

Abstract
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in Americans and is the second leading cause of cancer mortality. Only a minority (∼5%) of familial CRC can be explained by known genetic variants. To identify susceptibility genes for familial colorectal neoplasia, the colon neoplasia sibling study conducted a comprehensive, genome-wide linkage scan of 194 kindreds. Clinical information (histopathology, size and number of polyps, and other primary cancers) was used in conjunction with age at onset and family history for classification of the families into five phenotypic subgroups (severe histopathology, oligopolyposis, young, colon/breast, and multiple cancer) prior to analysis. By expanding the traditional affected-sib-pair design to include unaffected and discordant sib pairs, analytical power and robustness to type I error were increased. Sib-pair linkage statistics and Haseman-Elston regression identified 19 linkage peaks, with interesting results for chromosomes 1p31.1, 15q14-q22, 17p13.3, and 21. At marker D1S1665 (1p31.1), there was strong evidence for linkage in the multiple-cancer subgroup (p = 0.00007). For chromosome 15q14-q22, a linkage peak was identified in the full-sample (p = 0.018), oligopolyposis (p = 0.003), and young (p = 0.0009) phenotypes. This region includes the HMPS/CRAC1 locus associated with hereditary mixed polyposis syndrome (HMPS) in families of Ashkenazi descent. We provide compelling evidence linking this region in families of European descent with oligopolyposis and/or young age at onset (≤51) phenotypes. We found linkage to BRCA2 in the colon/breast phenotypic subgroup and identified a second locus in the region of D21S1437 segregating with, but distinct from, BRCA2. Linkage to 17p13.3 at marker D17S1308 in the breast/colon subgroup identified HIC1 as a candidate gene. We demonstrated that using clinical information, unaffected siblings, and family history can increase the analytical power of a linkage study.
Introduction
Of the 145,290 CRC cancer cases diagnosed every year in the United States, 20%–25% have a family history of colon cancer.1 Studies have shown that the risk for CRC in first-degree relatives (FDR) of patients with either CRC or adenomatous polyps is 2- to 4-fold greater than that for the general population.2 Studies with twins suggest that up to 35% of CRC is genetic,3 whereas only a small minority (2%–6%) of familial CRC cases can be explained by known genetic variants.4,5 To identify susceptibility genes for familial colorectal neoplasia, we located and recruited into our colon neoplasia sibling study (CNSS) kindreds demonstrating familial clustering of colon cancers and colon adenomas and polyps.
Colon neoplasia is a heterogeneous disease both in its genetic (allelic and locus) origins and in phenotypic presentation and thus offers significant challenges both in designing and in analyzing a linkage study. Frequently, all families with affected individuals (CRC or colon neoplasia) are analyzed together, the rationale being that increasing sample size also increases the analytical power. This is correct and appropriate in the absence of genetic heterogeneity. However, in the presence of such heterogeneity, pooling families with diverse phenotypic expression of disease may actually serve to increase genetic heterogeneity, with a resulting loss in analytical power. Therefore, we hypothesized that clinical information and family history could be used to identify families with both similar phenotypes and genetic homogeneity, hence increasing analytical power. We stratified the complete set of 194 families, which had fulfilled our criteria for inclusion in this study, into five phenotypic subgroups (severe histopathology, oligopolyposis, young, colon/breast, and multiple cancer) prior to performing any statistical analysis. Classification of phenotypic subgroups was not exclusive, and families were classified into more than one phenotypic subgroup.
In addition to the classification and implementation of innovative phenotypes, we expanded upon the traditional affected-sib-pair design by incorporating both concordant and discordant sib pairs into the analysis. The CNNS study recruits all siblings (affected and unaffected) as well as parents. Traditionally, in affected-sib-pair designs, information from unaffected siblings is used only to help determine identity by descent (IBD) allele sharing when the parental marker genotypes are incompletely known; thus, phenotypic information that could be used to increase the robustness and analytical power of the study is discarded.6 An analysis that is based only on affected sib pairs can result in incorrect inferences about linkage between a marker and a disease trait because such analysis does not include an appropriate control group of discordant sibs.6 Regions of the genome may demonstrate excess allele sharing among all types of sibling pairs, and these observations are better explained by transmission distortion and relative fitness.6–8 Matched-sibling controls provide protection against these problems, making the test statistics more robust to type I error.6 Our objective was to use not only the information from unaffected individuals but also specific phenotype definitions to lessen genetic heterogeneity, thereby increasing analytical power in our search for CRC susceptibility genes.
