

Abstract
Parkinson disease (PD) is a common neurodegenerative disorder caused by environmental and genetic factors. We have previously shown linkage of PD to chromosome 8p. Subsequently, fibroblast growth factor 20 (FGF20) at 8p21.3–22 was identified as a risk factor in several association studies. To identify the risk-conferring polymorphism in FGF20, we performed genetic and functional analysis of single-nucleotide polymorphisms within the gene. In a sample of 729 nuclear families with 1089 affected and 1165 unaffected individuals, the strongest evidence of association came from rs12720208 in the 3′ untranslated region of FGF20. We show in several functional assays that the risk allele for rs12720208 disrupts a binding site for microRNA-433, increasing translation of FGF20 in vitro and in vivo. In a cell-based system and in PD brains, this increase in translation of FGF20 is correlated with increased α-synuclein expression, which has previously been shown to cause PD through both overexpression and point mutations. We suggest a novel mechanism of action for PD risk in which the modulation of the susceptibility gene's translation by common variations interfere with the regulation mechanisms of microRNA. We propose this is likely to be a common mechanism of genetic modulation of individual susceptibility to complex disease.
Introduction
The major causes of Parkinson disease (PD [MIM 168600]) are still largely unknown.1,2 The exciting discovery of several causative monogenic mutations in PD families can only explain a very limited number of PD cases.3 In contrast, the genetic contribution in approximately 90% of PD cases is more complex.4 To this end, we have been actively seeking functionally relevant polymorphic variations associated with the risk of developing PD.
We previously reported that one such polymorphism, rs1989754, in the fibroblast growth factor 20 (FGF20 [MIM 605558]) gene, was associated with an increased risk for PD.5 Several lines of evidence support this association: (1) FGF20 is located within the chromosome 8 linkage peak in our previous linkage analysis.6 (2) FGF20 is preferentially expressed in the substantia nigra compared to other brain regions.7 (3) A recent report showed that FGF20 has neurotrophic properties and promotes survival of dopaminergic neurons.8 (4) Clarimon et al. examined FGF20 in a small case-control dataset and confirmed the significance of the same single-nucleotide polymorphism (SNP) rs1989754 in a Finnish sample, although it did not meet stringent Bonferroni correction requirements when adjusting for multiple comparisons were adjusted for.9 (5) Maraganore et al. showed association between rs17515020 within the FGF20 gene and PD in the first-tier analysis (p = 0.03) of their whole-genome association study of PD but did not consider the SNP in their second-stage analysis.10 Given these multiple lines of evidence, we sought to extend our initial association in an expanded dataset and identify the biological basis for this association.
Material and Methods
Samples
Affected individuals and family members were collected by the Duke Center for Human Genetics (DCHG) Morris K. Udall Parkinson Disease Research Center of Excellence ascertainment core and the 13 centers of the Parkinson Disease Genetics Collaboration. A standard clinical evaluation involves a neurological examination, including assessment by use of the Unified Parkinson's Disease Rating Scale. Our total dataset includes 729 independent families with 1089 affected and 1165 unaffected individuals. Unaffected participants demonstrated no signs of the disease. So that diagnostic consistency could be maintained across participating clinics, a dually board-certified neurologist and Ph.D. medical geneticist (Jeffery M. Vance) reviewed the clinical data for all participants. Parkin mutation carriers were excluded from this study. All participants signed informed consents prior to blood and data collection. Study protocols and consent forms were approved by institutional review board. All subjects analyzed were white.
DNA Extraction and Genotyping
DNA samples were prepared and stored by the DCHG DNA bank. Genomic DNA was extracted from whole blood with the PureGene system (Gentra Systems). Primers and probes were designed with the Primer Express 2.0 program (Applied Biosystems). The fluorescence generated during the PCR amplification was detected with the ABI Prism 7900HT Sequence Detection System and was analyzed with SDS software (Applied Biosystems). Stringent quality-control measures were taken so that data consistency could be ensured. Internal controls consisted of 24 duplicated individuals per 384-well plate. Data were stored and managed by the PEDIGENE system.
