

Abstract
Patients with dyskeratosis congenita (DC), a heterogeneous inherited bone marrow failure syndrome, have abnormalities in telomere biology, including very short telomeres and germline mutations in DKC1, TERC, TERT, or NOP10, but ∼60% of DC patients lack an identifiable mutation. With the very short telomere phenotype and a highly penetrant, rare disease model, a linkage scan was performed on a family with autosomal-dominant DC and no mutations in DKCI, TERC, or TERT. Evidence favoring linkage was found at 2p24 and 14q11.2, and this led to the identification of TINF2 (14q11.2) mutations, K280E, in the proband and her five affected relatives and TINF2 R282H in three additional unrelated DC probands, including one with Revesz syndrome; a fifth DC proband had a R282S mutation. TINF2 mutations were not present in unaffected relatives, DC probands with mutations in DKC1, TERC, or TERT or 298 control subjects. We demonstrate that a fifth gene, TINF2, is mutated in classical DC and, for the first time, in Revesz syndrome. This represents the first shelterin complex mutation linked to human disease and confirms the role of very short telomeres as a diagnostic test for DC.
Main Text
Dyskeratosis congenita (DC) is an inherited bone marrow failure syndrome (IBMFS) characterized by the diagnostic triad of abnormal nails, lacey reticular pigmentation, and oral leukoplakia. Patients with DC are at high risk of developing aplastic anemia, myelodysplastic syndrome, and leukemia. Less common physical findings include epiphora, premature gray hair, developmental delay, short stature, cerebellar hypoplasia, microcephaly, esophageal stenosis, urethral stenosis, pulmonary fibrosis, liver disease, avascular necrosis of hips or shoulders, and epithelial cancers.1–3 Hoyeraal-Hreidarsson (HH) syndrome is a severe form of DC with bone-marrow failure, immunodeficiency, microcephaly, cerebellar hypoplasia, intrauterine growth retardation, and developmental delay.2,4 Revesz syndrome, characterized by bilateral exudative retinopathy, bone-marrow hypoplasia, nail dystrophy, fine hair, cerebellar hypoplasia, and growth retardation, also appears to be in the DC disease spectrum.5,6
This clinical heterogeneity has made the diagnosis of DC challenging; many cases of DC are not diagnosed until the development of cytopenias. DC patients with aplastic anemia do not respond to immunosuppressive therapy,7 and if bone-marrow transplantation is required, they are at high risk of treatment-related complications because of underlying pulmonary and liver disease.8–11 Selection of related bone-marrow donors may be complicated by the variable phenotype seen in DC, including silent carriers.
The unifying feature present in all patients with DC is abnormally short telomere lengths and defects in telomere biology.1,12 Short telomeres were first noted in primary fibroblasts and lymphoblasts of males with DKC1 mutations.13 Subsequent studies have confirmed that telomeres are extremely short in DC patients and appear to shorten further in successive generations of affected individuals from dominant families.14,15 Recently, very short telomere lengths in leukocyte subsets, defined as less than the first percentile for age, were found to be diagnostic of DC in comparison to other IBMFS.12
Telomeres, which consist of long TTAGGG nucleotide repeats and associated proteins at chromosome ends, are essential for the maintenance of chromosomal integrity.16 In order to preserve the chromosome end, the telomerase reverse transcriptase (TERT, MIM #187270), its RNA component, TERC (also known as hTR, 3q21–q28, MIM #602322), and an ordered protein complex, termed shelterin, consisting of six proteins (gene names TERF1 [MIM #600951], TERF2 [MIM #602027], TINF2 [MIM #604319], TERF2IP [MIM #605061], ACD [MIM #609377], and POT1 [MIM #606478]) protect the telomere from end-to-end fusion.16–18 Telomeric repeats are lost with each cell division, in part because of incomplete replication of the 3′ end of the chromosome. Telomeric attrition can result in critically short telomeres prompting cellular senescence or cellular crisis, including apoptosis, genomic instability, or a reduction in cellular lifespan.19,20
DC family pedigrees indicate X-linked recessive (XLR), autosomal dominant (AD), and autosomal recessive (AR) inheritance.15 X-linked mutations have been identified in dyskerin, DKC1 (MIM #300126). AD DC has been linked to mutations in the RNA subunit of telomerase, TERC.21,22 Mutations in TERT have been identified in patients with DC and those with aplastic anemia without the classical DC triad.14,23,24 AR inheritance of mutations in NOP10 (gene name NOLA3, MIM #606471), a component of H/ACA snoRNP complexes, was identified in one consanguineous family with DC.25 However, in a series of almost 300 DC patients, ∼60% remain unclassified at the molecular level.26
We identified a family with AD inheritance, variable clinical phenotype, and no mutations in DKCI, TERC, or TERT (family A) among the families with DC followed at the National Cancer Institute (NCI) as part of the IRB-approved study entitled “Etiologic Investigation of Cancer Susceptibility in Inherited Bone Marrow Failure Syndromes.” Evaluation of this family and of all others described in this report included review of medical records, careful physical examination, complete blood counts, bone marrow studies, measurement of telomere length,12,27 and mutation analyses. The diagnosis of DC, HH, or Revesz syndrome was made according to the definitions of Vulliamy et al.2 In brief, DC patients had at least two of the three components of the triad or one of the triad with either a hypoplastic bone marrow or at least two of the other somatic features.