Severe histopathology was our primary analysis phenotype used in our previous study identifying chromosome 9q22.2-31.2 as harboring a susceptibility gene for familial colon neoplasia.9 Evidence for linkage to this region has been confirmed in two other studies of familial colorectal cancer and is designated as Colorectal Cancer Susceptibility 1 (CRCS1; MIM 608812).10,11 Here, we report complete results for all the phenotypic subgroups and, in particular, strong evidence for linkage to chromosome 1 for kindreds with multiple cancers.
Subjects and Methods
Ascertainment and Collection of Familial Colorectal Neoplasia Kindreds
Kindreds were enrolled in the CNSS, after review and approval of the study design and of all informed-consent documents by the Institutional Review Board of University Hospitals of Cleveland. Initial ascertainment was nationwide as previously described9 and includes kindreds of European American (91%), Jewish American (7%), and African American (2%) descent: We enrolled families (1) if at least one sibling was affected with colorectal cancer, or had adenomatous polyps, at or before the age of 65 and (2) if there was a second living sibling who was willing to participate. From that sample, we selected for inclusion in this study 200 kindreds that met the following criteria: (1) the presence of an index case and a full sibling both of whom had been diagnosed with colorectal cancer, or with colon adenomatous polyps, by age 65 years; (2) histological verification of colonic adenomatous polyps or colorectal cancer; (3) no histological evidence for inflammatory-bowel disease; (4) no evidence of known hereditary forms of colorectal cancer (e.g., familial adenomatous polyposis (FAP [MIM 175100]) and hereditary nonpolyposis colorectal cancer (HNPCC [MIM 124035, 124036, 60078, 609309, 600259, 609310, and 609395])), which were excluded from this analysis by a combination of pedigree review and molecular testing, as previously described;9 and (5) donation of a blood sample for genetic analysis from two or more affected siblings. In addition, we requested blood samples from all parents and siblings who were available and willing to participate in the study.
Individuals of any age, who had undergone an endoscopic colon examination with no finding of either colon cancer or adenomas, were classified as unaffected—this criterion was used for determining unaffected status for all phenotypic subgroups unless otherwise indicated. We note that having a negative colon endoscopy places an individual at low risk of developing colon cancer or advanced colon adenomas for 5 to 10 years subsequent to the negative examination.12–14
Families were checked for pedigree and genotyping errors with the RELTEST and MARKERINFO programs in the S.A.G.E.15 package.9,15 As a result, six families were eliminated from the analysis for incorrectly specified relationships: In four cases, the affected pairs were most probably half siblings, and in two families, the affected pairs were monozygotic (MZ) twins. Ultimately, there were 194 families available (complete set) for analysis.
Phenotypic Subgroups
In recognition of the heterogeneous nature of colon neoplasia, we utilized clinical information to stratify the complete set of 194 kindreds into five clinical subgroups (severe histopathology, oligopolyposis, young, colon/breast, and multiple cancer) prior to performing any statistical analysis. Detailed definitions of these phenotypes are given in Table 1. Because of the Ashkenazi-specific I1307K variant in the APC gene,16 all kindreds with Ashkenazi Jewish heritage were tested and excluded from the analysis if the results were positive.9 However, it is speculated that there may be further Ashkenazi-specific mutations17,18 in the remaining families of Jewish descent in the complete (n = 13), young (n = 4), and oligopolyposis subgroups (n = 3); sensitivity analyses were conducted as appropriate.
Table 1.
Definition of Phenotypic Subgroups Used for the Colon Neoplasia Sibling Study
| Subgroup | Number of Kindreds | Agea | Selection Criteria | Tumor/Polyp Size | Definition of Individuals Classified as |
||
|---|---|---|---|---|---|---|---|
| Affected | Unaffected | Unknown | |||||
| Complete set | 194 | ≤65 | Two siblings with neoplasia at age ≤65 | All | Definition 1: Individuals with histologically verified colon neoplasiab at age ≤65 | Definition 2: Individuals with negative colonoscopy or flexible sigmoidoscopy | Definition 3: Individuals without colon screening or a history of colon cancer, and/or adenomatous polyps at age >65 |
| Severe histopathology | 53 | ≤65 | Two siblings with cancer, advanced adenomas | ≥1 cm | Individuals with colon cancer or advanced adenomas (≥1 cm) at age ≤65 | Definition 2 | Definition 3 and small adenomas <1 cm in size |
| Oligopolyposis (multiple polyps) | 32 | ≤65 | Two siblings with ≥4 polyps | All | Definition 1 | Definition 2 | Definition 3 |
| Young | 74 | ≤51 | Two siblings with neoplasia at age ≤51 | All | Definition 1 | Definition 2 | Definition 3 |
| Colon/breast | 33 | ≤65 | First degree relative with breast cancer and two affected siblings with colon neoplasia | All | Definition 1 or a history of breast cancer | Definition 2 and no history of breast cancer | Definition 3 and no history of breast cancer |
| Multiple cancer | 62 | ≤65 | One individual in the sibship with more than one primary cancer site | All | Definition 1 or a history of cancer of any primary site, including melanomas but excluding other skin cancers | Definition 2 and no history of other cancers | Definition 3 and no history of cancer at any other primary site |
Age is recorded as the age at diagnosis with cancer, colon adenomatous polyps, or colorectal cancer.