Human FGF20 3′ UTR Luciferase Constructs
The Renilla luciferase FGF20 3′ untranslated region (UTR) constructs were made by a previously reported method.11 The full length of human FGF20-3′ UTR containing both genotypes at rs12720208 was amplified with forward primer 5′-GATCTCTAGAAGTGCGATAGTGACATTATGGAAGAGTCAAAC-3′ and reverse primer 5′-CATGGATCCGCCAAAACACTGCATGAAAATGTTTAATTC-3′ from a heterozygous human genomic DNA sample. The polymerase chain reaction (PCR) product was separated in agarose gel and extracted, purified, and cloned with TA cloning Kit (Invitrogen, Carlsbad, CA). The inserts containing allele-C and allele-T were confirmed by sequencing. The 3′ UTR of Renilla luciferase in the vector pRL-SV40 (Promega, Madison, WI) was replaced with the full-length 3′ UTR of FGF20 by restriction enzymes XbaI and BamHI. Finally, the pRL-FGF20-3′ UTR deletion 150–171 construct was generated with the Quickchange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) with forward mutagenic deletion primer 5′-GAGAAAAAAAACCAAAAAAAATTTCACTCTTTATATGTGGATTAAGTTCCC-3′ and a complementary reverse mutagenic deletion primer, 5′-GGGAACTTAATCCACATATAAAGAGTGAAATTTTTTTTGGTTTTTTTTCTC-3′. The resulting constructs (pRL-FGF20-3′ UTR-C-allele, pRL-FGF20-3′ UTR-T-allele, and pRL-FGF20-3′ UTR-21bp deletion) were verified by sequencing.
Luciferase Assay
Neuro2A cells was maintained in minimum essential medium (Eagle) with 0.1% nonessential amino acids, Eagle's balanced salt solution (BSS), 1 mM sodium pyruvate, and 10% fetal bovine serum (FBS)at a 37°C incubator supplemented with 5% CO2. Cells were seeded at 3 × 105 cells per well in 24-well plates (BD Biosciences, Bedford, MA). Sixteen hours after the plating, cells were transfected by Lipofectamin 2000 according to manufacturer's suggestion. In each well, 3.28 fmol pRL-FGF20-3′ UTR vector and 0.73 fmol pGL3 control vector were cotransfected with 0, 0.1, 1, 10, 100 pmol pre-miR-433 accordingly. Five replicates for each group and the experiment repeated at least three times. After transfection for 24 hr, cells were harvested by the addition of 100 μl passive lysis buffer. Renilla luciferase activities in cell lysate were measured with the Dual Luciferase assay system (Promega, Madison, WI) in TD-20/20 luminometer (Turner Biosystems, Sunnyvale, CA) and were normalized with the firefly luciferase activities and analyzed with the t test.
Immunoblot Analysis
At 80% confluences in 6-well plates, human fibroblasts were transfected with 0, 5, and 50 pmol pre-miR-433, respectively, which is corresponding to 0, 1, and 10 pmol pre-miR-433 when cell cultured in 24-well plates. The fibroblast cells were lysed by radio-immunoprecipitation assay (RIPA) buffer (Pierce, Rockford, IL) with protease inhibitor cocktail (Sigma, St. Louis, MO) by dounce homogenizer after transfection for 24 hr. SH-SY5Y cells were grown in minimum essential medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1 mM sodium pyruvate, and 0.1 mM nonessential amino acids. After cells were incubated in starvation medium in which 10% FBS was replaced by 0.1% bovine serum albumin (BSA) for 16 hr, SH-SY5Y cells were stimulated with recombinant human FGF20 (R&D Systems, Minneapolis, MN) at 0, 5, and 50 ng/ml for 24 hr. Total SH-SY5Y cell lysate was prepared as it was for fibroblast cells. Human brain cortex was lysed as descried above. Both cell and tissue lysate were clarified by centrifugation. The protein concentration was determined by 2-D Quant Kit (GE Healthcare, Piscataway, NJ). The cell and tissue lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and immunoblotted with rat monoclonal anti-FGF20 antibody (R&D Systems, Minneapolis, MN), mouse monoclonal anti-α-synuclein ntibody (BD Biosciences, Bedford, MA), and mouse monoclonal anti-β-actin antibody (Sigma, St. Louis, MO). Proteins were visualized with chemiluminescence (Bio-Rad, Hercules, CA). Specific bands density were quantified with Fluor-S MultiImager and analyzed with the t test.