Telomere length was measured in white blood cell subsets by automated multicolor flow-FISH analysis of granulocytes, CD45RA+ lymphocytes (naive T cells), CD45RA− lymphocytes (memory T cells), CD20+ lymphocytes (B cells), CD57+ lymphocytes (NK/NKT cells), and total leukocytes and compared with age-matched controls.27 “Very short” was defined as less than the first percentile for age.12 Germline DNA was isolated from buccal cells or peripheral blood white cells by standard methods. Mutation analyses of DKC1 and TERC were performed by GeneDx (Gaithersburg, MD) on the basis of cDNA reference sequences NM_001363.2 and U86046.1, as previously described.28 The proximal promoter and all coding regions of TERT were sequenced as described.29 All sequence analyses were verified at least twice.
Family A is nonconsanguineous and of European ancestry (Figure 1). III-07 was the proband. The first cases of DC in this family were monozygotic twin brothers, II-01 and II-03. Telomere lengths were determined in all 22 living family members (Figure 2A). I-01 and I-02, the paternal grandparents of the proband, were clinically unaffected and had normal telomere lengths. Their other children (generation 2) included five daughters and three sons with normal telomere lengths. There was no other family history of cancer, anemia, skin abnormalities, or other features of DC. II-01 died at age 37 from aplastic anemia and pulmonary hemorrhage. He and his wife had five sons and one miscarriage. Two sons, III-01 and III-03, and their mother were clinically unaffected and had normal telomere lengths. His other three sons, III-02, III-04, and III-05, had telomere lengths less than the first percentile, all three features of the diagnostic triad, epiphora, and variable hematopoietic involvement (Table 1).
Figure 1.
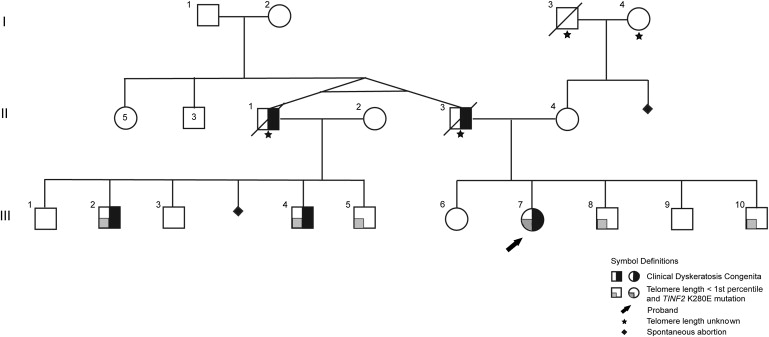
Pedigree of Family A
The monozygotic twins in generation II had five sisters and three brothers. Telomere length was determined on individuals except where indicated by a star.
Figure 2.
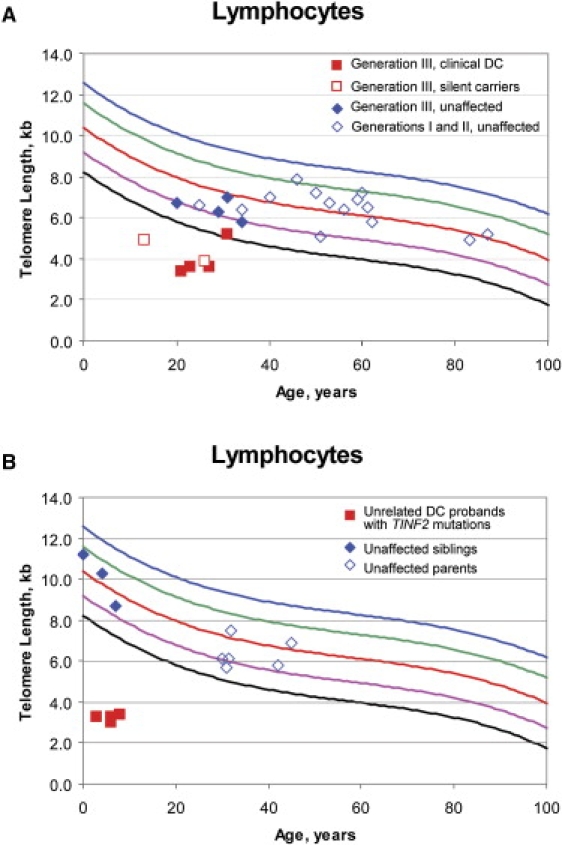
Lymphocyte Telomere Lengths
(A) Lymphocyte telomere lengths measured by flow-FISH of family A, including generation III affected individuals (red filled squares), generation III silent carriers (red open squares), unaffected members of generation III (blue filled diamonds), and unaffected members of generations I and II (blue open diamonds). The black line indicates the first percentile, and the blue line indicates the 99th percentile. Individual III-05 (filled red square on the first percentile line) had telomere lengths of 5.2 kb. The first percentile for age is 5 kb. However, his telomere lengths were less than the first percentile for age in naive T cells and granulocytes (data not shown).
(B) Lymphocyte telomere lengths of the four unrelated DC probands (filled red circles) with TINF2 mutations, their unaffected parents (open blue diamonds), and unaffected siblings (filled blue diamonds).
Table 1.
