Leonard C., Maunoury C., Prieur M., Rethoré M.O., Sinat P.M., Chery M. Molecular mapping of 21 features associated with partial monosomy 21: involvement of the APP-SOD1 region. Am. J. Hum. Genet. 1995;57:62–71. [PMC free article] [PubMed] [Google Scholar]
- 22.Osoegawa K., Susukida R., Okano S., Kudoh J., Minoshima S., Shimizu N., de Jong P.J., Groet J., Ives J., Lehrach H. An integrated map with cosmid/PAC contigs of a 4-Mb Down syndrome critical region. Genomics. 1996;32:375–387. doi: 10.1006/geno.1996.0132. [DOI] [PubMed] [Google Scholar]
- 23.Marschang P., Brich J., Weeber E.J., Sweatt J.D., Shelton J.M., Richardson J.A., Hammer R.E., Herz J. Normal development and fertility of knockout mice lacking the tumor suppressor gene LRP1b suggest functional compensation by LRP1. Mol. Cell. Biol. 2004;24:3782–3793. doi: 10.1128/MCB.24.9.3782-3793.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carissimi C., Saieva L., Gabanella F., Pellizzoni L. Gemin8 is required for the architecture and function of the survival motor neuron complex. J. Biol. Chem. 2006;281:37009–37016. doi: 10.1074/jbc.M607505200. [DOI] [PubMed] [Google Scholar]