Statistical Analysis
A single affected and unaffected individual were selected at random from each family for tests of Hardy-Weinberg disequilibrium. These tests were conducted with Genetic Data Analysis software, through the application of a permutation test with 3200 permutations to estimate each p value (Lewis Lab Software website). Two measures of linkage disequilibrium (LD), squared correlation coefficient (r2), and Lewontin's standardized disequilibrium coefficient (D′) were computed between pairs of SNPs through use of the Graphical Overview of Linkage Disequilibrium (GOLD) software package.12 Single-locus association analysis was performed in the families through use of the pedigree disequilibrium test (PDT).13 We used the PDT sum version of the statistic to compare allele frequencies between affected individuals and their unaffected parents (when available) or siblings within families.14 We used the association in the presence of linkage (APL) program as a second test of association in families.15 APL correctly infers missing parental genotypes by estimating identity-by-descent parameters for multiple affected siblings even when linkage is present. All tests were considered statistically significant at α = 0.05.
Results
Association Study of Polymorphism in FGF20 with PD
To further test the influence of FGF20 on the risk of PD, we genotyped eight SNPs within the FGF20 gene in an expanded dataset. Due to the ongoing collection of samples throughout the duration of the study, the family counts differ slightly for each SNP genotyped. Our expanded dataset includes 729 independent families with 1089 affected and 1165 unaffected individuals. No family that was found to carry a Parkin mutation was included in this dataset.
Five SNPs presented here were not previously genotyped in our initial dataset, and four of them are located within the 3′ UTR (Figure 1). No SNP genotyped showed significant evidence for deviation from Hardy-Weinberg equilibrium. The LD analyses based on r2 revealed strong LD between SNPs rs1989754 and rs11203822, between SNPs rs1989754 and rs10888125, and between rs11203822 and rs10888125 in the affected individuals (Table 1). Values of D′ were large for most SNP pairs, suggesting that there has been little historic recombination in the region over time. Levels of LD were similar in the unaffected set (data not shown).
Figure 1.
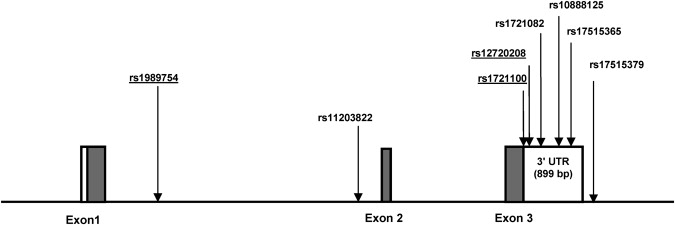
Gene Structure of FGF20
Exons are denoted by blocks; coding regions are shown in gray, and 3′ UTRs are shown in white. Locations of SNPs are indicated by arrows. Underlined SNPs were previously genotyped in the initial study.
Table 1.
Measures of Linkage Disequilibrium between FGF20 SNPs
| rs1989754 | rs11203822 | rs1721100 | rs12720208 | rs1721082 | rs10888125 | rs17515365 | rs17515379 | |
|---|---|---|---|---|---|---|---|---|
| rs1989754 | 0.96 | 0.295 | 0.067 | 0.218 | 0.882 | 0.108 | 0.004 | |
| rs11203822 | 0.991 | 0.305 | 0.075 | 0.231 | 0.911 | 0.114 | 0.004 | |
| rs1721100 | 0.959 | 0.983 | 0.25 | 0.607 | 0.334 | 0.265 | 0.012 | |
| rs12720208 | 0.899 | 0.964 | 0.987 | 0.334 | 0.075 | 0.018 | 0.046 | |
| rs1721082 | 0.955 | 0.989 | 0.904 | 0.989 | 0.253 | 0.515 | 0.014 | |
| rs10888125 | 0.968 | 0.993 | 0.992 | 0.938 | 1 | 0.129 | 0.003 | |
| rs17515365 | 0.953 | 0.977 | 0.841 | 0.999 | 1 | 1 | 0.001 | |
| rs17515379 | 1 | 1 | 1 | 1 | 1 | 1 | 0.205 |
r2 values are given in the upper diagonal, and the D′ values are below the diagonal.
Two methodologies were utilized for the analysis of genetic association with PD: (1) Results from the PDT13,14 in the expanded dataset were consistent with our previous report, i.e., single-locus tests demonstrated strong association with PD for several SNPs in the overall sample, including three 3′ UTR SNPs, rs10888125 (p = 0.0009, G allele positively associated), rs12720208 (p = 0.0019, T allele positively associated), and rs1721082 (p = 0.0029, T allele positively associated) (Table 2). (2) The APL test15 results were also significant. The strongest association was with the 3′ UTR SNP rs12720208 (p = 0.0001) (Table 3).
Table 2.