Clinical and Laboratory Features of Individuals with TINF2 Mutations
| Patient, Gender | Diagnosis | Age Presented,a Years | Age at Study, Years | DC Triad |
Hematology |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leuko-plakia | Nails | Skin | Other Clinical Features | WBC/mm3 | ANC/mm3 | Hbg/dl | MCVfL | Platelets ×103/mm3 | Mutation | ||||
| A-II-01M | DC | 13 | NA | + | + | + | SAA, bilateral hip AVN, urethral strictures, died from pulmonary hemorrhage at age 37 | 2100 | 945 | 9.7 | 105 | 23 | K280E |
| A-II-03M | DC | 16 | NA | NA | + | + | SAA bilateral hip AVN, died from NHL at age 43 | 1800 | 774 | 6 | 120 | 27 | K280E |
| A-III-02M | DC | 31 | 31 | + | + | + | epiphora | 4300 | 2209 | 15.1 | 96.2 | 142 | K280E |
| A-III-04M | DC | 24 | 24 | + | + | + | epiphora | 3400 | 1582 | 14.8 | 98.3 | 108 | K280E |
| A-III-05M | DC | 12 | 21 | + | + | + | epiphora, reduced DLCO | 3200 | 1909 | 11.6 | 109 | 23 | K280E |
| A-III-07F | DC | 10 | 27 | + | + | + | SAA, matched sibling BMT, epiphora | 2200 | 828 | 7.8 | 118 | 7 | K280E |
| A-III-08M | Silent Carrier | 26 | 26 | − | b | − | gray hair, penile leukoplakia | 3400 | 1816 | 14.6 | 94 | 148 | K280E |
| A-III-10M | Silent Carrier | 13 | 13 | − | − | − | reduced DLCO | 3290 | 1414 | 13.9 | 88.1 | 188 | K280E |
| B-1M | DC | 3.7 | 5.8 | + | + | + | SAA, epiphora, urethral stenosis, speech delay, microcephaly, intrauterine growth retardation, reduced DLCO | 4400 | 1496 | 9.9 | 89.7 | 28 | R282H |
| C-1M | DC, RS | 1.5 | 6.8 | + | + | + | SAA, died after BMT, bilateral exudative retinopathy, developmental delay, cerebellar hypoplasia | 2200 | 1453 | 7.9 | 111 | 36 | R282H |
| D-1M | DC | 2.8 | 3.7 | − | + | − | SAA, IgA deficiency | 2900 | 435 | 10.3 | 89.6 | 47 | R282H |
| E-1F | DC | 3.2 | 9.5 | + | + | + | SAA, epiphora, reduced DLCO, decreased NK cell number | 2000 | 629 | 10.4 | 87.9 | 21 | R282S |
All patients listed had telomere lengths less than the first percentile for age as measured by multicolor flow-FISH. Abbreviations are used as follows: DC, dyskeratosis congenita; RS, Revesz syndrome; M, male; F, female; NA, not available because patients died prior to study; WBC, white blood cell count; ANC, absolute neutrophil count; Hb, hemoglobin; MCV, mean corpuscular volume; SAA, severe aplastic anemia; AVN, avascular necrosis; NHL, Non-Hodgkin lymphoma; DLCO, diffusion capacity of the lung for carbon monoxide; BMT, bone-marrow transplant; and NK cell, natural killer cell.
Age when patient first presented to a physician with clinical findings that were suspicious for DC.
This patient had severe frostbite of fingers and toes at age 2, and thus we could not evaluate nail dystrophy.
II-03 died at age 43 because of aplastic anemia, non-Hodgkin lymphoma, and pulmonary failure. He and his wife had two daughters, III-06 and III-07, and three sons, III-08, III-09, and III-10. III-06, III-09, and their mother had no clinical signs of DC and normal telomere lengths (Figure 2A). The other three children had telomere lengths less than the first percentile and variable phenotypes (Table 1). The proband, III-07, had severe aplastic anemia since age 10, and thereby required a stem cell transplant at age 29, but only subtle fingernail ridging. III-08 had abnormal nails (but had a history of frostbite at age 2 years), premature gray hair, and penile leukoplakia but normal blood counts; he did not meet the clinical definition of DC but had very short telomere lengths. III-10 had no clinical signs of DC or bone marrow failure; however, his telomere lengths were less than the first percentile at age 13. Therefore, III-08 and III-10 were classified as silent carriers.
An SNP genome-wide linkage screen was conducted with genomic DNA samples from family A (Figure 1). Individuals genotyped were I-01, I-02, II-02, II-04, and III-01 through III-10. Genotyping was performed at the NCI's Core Genotyping Facility. All samples were quantified and evaluated by Identifiler (Applied Biosystems, Carlsbad, CA) so that duplicated or contaminated samples could be ruled out. The 6008 SNP markers of the Human Linkage IVb Panel (Illumina, San Diego, CA) were genotyped according to the manufacturer's instructions. The overall genotype completion rate was ≥99% for all chromosomes.
Progeny Lab software (version 5, Progeny Software, LLC, South Bend, IN) was used for processing genotype data. With cutoffs of D′ = 0.7 and R2 = 0.4, SNPLINK was used for evaluating and removing SNPs that were in linkage disequilibrium with each other.30 After SNPLINK processing, GeneHunter31 was used for analysis of the data under a parametric, AD, rare, highly penetrant disease (0.00001 disease frequency, penetrance of 1.0) model with telomere length less than the first percentile as the affected phenotype. Evidence favoring linkage was found at two locations, a 17.6 megabase (Mb) region on chromosome 2p24 and a 2.8 Mb region on chromosome 14q11.2 (LOD score = 2.62 at both sites) (Table 2 and Figure 3).
Table 2.
Linkage-Scan Results
| Chromosome | Maximum LOD Score |
|---|---|
| 1 | 0.03 |
| 2 | 2.62 |
| 3 | 0.00 |
| 4 | 0.00 |
| 5 | 0.02 |
| 6 | 0.98 |
| 7 | 0.00 |
| 8 | 0.00 |
| 9 | 0.00 |
| 10 | 0.00 |
| 11 | 0.00 |
| 12 | 0.00 |
| 13 | 1.32 |
| 14 | 2.62 |
| 15 | 0.00 |
| 16 | 0.00 |
| 17 | 0.00 |
| 18 | 0.00 |
| 19 | 0.00 |
| 20 | 0.00 |
| 21 | 0.00 |
| 22 | 0.35 |
| X | 0.98 |
A SNP linkage scan was performed on family A as described. Linkage disequilibrium between SNPs was removed with SNPLINK, and GeneHunter was used for analysis of the data under an autosomal-dominant, rare disease model with telomere length less than the first percentile as the affected phenotype. The maximum LOD scores for each chromosome are shown. Statistically significant scores are shown in bold type.