Colon neoplasia is defined as all individuals with adenomatous polyps of any size and/or colorectal cancer. Note that we do not consider individuals with hyperplastic polyps to be affected for any of our analyses, and our polyp/histology does not differentiate between adenomas and serrated adenomas.
Complete Analysis
The complete data set is the union of all of the phenotypic subgroups and contains 194 kindreds diverse in histopathology, family history, and age at onset. This analysis has the greatest power to detect CRC and colon neoplasia susceptibility genes present in all phenotypes.
Severe Histopathology
This subgroup was defined for capturing the variability present within the pathology of adenomatous polyps. Individuals with polyps that are large in size or that display high-grade dysplasia are more likely to develop cancer.19,20 We identified 53 kindreds with severe histopathology that was defined as CRC, high-grade dysplasia (HGD), or adenomatous polyps (≥1 cm in size) and that was diagnosed before the age of 65 for all phenotypes (CRC, HGD, and adenomatous polyps). Because we have published results for this subgroup,9 only summary information is presented here for full disclosure of the tests that were performed on the data and for ease of reference.
Oligopolyposis
This subgroup defined 32 families with multiple colorectal adenomas (Table 1). Families were identified because at least one individual in the family had greater than or equal to four adenomatous polyps. It is well recognized that families with multiple polyps are representative of a separate and distinct phenotypic subgroup of colorectal neoplasia, which has a familial inheritance pattern,5,21 although there is no consensus agreement upon the phenotypic definition of these families, and they have been rather arbitrarily defined in the literature as patients with between 5 and 100 adenomas.21
Young
One of the hallmarks of inherited predisposition to cancer is an early age at onset. Our young phenotype defined 74 kindreds with a family mean age at diagnosis ≤51 years of age (Table 1). We chose 51 as a cutoff point because (1) the mean age at diagnosis in our sample was 51.4 years; (2) the impact of having a family history of colon cancer is greater among those diagnosed before the age of 50;22 and (3) the population prevalence of adenomatous polyps in individuals between the ages of 40 and 49 is 1.2%.23 Individuals affected at age ≤51 are in the tail of the population distribution and are more likely to have “hereditary colon neoplasia.”
Colon/Breast
This phenotypic subgroup examines the strong clustering of breast and colon cancer noted in population-based studies.24,25 Cancer geneticists have speculated that there are likely to be “breast-colon” susceptibility genes.26 Families segregating Breast Cancer 1 (BRCA1 [MIM 113705]), Breast Cancer 2 (BRCA2 [MIM 600185]), and Checkpoint Kinase 2 (CHEK2 [MIM 604373]) mutations have an increased risk for colon cancer.27–32 However, these relatively rare genes cannot explain the strong clustering of breast and colon cancer.24 We identified 33 families with a case of breast cancer in a FDR and 172 families in which there was an individual affected with breast cancer in either a FDR or second-degree relative. Our analysis included only families with a FDR affected with breast cancer because this allowed us to compare the allele sharing between breast and colon neoplasia cases (Table 1). Families in this subgroup had at least two sibs who were affected with adenomatous polyps or colon cancer and a sib or mother with breast cancer.
Multiple Cancer
We identified 62 families in which at least one individual in the nuclear family had been diagnosed with cancer at two or more primary sites (Table 1). Individuals affected with both colon cancer and colon polyps, but who had no other primary-site cancers, were not considered to be affected with multiple primary-site cancers. Melanomas were considered to be a primary site, but common skin cancers such as basal and squamous cell carcinomas were excluded. The rationale behind this subset was to identify families with a general propensity for developing cancer because one of the clinical hallmarks of hereditary cancer is an excess of multiple primary malignancies.33 An analysis25 of the Swedish family-cancer database determined that family history is a major risk factor for occurrence of multiple primary cancers of the colon (standardized incidence ratio [SIR] = 59.1), breast (SIR = 7.9), and skin (SIR = 7.9).