Allele Frequencies and p Value of Single-Locus Association Analysis
| Family Triads |
Discordant Siblings |
||||
|---|---|---|---|---|---|
| SNP Allele | Parental Transmission | Parental Nontransmission (na) | Affected Frequency (nb) | Unaffected Frequency (nc) | PDT p Value |
| rs1989754C | 0.387 | 0.481 (111) | 0.41 (665) | 0.432 (902) | 0.0029 |
| rs11203822A | 0.378 | 0.49 (103) | 0.405 (648) | 0.436 (872) | 0.0013 |
| rs1721100C | 0.324 | 0.193 (111) | 0.3 (670) | 0.285 (912) | 0.033 |
| rs12720208T | 0.1 | 0.045 (110) | 0.105 (720) | 0.092 (967) | 0.0019 |
| rs1721082T | 0.283 | 0.16 (97) | 0.25 (701) | 0.221 (935) | 0.0029 |
| rs10888125G | 0.383 | 0.495 (103) | 0.42 (710) | 0.447 (950) | 0.0009 |
| rs17515365T | 0.155 | 0.108 (106) | 0.14 (705) | 0.122 (957) | 0.19 |
| rs17515379G | 0.01 | 0 (100) | 0.005 (678) | 0.004 (930) | 0.096 |
Number of triads used from nuclear families.
Number of affected siblings from discordant sibships.
Number of unaffected siblings from discordant sibships.
Table 3.
Allele Frequencies and p Value of Association in the Presence of Linkage
| SNP | na | Observed Transmitted Frequency | Expected Transmitted Frequency | APL p Value |
|---|---|---|---|---|
| rs1989754C | 689 | 0.42 | 0.44 | 0.02 |
| rs11203822A | 684 | 0.416 | 0.436 | 0.003 |
| rs1721100C | 693 | 0.29 | 0.278 | 0.02 |
| rs12720208T | 729 | 0.096 | 0.083 | 0.0001 |
| rs1721082T | 717 | 0.24 | 0.223 | 0.004 |
| rs10888125G | 723 | 0.43 | 0.452 | 0.003 |
| rs17515365T | 729 | 0.86 | 0.863 | 0.55 |
| rs17515379G | 715 | 0.005 | 0.003 | 0.04 |
Total nuclear families with at least one affected individual genotyped.
miR-433 Inhibits FGF20 Translation at SNP rs12720208
SNP rs12720208 is located 166 bp downstream of the terminating codon of FGF20 and lies within a predicted binding site for microRNA (miRNA) miR-433. miRNAs are small noncoding transcripts of approximately 22 nucleotides that negatively regulate the translation of complementary messenger RNA (mRNA), usually by binding the 3′ UTR. It is believed that miRNAs play important roles in development, cell death, and cell proliferation.16,17
miR-433 is highly expressed in brain but not in heart, liver, or kidney.18 The allele C of rs12720208 matches the predicted miR-433 binding domain, whereas the T allele represents a G:U wobble base pairing (Figures 2A and 2B) and thus a mismatch. It has been shown that the G:U wobble base pairing has an inhibitory effect on miRNA-mediated repression of gene translation.19 Therefore, we hypothesized that miR-433 would bind tightly to FGF20 mRNA transcripts containing the C allele, negatively regulating FGF20 protein translation. Conversely, the binding with mRNA transcripts containing the T allele would be disrupted, allowing a higher translation of FGF20 into protein.
Figure 2.
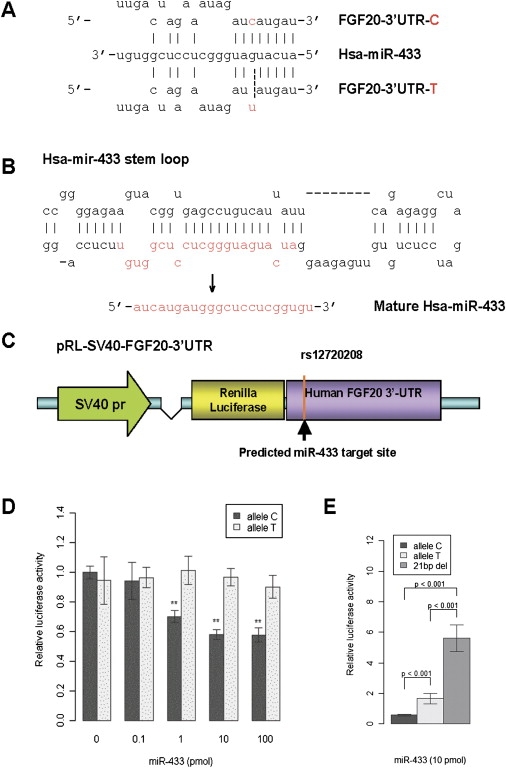
Characterization of miR-433 and Functional Analysis of miR-433 Targeting at the 3′ UTR of FGF20 Gene
(A) The predicted binding site for miR-433 at 3′ UTR of FGF20 gene. At rs12720208, allele C base paired with G in Watson-Crick mode (as shown with a solid line), whereas allele T wobble base paired with G (as shown with a dashed line).