Figure 3.
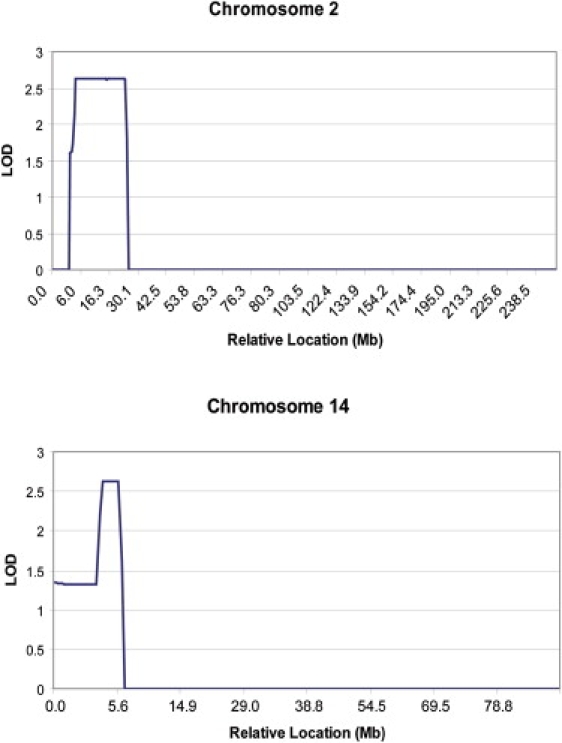
LOD Scores for Chromosomes 2 and 14
LOD scores were determined under an autosomal-dominant (parametric), rare disease model with telomere length less than the first percentile as the affected phenotype as described in the text. The relative location of the markers is noted on the x axis.
(Top) A total of 356 SNP markers on chromosome 2 were evaluated after removal of SNPs in linkage disequilibrium as described. The maximum LOD score of 2.62 was located between SNP markers rs6767 and rs520354 on chromosome 2 at positions 3.5 Mb and 21.1 Mb, respectively. DDX1 was the best candidate gene in this location.
(Bottom) A total of 161 SNP markers on chromosome 14 were evaluated after removal of SNPs in linkage disequilibrium as described. The maximum LOD score of 2.62 was located between SNP markers rs1570342 and rs195677 on chromosome 14 at positions 22.4 Mb and 25.2 Mb, respectively. TINF2 was the best candidate gene in this location.
The strongest candidate gene at chromosome 2p24 was DDX1 (DEAD [Asp-Glu-Ala-Asp] box polypeptide 1, MIM #601257), an RNA helicase that is involved in pre-mRNA processing and RNA unwinding.32 Primers for DDX1 were designed with the NCI's Oncogenomics database. Primer sequences and annealing temperatures are available upon request. Genomic DNA was amplified by PCR with MJ Research model PTC-225 thermal cyclers (Bio-Rad Laboratories, Waltham, MA) with the following conditions: 10 ng of genomic DNA, 0.2 μM of each primer (appended with M13 tags), 200 μM of each dNTP, 2 mM MgCl2, 0.5 units AmpliTaq Gold DNA polymerase (ABI-Perkin Elmer, Foster City, CA), and the manufacturer's buffer. Bidirectional sequencing of amplified DNA with the Dye Terminator method (ABI-Perkin Elmer) and M13 forward and reverse primers was analyzed on ABI-Perkin Elmer platforms (models 3730 or 3700) with Sequence Analysis 3.7 software and Sequencher v4.6 software (Gene Codes Corporation, Ann Arbor, MI).
No mutations were identified in the coding regions of all 20 exons of DDX1 in the proband III-07. Three common SNPs were identified, rs2302929, rs10929378, and rs10221770, in III-07 and were also present in the dbSNP database.
The strongest candidate gene at chromosome 14q11.2 was TINF2 (MIM #604319), TERF1 (TRF1)-interacting nuclear factor 2 (protein name TIN2), a 6 exon gene whose protein product (TIN2) binds to telomeric repeat binding factors 1 and 2 (TRF1 and TRF2). Therefore, bidirectional sequence analysis on genomic DNA from the proband was performed on TINF2 as described.29 Sequence analysis of 298 healthy control individuals (596 chromosomes) was performed when a possible mutation was identified. Controls were derived from the SNP500Cancer Panel33 and Human Variation Panels HD100AA and HD100CA from Coriell Cell Repositories. Their self-identified ethnicities were 118 African Americans, 125 Caucasians, 32 Asian/Pacific Rim, and 23 Hispanic.
The proband, III-07, was heterozygous for an A-to-G mutation at Ex6+234 in TINF2, and this results in a change from lysine to glutamic acid at amino acid 280 (K280E) (Figure 4). Sequence analysis of the individuals who were part of the linkage scan from family A showed that this mutation was present only in individuals who had telomere lengths less than the first percentile, III-02, III-04, III-05, III-07, III-08, and III-10. Specifically, I-01 and I-02, the paternal grandparents of the proband, did not have this mutation. It probably arose as a previously unrecognized mutation in their monozygotic twin sons, II-01 and II-03; however, we were unable to verify this because of the absence of archival tissue from the deceased twins. This mutation was not identified in the sequence analysis of genomic DNA from 298 healthy individuals.
Figure 4.