Statistical Methods
We used genotypes from all available siblings and parents to estimate the proportion of alleles shared IBD for each sib pair, denoted by the symbol , at 2 cM intervals in a multipoint analysis by using GENIBD in the program package S.A.G.E.9,15 Markers were polymorphic dinucleotide repeat markers generated by the Center for Inherited Disease Research (CIDR); genotyping details9 are published and are also available from the authors. The mean tests are the foundation of the traditional affected-only analyses, which uses the allele sharing between concordantly affected sib pairs, to determine the evidence for linkage. In our analyses, conducted with mean tests and the original Haseman-Elston (H-E) regression method, we used the information available from all sib pairs, with each sib being scored x = 1 if affected or x = 0 if unaffected, as implemented in the S.A.G.E. program SIBPAL. Although it has been shown that weighted H-E is more powerful asymptotically,34 weights estimated from the data may demonstrate numerical instability with smaller size samples. Because some of our subgroups have a small number of concordantly affected sib pairs (<20), we used the original H-E regression equation that regresses the squared sib-pair-trait difference y (0, for concordant pairs; 1, for discordant pairs) on , in the form of y = α+β. The degree to which allele sharing is associated with concordance or discordance in affection status determines the significance of the regression coefficient β. Reported p values were confirmed by comparison to a Monte Carlo sample of the permutation distribution created by permutation of the allele-sharing values relative to the pair labels (concordant or discordant). Permutations were done both within sibships and across sibships of the same size, sufficient in number for assuring with 95% confidence that the estimated p value was within 5% of the true p value. The number of permutations ranged from 0 to 1 million (chromosome 1, multiple-cancer subgroup). We used matrix spectral decomposition (matSpD), a variant of SNPSpD,35 to estimate the number of independent phenotypes being analyzed (n = 4.23). This was applied as a Sidak correction,36,37 which is less conservative than the Bonferroni correction, to the p value obtained from the permutation distribution. Throughout this work, this corrected p value is denoted as cP; otherwise all reported p values were uncorrected for multiple testing. All p values were estimated from the permutation distribution.
Association testing was performed on peak marker locations with ASSOC in the S.A.G.E. programming package. We tested all alleles at the microsatellites closest to the peak locations listed in Table 2 for association with the corresponding phenotype by using ASSOC, which can test for association by using a likelihood ratio test under a logistic model allowing for the presence of familial correlations. We found no evidence for association (minimum p = 0.07).
Table 2.
Chromosomal Regions Suggestive of Linkage by the Haseman-Elston Regression Analysis
| Group | Chromosome (cM) | Concordantly Affected Sib Pairs |
Discordantly Affected Sib Pairs |