(B) The precursor molecule pre-miR-433 proceeds to matured miR-433.
(C) The construct of pRL-SV40-FGF20-3′ UTR contains SV40 promoter, renilla luciferase gene, and full-length 3′ UTR of FGF20 gene with different alleles.
(D) The expression of renilla luciferase in Neuro2A cells transfected by pRL-SV40-FGF20-3′ UTR was measured by relative luciferase activity. Relative luciferase activity stands for renilla luciferase activity normalized by firefly luciferase activity which is the internal control. MiR-433 (1–100 pmol) decreased the expression level of renilla luciferase significantly in allele C constructs but not in allele T constructs (∗∗ indicates p < 0.01). Data are mean ± standard error (SE).
(E) The expression of renilla luciferase is significantly higher in neuro2A cells transfected by pRL-SV40-FGF20-21bp deletion than the constructs containing either C or T alleles under miR-433 (10 pmol). Data are mean ± SE.
To test this hypothesis, we replaced the 3′ UTR of a Renilla luciferase reporter gene with the full-length FGF20 3′ UTR containing either the T or the C alleles of rs12720208 (Figure 2C). In transiently transfected Neuro2A cells, the translation of Renilla luciferase (representing FGF20 protein) from constructs containing the C allele was dramatically reduced in the presence miR-433 in a concentration-dependent manner. However, no repression of Renilla luciferase translation was seen for the T allele construct when miR-433 was added (Figure 2D). After complete deletion of the miR-433 binding site (21 bp), we observed significantly higher Renilla luciferase translation compared to the constructs containing either C or T alleles (Figure 2E). In addition, the larger complete deletion demonstrates more translation than the smaller deletion, as would be expected (data not shown). This suggests that indeed, miR-433 binds and functionally negatively regulates the protein translation of FGF20, and that this regulation is disrupted by the presence of the T allele, presumably affecting miR-433 binding to the FGF20 transcript.
To further explore the effects of miR-433 on the translation of FGF20, we used two fibroblast lines, one homozygous for the C allele, the other heterozygous. Immunoblot analysis showed that miR-433 (1 and 10 pmol) repressed FGF20 translation in both cell lines, with greater inhibitory effects found in the fibroblast line homozygous for allele C (Figure 3A) than in the heterozygous line.
Figure 3.
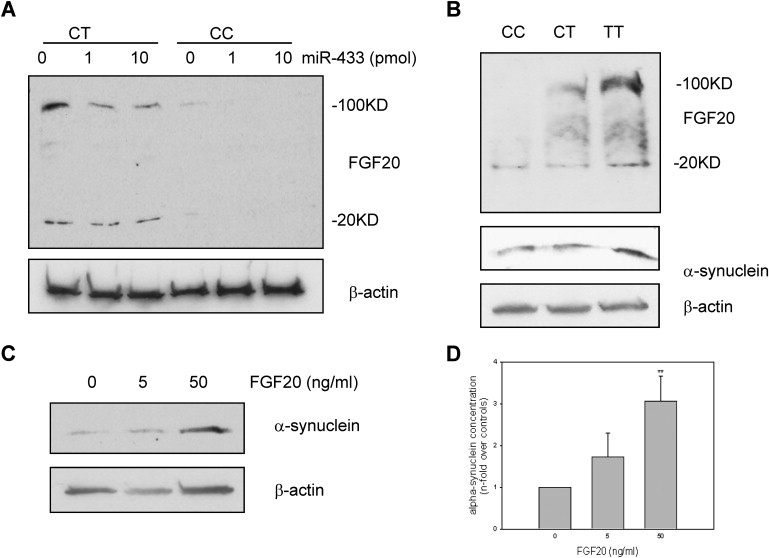
Immunoblot Analysis of Translation Levels of FGF20 and α-Synuclein in Different Genotypic Fibroblasts and Human Brains
(A) MiR-433 at 1 and 10 pmol significantly suppresses FGF20 protein level in the homozygote allele C and heterozygote at rs12720208 fibroblasts. FGF20 level is much higher in heterozygote cells than in homozygote allele C cells. In the heterozygote at rs12720208 fibroblasts, treatment with miR-433 for 24 hr does not change much of monomer form FGF20 (20 KD) but dramatically decreases the oligomer form of FGF20 (100 KD). The blot was re-probed with β-actin antibody showing the same amount loading.