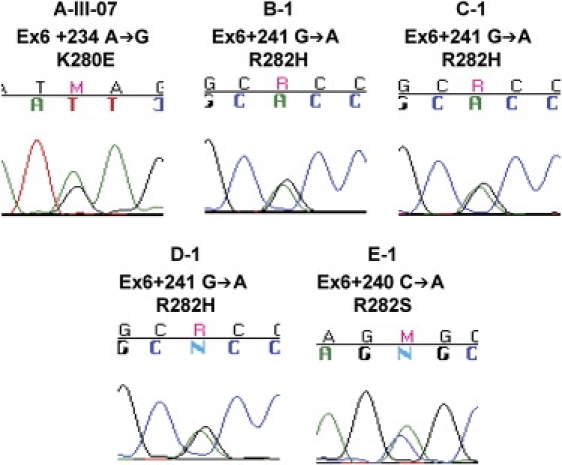
Representative Sequence Tracings of TINF2 Mutations
The proband and mutation are noted above the sequence tracing.
The presence of TINF2 mutations only in individuals from family A with very short telomeres prompted the evaluation of all DC patients that were available for analyses. TINF2 was sequenced in an additional 15 DC probands with very short telomeres. Eight of these patients had no detectable mutations in DKC1, TERC, or TERT, three had DKC1 mutations, three had TERC mutations and one had a TERT mutation. Half (8 out of 16 total) of the DC patients were molecularly uncharacterized, consistent with previous estimates by others.26 TINF2 was not mutated in DC probands with mutations in DKC1, TERC, or TERT. In addition, common SNPs in TINF2 were not identified in any DC probands.
Heterozygous TINF2 mutations were identified in four of the eight previously molecularly uncharacterized probands (normal DKC1, TERC, or TERT) with very short telomeres (Table 1 and Figures 2B and 3) but not in any of the 298 controls. Three probands, B-1, C-1, and D-1, shared the same TINF2 mutation, arginine to histidine at amino acid 282 (R282H, Ex6+241G→A). The fourth proband, E-1, had a different mutation at amino acid 282, arginine to serine (R282S, Ex6+240C→A).
Proband B-1 had the DC triad, very short telomeres, thrombocytopenia, intrauterine growth retardation, and speech delay. He developed severe aplastic anemia at 3.7 years of age. His unaffected mother had normal telomere lengths and no mutation in TINF2. His father and brother were clinically unaffected, but blood was not available for telomere length measurement or mutation testing.
Proband C-1 had Revesz syndrome with the characteristic bilateral exudative retinopathy, the DC triad, developmental delay, cerebellar hypoplasia, and very short telomere lengths. Severe aplastic anemia developed at age 1.5 years. His unaffected parents and one sister had normal telomere lengths and no mutation in TINF2. A second sister had normal telomere lengths, but DNA was not available for sequence analysis.
Proband D-1 did not have the DC diagnostic triad but did have dystrophic nails, very short telomeres, and IgA deficiency. Severe aplastic anemia developed at 2.8 years of age. He had no siblings; his parents had normal telomere lengths with no mutation in TINF2.
The proband with the R282S TINF2 mutation, E-1, was unsuccessfully treated with two courses of immunosuppressive therapy for acquired severe aplastic anemia, first diagnosed at age 3.2 years, before the classic nail, skin, and oral findings of DC were identified. She had very short telomeres and reduced numbers of natural killer cells. Her parents and sister were healthy, did not have a TINF2 mutation, and had normal telomere lengths.
TINF2, like many genes important in telomere biology, has limited nucleotide diversity and is highly evolutionarily conserved.29 It has been suggested that mutations in highly conserved genomic regions are more likely to be pathogenic.34 Therefore, the degree of evolutionary conservation in the mutated region of TINF2 was further evaluated with mVISTA alignments.35 Figure 5A shows the high level of evolutionary conservation between human and mouse TINF2 genomic sequences. Comparisons of the genomic and protein sequences of TINF2 between humans and five other species, chimpanzee (Pan troglodytes), mouse (Mus musculus), rat (Rattus norvegicus), dog (Canis lupus familiaris), and cow (Bos taurus), are shown in Figures 5B and 5C, respectively. The nucleotides that were mutated in DC patients, Ex6+234A→G, Ex6+240C→A, and Ex6+241G→A, are conserved between all six species. Amino acids 279–287 are also conserved between the six species evaluated.
Figure 5.
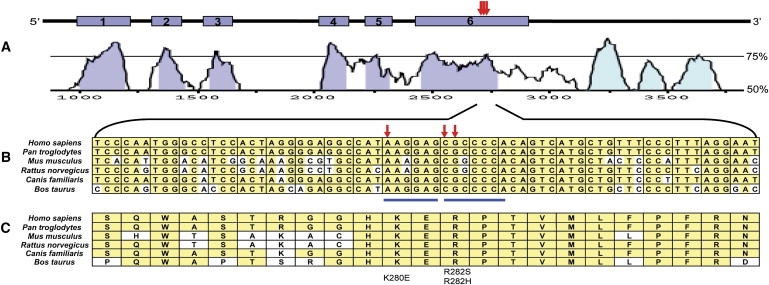
Genomic Structure and Evolutionary Conservation of TINF2
(A) The genomic region of TINF2 consists of 2686 base pairs. The red arrows show the relative positions of the mutations identified in the DC patients in this study, Ex6+234A→G (K280E), Ex6+240C→A (R282S), and Ex6+241G→A (R282H). Curves showing conservation between human and mouse genomic sequences were generated with VISTA. On the right, 50% and 75% conservation are noted. Exons are shown in blue, and the conserved 3′ untranslated region is shown in light blue.
(B) Comparison of nucleotide conservation between Homo sapiens (Entrez Gene Annotation, NC_000014.7), Pan troglodytes (NC_006481.2), Mus musculus (NC_00080.5), Rattus norvegicus (NC_005114.2), Canis lupus familiaris (NC_006590.2), and Bos taurus (NC_007308.2) TINF2 genomic sequences between Ex6+204 through Ex6+272. Conserved nucleotides are shown in yellow. Mutated nucleotides are noted with red arrows. The blue lines above the nucleotides indicate putative exonic splice enhancer (ESE) sequences.