(B) The human brain of homozygous allele T at rs12720208 has the highest level of both FGF20 and α-synuclein, comparing with the human brains of homozygous allele C or heterozygous at rs12720208. The blot was re-probed with β-actin antibody showing the same amount loading.
(C) α-synuclein translation amount is dramatically increased in SH-SY5Y cells after incubation with 50ng/ml FGF20 for 24 hr. The blot was re-probed with β-actin antibody showing the same amount loading.
(D) Quantification of immunoblotted α-synuclein in SH-SY5Y cells treated by FGF20. α-synuclein-band densities were normalized to their counterpart β-actin-band densities and represented the mean ± SE of three independent experiments (∗∗ indicates p < .01).
These experiments strongly suggest that miR-433 represses the FGF20 translation in carriers of allele C in vitro. To explore this in vivo, we compared FGF20 protein levels in human brain in three subjects with different genotypes. As a membrane-bound growth factor, both monomer (about 20 KD) and oligomer (about 100 KD) forms of FGF20 were observed in the immunoblot assay. Human brain homozygous for allele T has the highest FGF20 level of both monomer and oligomer, and brains carrying the C allele have substantially lower levels of both forms. This is particularly observed for the oligomer form of FGF20 (Figure 3B). Thus, both in vitro and in vivo studies suggest that SNP rs12720208 influences FGF20 translation level by alternating miR-433 targeting.
Increased FGF20 Leads to α-Synuclein Overexpression
FGF20 belongs to the fibroblast growth factor (FGF) family, which has at least 22 members. FGFs exert their specific biological functions through four different types of fibroblast growth factor receptors (FGFR1–4).20 By binding to FGFR1, FGF20 has been reported to promote differentiation of neural stem cells into tyrosine-hydroxylase-positive cells and enhance dopaminergic neuron survival.7,21 Interestingly, FGF2, another member in the FGF family that acts through FGFR1, has been shown to upregulate the expression of α-synuclein [MIM 163890] in cultured rat ventral midbrain dopaminergic neurons.22 Beucase FGF2 and FGF20 both bind to FGFR1, it seemed possible that FGF20 might also increase α-synuclein levels in dopaminergic neurons. To test this hypothesis, we used the dopaminergic neuroblastoma cell line SH-SY5Y, which has endogenous α-synuclein expression.23 After incubating SH-SY5Y cells in recombinant human FGF20 for 24 hr, the α-synuclein protein level was significantly increased compared that of to controls (Figures 3C and 3D). Human brain homozygous for allele T not only has the highest level of FGF20 translation but also the highest α-synuclein protein level in three brains with different genotypes (Figure 3B).
Discussion
Previous studies have shown that mutations of α-synuclein cause autosomal-dominant PD.24 α-synuclein is the principal component of filamentous Lewy bodies, the defining pathological hallmark of PD.25 Although the exact function of α-synuclein remains to be understood, pertinent to this report is that triplication and duplication of the α-synuclein gene can also cause dominant PD,26–28 and overexpression of exogenous α-synuclein is involved in the pathogenesis of PD.29,30 It has been shown that overexpression of wild-type α-synuclein reduces the number of tyrosine-hydroxylase-positive neurons differentiated from neural progenitors and increases the vulnerability of human dopaminergic neurons to neurotoxic insults, potentially resulting in neuronal death.31,32 Primates overexpressing exogenous α-synuclein develop PD-like neuropathological features, such as α-synuclein-positive inclusions and dystrophic neuritis.29
Thus, we suggest that FGF20 increases susceptibility to PD via chronically elevated levels of α-synuclein in human brain. During early stages of life, dopaminergic neurons in midbrain might benefit from FGF20 for proliferation, differentiation, and even protection from stress assault. But at later stages of life, the chronically increased level of FGF20 can indirectly contribute to dopaminergic neuron death.