(C) Comparison of amino acid conservation between Homo sapiens (Entrez Protein Annotation, NP_036593.1), Pan troglodytes (CAH89509.1, hypothetical protein TIN2), Mus musculus (NP_663751.2), Rattus norvegicus (NP_001006963.1), Canis lupus familiaris (XP_850486.1, predicted protein), and Bos taurus (XP_593926.3, predicted protein) between amino acids 270 and 292. Conserved amino acids are noted in yellow. Mutated amino acids K280E, R282S, and R292H are noted at the bottom.
In silico analyses were performed for predicting the possible functional effects of TINF2 mutations with sorting intolerant from tolerant (SIFT).36 SIFT predicted that all three mutations, K280E, R282H, and R282S, would not be tolerated. Exonic splicing enhancers (ESEs) were identified with ESEFinder37 and RESCUE-ESE38 (Figure 5B). An AAGGAG hexamer is present, beginning at Ex6+234 (start of codon 280). The mutation at that site changes the putative ESE to GAGGAG in addition to the K280E mutation in family A. The Ex6+240C→A (start of codon 282) mutation was predicted to potentially alter the SF2/ASF motif by changing CGCCCCA to AGCCCCA in addition to the R282S coding mutation. Ex6+241G→A, at the second site of this motif and the R282H mutation, may have a weaker effect.
It is also notable that the mutated amino acids are located in extremely close proximity, positions 280 and 282; amino acid 282 was mutated in four patients. A recombination or mutation “hot spot” that is undefined at this time is theoretically possible in this region. This possibility, in combination with in silico predictions of the deleterious effects of these mutations, and the high degree of evolutionary conservation of this region of TINF2, suggests that these mutations are deleterious and probably contribute to the telomere abnormalities and phenotypes seen in our DC patients with TINF2 mutations.
The protein product of TINF2, TIN2, is a central component of shelterin, the protein complex that stabilizes telomeres.17 TIN2 connects the three primary DNA-binding proteins, TRF1, TRF2, and POT1. The POT1 connection occurs via TPP1 (gene name ACD).17,39–42 TIN2, POT1, and TPP1 interactions in the cytoplasm were also found to regulate the assembly and function of the shelterin complex (also referred to as the telosome) in the nucleus.43 In that study, Chen et al. proposed that TIN2 could be a limiting factor in telosome formation, in terms of both bridging different subcomplexes and regulating their localization.
The mutated region of TIN2 in DC patients is near the end of its TRF1-binding domain.40,42,44 Kim et al. showed that the TIN2-15C mutant, containing only amino acids 1–257, resulted in loss of TRF1 binding to TIN2.40 Therefore, it is possible that disruption of this domain by the mutations in amino acids 280 and 282 will lead to altered TRF1 binding to TIN2, thereby disrupting the TIN2-TRF1-TRF2 complex and resulting in telomeric instability. TIN2 binding partners and its 3D structure are active areas of investigation.
Other than a genetic association study that suggested an increased risk for acquired aplastic anemia in individuals with a variant allele in intron 9 of the TRF1 gene (TERF1),45 the role of germline polymorphisms or mutations in proteins of the shelterin telomere protection complex is predominantly unexplored.
The clinical spectrum of DC is broad, ranging from the diagnosis of a severe phenotype during infancy to more subtle cases that present in adulthood.2,3 Therefore, we chose to define the affected phenotype for the linkage study as individuals with telomeres in leukocyte subsets less than the first percentile because this was shown to be a sensitive and specific test to differentiate DC patients from healthy relatives and other IBMFS patients.12 This approach, in a clinically heterogeneous family, identified a previously unreported DC susceptibility gene, TINF2. This further illustrates the utility of telomere lengths that are less than the first percentile as a diagnostic test for DC in the setting of variable clinical phenotypes.
Four of eight additional unrelated probands with clinical DC and very short telomere lengths but no mutations in DKC1, TERC, or TERT were heterozygous for mutations in TINF2 (R282H and R282S). In total, TINF2 mutations were present in five out of nine (56%) previously molecularly uncharacterized DC probands, including the probands, family A III-07, B-1, C-1, D-1, and E-1. The mutations in DC probands represented in our patients are DKC1 (18.8%), TERC (18.8%), TERT (6.2%), and TINF2 (31.2%). Four patients (25%) remain unclassified at the molecular level. Larger studies that would determine the prevalence of TINF2 mutations in DC are warranted.
By focusing on very short telomere lengths in leukocyte subsets as the affected phenotype, instead of the heterogeneous clinical features present in DC patients, we achieved sufficient statistical power to permit our mapping and identifying a previously unreported DC gene, TINF2. This strategy also provided important further validation of telomere length as a diagnostic test for DC. This study demonstrates that TINF2 is the fifth gene mutated in DC, the first in Revesz syndrome, and the first shelterin complex gene mutated in human disease.
Acknowledgments
We are grateful to the patients and their families for their invaluable contributions to this study. We would like to thank Dr. Rodrigo Calado and Dr. Neal Young for performing the initial TERT sequence analysis on the family A proband. We would also like thank Dr. Lynn Goldin, Dr. Mark H. Greene, and Dr. Stephen J. Chanock for helpful advice and discussion. Lisa Leathwood, Ann Carr, and Luda Brener of Westat and June Peters of NCI provided outstanding clinical support. Irma Vulto of the British Columbia Cancer Research Center provided excellent technical support. We thank Tara Namey of Lehigh Valley Hospital for referral of family A. This research was supported in part by the Intramural Research Program of the National Institutes of Health and the National Cancer Institute and by contracts N02-CP-91026, N02-CP-11019, and HHSN261200655001C with Westat. G.M.B. was supported by the Swiss National Foundation and the Bernese Cancer League. Work in the laboratory of P.M.L. is supported by grants from the National Institutes of Health (AI29524), the Canadian Institute of Health Research (MOP38075 and GMH79042), and the National Cancer Institute of Canada with funds from the Terry Fox Foundation. P.M.L. is a founding shareholder in Repeat Diagnostic, a company specializing in leukocyte telomere length measurements with flow FISH.
Web Resources
The URLs for data presented herein are as follows:
Coriell Cell Repository, http://ccr.coriell.org
NCBI SNP Database, http://www.ncbi.nlm.nih.gov/SNP/index.html
NCI Core Genotyping Facility, http://cgf.nci.nih.gov
NCI Inherited Bone Marrow Failure Syndromes Study Web Site, www.marrowfailure.cancer.gov
Oncogenomics Primer Database, http://ntddb.abcc.ncifcrf.gov/cgi-bin/Primers.pl
Online Mendelian Inheritance in Man (OMIM), http://ncbi.nlm.nih.gov/Omim
SNP500Cancer, http://snp500cancer.nci.nih.gov
References
- 1.Walne A.J., Marrone A., Dokal I. Dyskeratosis congenita: A disorder of defective telomere maintenance? Int. J. Hematol. 2005;82:184–189. doi: 10.1532/IJH97.05067. [DOI] [PubMed] [Google Scholar]
- 2.Vulliamy T.J., Marrone A., Knight S.W., Walne A., Mason P.J., Dokal I. Mutations in dyskeratosis congenita: Their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–2685. doi: 10.1182/blood-2005-07-2622. [DOI] [PubMed] [Google Scholar]
- 3.Tamary H., Alter B.P. Current diagnosis of inherited bone marrow failure syndromes. Pediatr. Hematol. Oncol. 2007;24:87–99. doi: 10.1080/08880010601123240. [DOI] [PubMed] [Google Scholar]
- 4.Sznajer Y., Baumann C., David A., Journel H., Lacombe D., Perel Y., Blouin P., Segura J.F., Cezard J.P., Peuchmaur M. Further delineation of the congenital form of X-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome) Eur. J. Pediatr. 2003;162:863–867. doi: 10.1007/s00431-003-1317-5. [DOI] [PubMed] [Google Scholar]
- 5.Revesz T., Fletcher S., al Gazali L.I., DeBuse P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: A new syndrome? J. Med. Genet. 1992;29:673–675. doi: 10.1136/jmg.29.9.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kajtar P., Mehes K. Bilateral coats retinopathy associated with aplastic anaemia and mild dyskeratotic signs. Am. J. Med. Genet. 1994;49:374–377. doi: 10.1002/ajmg.1320490404. [DOI] [PubMed] [Google Scholar]
- 7.Al Rahawan M.M., Giri N., Alter B.P. Intensive immunosuppression therapy for aplastic anemia associated with dyskeratosis congenita. Int. J. Hematol. 2006;83:275–276. doi: 10.1532/IJH97.06030. [DOI] [PubMed] [Google Scholar]
- 8.Brazzola P., Duval M., Fournet J.C., Gauvin F., Dalle J.H., Champagne J., Champagne M.A. Fatal diffuse capillaritis after hematopoietic stem-cell transplantation for dyskeratosis congenita despite low-intensity conditioning regimen. Bone Marrow Transplant. 2005;36:1103–1105. doi: 10.1038/sj.bmt.1705171. [DOI] [PubMed] [Google Scholar]
- 9.Dror Y., Freedman M.H., Leaker M., Verbeek J., Armstrong C.A., Saunders F.E., Doyle J.J. Low-intensity hematopoietic stem-cell transplantation across human leucocyte antigen barriers in dyskeratosis congenita. Bone Marrow Transplant. 2003;31:847–850. doi: 10.1038/sj.bmt.1703931. [DOI] [PubMed] [Google Scholar]
- 10.Ostronoff F., Ostronoff M., Calixto R., Florencio R., Domingues M.C., Souto Maior A.P., Sucupira A., Tagliari C. Fludarabine, cyclophosphamide, and antithymocyte globulin for a patient with dyskeratosis congenita and severe bone marrow failure. Biol. Blood Marrow Transplant. 2007;13:366–368. doi: 10.1016/j.bbmt.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Yabe M., Yabe H., Hattori K., Morimoto T., Hinohara T., Takakura I., Shimizu T., Shimamura K., Tang X., Kato S. Fatal interstitial pulmonary disease in a patient with dyskeratosis congenita after allogeneic bone marrow transplantation. Bone Marrow Transplant. 1997;19:389–392. doi: 10.1038/sj.bmt.1700674. [DOI] [PubMed] [Google Scholar]
- 12.Alter B.P., Baerlocher G.M., Savage S.A., Chanock S.J., Weksler B.B., Willner J.P., Peters J.A., Giri N., Lansdorp P.M. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110:1439–1447. doi: 10.1182/blood-2007-02-075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell J.R., Wood E., Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 14.Armanios M., Chen J.L., Chang Y.P., Brodsky R.A., Hawkins A., Griffin C.A., Eshleman J.R., Cohen A.R., Chakravarti A., Hamosh A. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl. Acad. Sci. USA. 2005;102:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mason P.J., Wilson D.B., Bessler M. Dyskeratosis congenita–a disease of dysfunctional telomere maintenance. Curr. Mol. Med. 2005;5:159–170. doi: 10.2174/1566524053586581. [DOI] [PubMed] [Google Scholar]
- 16.Collins K., Mitchell J.R. Telomerase in the human organism. Oncogene. 2002;21:564–579. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 17.de Lange T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura T.M., Cech T.R. Reversing time: Origin of telomerase. Cell. 1998;92:587–590. doi: 10.1016/s0092-8674(00)81123-x. [DOI] [PubMed] [Google Scholar]
- 19.Shay J.W., Zou Y., Hiyama E., Wright W.E. Telomerase and cancer. Hum. Mol. Genet. 2001;10:677–685. doi: 10.1093/hmg/10.7.677. [DOI] [PubMed] [Google Scholar]