miRNAs, as regulatory factors in large numbers, have great potential to be important in contributing to disease risk. A mutation in the 3′ UTR of the myostatin gene has been shown to create a de novo miRNA target site, allowing miRNA to suppress the translation of the gene and leading to the characteristic muscular hypertrophy of Texel sheep.33 Abelson et al. also reported that rare mutations in the miR-189 targeting site of the SLITRK1 gene can lead to Tourette syndrome.11 However, in this report, the variation is not a rare mutation but a polymorphism, demonstrating the enormous potential of normal polymorphic variation in miRNA targeting sites to provide a mechanism for susceptibility genes for a common complex disease. We predict that this is just the tip of the iceberg for future research in this area. Because of elevated FGF20 protein level correlating with enhanced α-synuclein expression, these results also reinforce the importance of α-synuclein in the pathogenesis of PD. It also suggests that this mechanism of increasing α-synuclein expression could be common for many susceptibility genes in PD.
Extending our previous linkage analysis and genetic association study, we have successfully identified the risk-conferring polymorphism in the FGF20 gene and a likely biological mechanism for increasing risk of PD. Next steps will be to explore whether miR-433 and FGF20 will be potentially useful for PD diagnosis and treatment.
Web Resources
The URLs for data presented herein are as follows:
Lewis Lab Software website, http://hydrodictyon.eeb.uconn.edu/people/plewis/software.php
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We thank all the families who participated in this study. We thank the members of the PD Genetics collaboration—Martha A. Nance, Ray L. Watts, Jean P. Hubble, William C. Koller, Kelly Lyons, Rajesh Pahwa, Matthew B. Stern, Amy Colcher, Bradley C. Hiner, Joseph Jankovic, William G. Ondo, Fred H. Allen Jr., Christopher G. Goetz, Gary W. Small, Donna Masterman, Frank Mastaglia, and Jonathan L. Haines—who contributed families to the study. This research was supported, in part, by National Institutes of Health Program Project grants 2 P50 NS39764-03 and P01 NS26630. Additional funding was received from National Institute on Aging grant 1R01-AG-20135-01.
References
- 1.Moore D.J., West A.B., Dawson V.L., Dawson T.M. Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 2.Farrer M.J. Genetics of Parkinson disease: Paradigm shifts and future prospects. Nat. Rev. Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 3.Dekker M.C., Bonifati V., van Duijn C.M. Parkinson's disease: Piecing together a genetic jigsaw. Brain. 2003;126:1722–1723. doi: 10.1093/brain/awg172. [DOI] [PubMed] [Google Scholar]
- 4.de Lau L.M.L., Breteler M.M.B. Epidemiology of Parkinson's disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 5.van der Walt J.M., Noureddine M.A., Kittappa R., Hauser M.A., Scott W.K., McKay R., Zhang F., Stajich J.M., Fujiwara K., Scott B.L. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am. J. Hum. Genet. 2004;74:1121–1127. doi: 10.1086/421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott W.K., Nance M.A., Watts R.L., Hubble J.P., Koller W.C., Lyons K., Pahwa R., Stern M.B., Colcher A., Hiner B.C. Complete genomic screen in Parkinson disease: Evidence for multiple genes. JAMA. 2001;286:2239–2244. doi: 10.1001/jama.286.18.2239. [DOI] [PubMed] [Google Scholar]
- 7.Ohmachi S., Watanabe Y., Mikami T., Kusu N., Ibi T., Akaike A., Itoh N. FGF-20, a novel neurotrophic factor, preferentially expressed in the substantia nigra pars compacta of rat brain. Biochem. Biophys. Res. Commun. 2000;277:355–360. doi: 10.1006/bbrc.2000.3675. [DOI] [PubMed] [Google Scholar]
- 8.Murase S., McKay R.D. A specific survival response in dopaminergic neurons at most risk in Parkinson's disease. J. Neurosci. 2006;26:9750–9760. doi: 10.1523/JNEUROSCI.2745-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarimon J., Xiromerisiou G., Eerola J., Gourbali V., Hellstrom O., Dardiotis E., Peuralinna T., Papadimitriou A., Hadjigeorgiou G.M., Tienari P.J. Lack of evidence of genetic association between FGF20 and Parkinson's disease in Finnish and Greek patients. BMC Neurol. 2005;5:11. doi: 10.1186/1471-2377-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maraganore D.M., de Andrade M., Lesnick T.G., Strain K.J., Farrer M.J., Rocca W.A., Pant P.V., Frazer K.A., Cox D.R., Ballinger D.G. High-resolution whole-genome association study of Parkinson disease. Am. J. Hum. Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abelson J.F., Kwan K.Y., O'Roak B.J., Baek D.Y., Stillman A.A., Morgan T.M., Mathews C.A., Pauls D.L., Rasin M.R., Gunel M. Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- 12.Abecasis G.R., Cookson W.O. GOLD—graphical overview of linkage disequilibrium. Bioinfomatics. 2000;16:182–183. doi: 10.1093/bioinformatics/16.2.182. [DOI] [PubMed] [Google Scholar]
- 13.Martin E.R., Monks S.A., Warren L.L., Kaplan N.L. A test for linkage and association in general pedigrees: The pedigree disequilibrium test. Am. J. Hum. Genet. 2000;67:146–154. doi: 10.1086/302957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin E.R., Bass M.P., Kaplan N.L. Correcting for a potential bias in the pedigree disequilibrium test. Am. J. Hum. Genet. 2001;68:1065–1067. doi: 10.1086/319525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin E.R., Bass M.P., Hauser E.R., Kaplan N.L. Accounting for linkage in family-based tests of association with missing parental genotypes. Am. J. Hum. Genet. 2003;73:1016–1026. doi: 10.1086/378779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao X., Yeo G., Muotri A.R., Kuwabara T., Gage F.H. Noncoding RNAs in the mammalian central nervous system. Annu. Rev. Neurosci. 2006;29:77–103. doi: 10.1146/annurev.neuro.29.051605.112839. [DOI] [PubMed] [Google Scholar]
- 17.Kosik K.S. The neuronal microRNA system. Nat. Rev. Neurosci. 2006;7:911–920. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- 18.Davis E., Caiment F., Tordoir X., Cavaille J., Ferguson-Smith A., Cockett N., Georges M., Charlier C. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr. Biol. 2005;15:743–749. doi: 10.1016/j.cub.2005.02.060. [DOI] [PubMed] [Google Scholar]
- 19.Doench J.G., Sharo P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh N., Ornitz D.M. Evolution of the FGF and FGFR gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 21.Ohmachi S., Mikami T., Konishi M., Miyake A., Itoh N. Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J. Neurosci. Res. 2003;72:436–443. doi: 10.1002/jnr.10592. [DOI] [PubMed] [Google Scholar]
- 22.Rideout H.J., Dietrich P., Savalle M., Dauer W.T., Stefanis L. Regulation of alpha-synuclein by bFGF in cultured ventral midbrain dopaminergic neurons. J. Neurochem. 2003;84:803–813. doi: 10.1046/j.1471-4159.2003.01574.x. [DOI] [PubMed] [Google Scholar]
- 23.Leng Y., Chase T.N., Bennett M.C. Muscarinic receptor stimulation induces translocation of an alpha-synuclein oligomer from plasma membrane to a light vesicle fraction in cytoplasm. J. Biol. Chem. 2001;276:28212–28218. doi: 10.1074/jbc.M011121200. [DOI] [PubMed] [Google Scholar]
- 24.Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 25.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 26.Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R. α-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 27.Chartier-Harlin M.C., Kachergus J., Roumier C., Mouroux V., Douay X., Lincoln S., Levecque C., Larvor L., Andrieux J., Hulihan M. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 28.Ibanez P., Bonnet A.M., Debarges B., Lohmann E., Tison F., Pollak P., Agid Y., Durr A., Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 29.Kirik D., Annett L.E., Burger C., Muzyczka N., Mandel R.J., Bjorklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: A new primate model of Parkinson's disease. Proc. Natl. Acad. Sci. USA. 2002;100:2884–2889. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirik D., Rosenblad C., Burger C., Lundberg C., Johansen T.E., Muzyczka N., Mandel R.J., Bjorklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider B.L., Seehus C.R., Capowski E.E., Aebischer P., Zhang S.C., Svendsen C.N. Over-expression of alpha-synuclein in human neural progenitors leads to specific changes in fate and differentiation. Hum. Mol. Genet. 2007;16:651–666. doi: 10.1093/hmg/ddm008. [DOI] [PubMed] [Google Scholar]
- 32.Zhou W., Schaack J., Zawada W.M., Freed C.R. Overexpression of human alpha-synuclein causes dopamine neuron death in primary human mesencephalic culture. Brain Res. 2002;926:42–50. doi: 10.1016/s0006-8993(01)03292-9. [DOI] [PubMed] [Google Scholar]
- 33.Clop A., Marcq F., Takeda H., Pirottin D., Tordoir X., Bibe B., Bouix J., Caiment F., Elsen J.M., Eychenne F. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]