- 20.Maser R.S., DePinho R.A. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- 21.Yamaguchi H., Baerlocher G.M., Lansdorp P.M., Chanock S.J., Nunez O., Sloand E., Young N.S. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102:916–918. doi: 10.1182/blood-2003-01-0335. [DOI] [PubMed] [Google Scholar]
- 22.Knight S.W., Heiss N.S., Vulliamy T.J., Greschner S., Stavrides G., Pai G.S., Lestringant G., Varma N., Mason P.J., Dokal I. X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am. J. Hum. Genet. 1999;65:50–58. doi: 10.1086/302446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vulliamy T.J., Walne A., Baskaradas A., Mason P.J., Marrone A., Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol. Dis. 2005;34:257–263. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi H., Calado R.T., Ly H., Kajigaya S., Baerlocher G.M., Chanock S.J., Lansdorp P.M., Young N.S. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 25.Walne A.J., Vulliamy T., Marrone A., Beswick R., Kirwan M., Masunari Y., Al Qurashi F.H., Aljurf M., Dokal I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 2007;16:1619–1629. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marrone A., Dokal I. Dyskeratosis congenita: Molecular insights into telomerase function, ageing and cancer. Expert Rev. Mol. Med. 2004;6:1–23. doi: 10.1017/S1462399404008671. [DOI] [PubMed] [Google Scholar]
- 27.Baerlocher G.M., Vulto I., de Jong G., Lansdorp P.M. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat. Protoc. 2006;1:2365–2376. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- 28.Savage S.A., Stewart B.J., Weksler B.B., Baerlocher G.M., Lansdorp P.M., Chanock S.J., Alter B.P. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol. Dis. 2006;37:134–136. doi: 10.1016/j.bcmd.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Savage S.A., Stewart B.J., Eckert A., Kiley M., Liao J.S., Chanock S.J. Genetic variation, nucleotide diversity, and linkage disequilibrium in seven telomere stability genes suggest that these genes may be under constraint. Hum. Mutat. 2005;26:343–350. doi: 10.1002/humu.20226. [DOI] [PubMed] [Google Scholar]
- 30.Webb E.L., Sellick G.S., Houlston R.S. SNPLINK: Multipoint linkage analysis of densely distributed SNP data incorporating automated linkage disequilibrium removal. Bioinformatics. 2005;21:3060–3061. doi: 10.1093/bioinformatics/bti449. [DOI] [PubMed] [Google Scholar]
- 31.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: A unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 32.Abdelhaleem M. Do human RNA helicases have a role in cancer? Biochim. Biophys. Acta. 2004;1704:37–46. doi: 10.1016/j.bbcan.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Packer B.R., Yeager M., Burdett L., Welch R., Beerman M., Qi L., Sicotte H., Staats B., Acharya M., Crenshaw A. SNP500Cancer: A public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res. 2006;34:D617–D621. doi: 10.1093/nar/gkj151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y., Spitz M.R., Amos C.I., Lin J., Schabath M.B., Wu X. An evolutionary perspective on single-nucleotide polymorphism screening in molecular cancer epidemiology. Cancer Res. 2004;64:2251–2257. doi: 10.1158/0008-5472.can-03-2800. [DOI] [PubMed] [Google Scholar]
- 35.Frazer K.A., Pachter L., Poliakov A., Rubin E.M., Dubchak I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fairbrother W.G., Yeh R.F., Sharp P.A., Burge C.B. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 39.Liu D., Safari A., O'Connor M.S., Chan D.W., Laegeler A., Qin J., Songyang Z. PTOP interacts with POT1 and regulates its localization to telomeres. Nat. Cell Biol. 2004;6:673–680. doi: 10.1038/ncb1142. [DOI] [PubMed] [Google Scholar]
- 40.Kim S.H., Beausejour C., Davalos A.R., Kaminker P., Heo S.J., Campisi J. TIN2 mediates functions of TRF2 at human telomeres. J. Biol. Chem. 2004;279:43799–43804. doi: 10.1074/jbc.M408650200. [DOI] [PubMed] [Google Scholar]
- 41.O'Connor M.S., Safari A., Xin H., Liu D., Songyang Z. A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc. Natl. Acad. Sci. USA. 2006;103:11874–11879. doi: 10.1073/pnas.0605303103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye J.Z., Donigian J.R., van Overbeek M., Loayza D., Luo Y., Krutchinsky A.N., Chait B.T., de Lange T. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J. Biol. Chem. 2004;279:47264–47271. doi: 10.1074/jbc.M409047200. [DOI] [PubMed] [Google Scholar]
- 43.Chen L.Y., Liu D., Songyang Z. Telomere maintenance through spatial control of telomeric proteins. Mol. Cell. Biol. 2007;27:5898–5909. doi: 10.1128/MCB.00603-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim S.H., Kaminker P., Campisi J. TIN2, a new regulator of telomere length in human cells. Nat. Genet. 1999;23:405–412. doi: 10.1038/70508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savage S.A., Calado R.T., Xin Z.T., Ly H., Young N.S., Chanock S.J. Genetic variation in telomeric repeat binding factors 1 and 2 in aplastic anemia. Exp. Hematol. 2006;34:664–671. doi: 10.1016/j.exphem.2006.02.008. [DOI] [PubMed] [Google Scholar]