Concordantly Unaffected Sib Pairs |
H-E p Value | H-E Corrected (cP) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | ||||
| Complete | ||||||||||||
| 1 (171 cM D1S1679) | 0.5291 | 0.0169 | 0.0433 | 0.4798 | 0.0171 | 0.1215 | 0.4949 | 0.0411 | 0.5486 | 0.0304 | 0.1286 | |
| 3 (206 cM) | 0.5227 | 0.0140 | 0.0530 | 0.4846 | 0.0128 | 0.1155 | 0.4998 | 0.0328 | 0.5015 | 0.0311 | 0.1316 | |
| 4 (13 cM at D4S2366)a | 0.5416 | 0.0155 | 0.0038 | 0.4905 | 0.0147 | 0.2607 | 0.5324 | 0.0341 | 0.1727 | 0.0080 | 0.0338 | |
| 4 (93 cM D4S2361) | 0.5399 | 0.0167 | 0.0088 | 0.4832 | 0.0167 | 0.1468 | 0.4785 | 0.0380 | 0.7125 | 0.0187 | 0.0791 | |
| 5 (96 cM) | 0.5064 | 0.0166 | 0.3510 | 0.4847 | 0.0165 | 0.1759 | 0.5470 | 0.0365 | 0.1009 | 0.0431 | 0.1823 | |
| 7 (163 cM D7S3070) | 0.5267 | 0.0174 | 0.0624 | 0.4565 | 0.0171 | 0.0056 | 0.5361 | 0.0408 | 0.1891 | 0.0011 | 0.0047 | |
| 10 (114 cM) | 0.5104 | 0.0179 | 0.2808 | 0.5021 | 0.0170 | 0.5497 | 0.5915 | 0.0397 | 0.0122 | 0.0302 | 0.1277 | |
| 12 (58 cM) | 0.5228 | 0.0167 | 0.0807 | 0.4713 | 0.0157 | 0.0351 | 0.5520 | 0.0395 | 0.0960 | 0.0077 | 0.0326 | |
| 15 (42 cM) | 0.5318 | 0.0154 | 0.0197 | 0.4884 | 0.0153 | 0.2275 | 0.5173 | 0.0336 | 0.3039 | 0.0205 | 0.0867 | |
| 16 (39 cM) | 0.5268 | 0.0159 | 0.0469 | 0.4707 | 0.0150 | 0.0262 | 0.4867 | 0.0358 | 0.6436 | 0.0098 | 0.0415 | |
| 16 (111 cM D16S3091) | 0.5443 | 0.0162 | 0.0033 | 0.4778 | 0.0158 | 0.0911 | 0.4696 | 0.0363 | 0.7960 | 0.0075 | 0.0317 | |
| 17 (78 cM) | 0.5222 | 0.0152 | 0.0726 | 0.4769 | 0.0154 | 0.0681 | 0.4701 | 0.0340 | 0.8079 | 0.0489 | 0.2068 | |
| 19 (40 cM) | 0.5123 | 0.0159 | 0.2197 | 0.4809 | 0.0150 | 0.1044 | 0.6021 | 0.0356 | 0.0028 | 0.0155 | 0.0656 | |
| Oligo | ||||||||||||
| 1 (259 cM) | 0.5189 | 0.0328 | 0.2828 | 0.4247 | 0.0279 | 0.0044 | 0.6198 | 0.0763 | 0.0687 | 0.0078 | 0.0330 | |
| 2 (155 cM D2S1399) | 0.5313 | 0.0370 | 0.1997 | 0.4574 | 0.0426 | 0.1609 | 0.6937 | 0.0892 | 0.0231 | 0.0160 | 0.0677 | |
| 3 (90 cM D3S4542) | 0.5695 | 0.0374 | 0.0336 | 0.4386 | 0.0375 | 0.0533 | 0.4752 | 0.0687 | 0.6380 | 0.0163 | 0.0689 | |
| 3 (216 cM D3S2418) | 0.5714 | 0.0346 | 0.0213 | 0.4111 | 0.0331 | 0.0045 | 0.4255 | 0.0601 | 0.8825 | 0.0040 | 0.0169 | |
| 4 (158 cM D4S1629) | 0.5188 | 0.0328 | 0.2836 | 0.4357 | 0.0388 | 0.0513 | 0.6392 | 0.0754 | 0.0423 | 0.0218 | 0.0922 | |
| 10 (59 cM D10S1426) | 0.4852 | 0.0336 | 0.6688 | 0.4211 | 0.0374 | 0.0193 | 0.5536 | 0.0907 | 0.2818 | 0.0475 | 0.2009 | |
| 12 (62 cM) | 0.5689 | 0.0348 | 0.0473 | 0.4674 | 0.0327 | 0.1616 | 0.5546 | 0.0693 | 0.2214 | 0.0301 | 0.1273 | |
| 12 (81 cM D12S375) | 0.5400 | 0.0414 | 0.1685 | 0.4514 | 0.0425 | 0.1287 | 0.5877 | 0.0680 | 0.1085 | 0.0345 | 0.1459 | |
| 14 (116 cM) | 0.5412 | 0.0359 | 0.1280 | 0.4094 | 0.0313 | 0.0025 | 0.5463 | 0.0601 | 0.2266 | 0.0018 | 0.0076 | |
| 15 (28 cM) | 0.5244 | 0.0299 | 0.2091 | 0.4610 | 0.0287 | 0.0901 | 0.6414 | 0.0640 | 0.0216 | 0.0077 | 0.0326 | |
| 16 (93 cM) | 0.5827 | 0.0295 | 0.0028 | 0.4415 | 0.0325 | 0.0382 | 0.4967 | 0.0785 | 0.5163 | 0.0026 | 0.0110 | |
| Young | ||||||||||||
| 4 (99 cM) | 0.5499 | 0.0230 | 0.0159 | 0.4863 | 0.0207 | 0.2560 | 0.5486 | 0.0512 | 0.1756 | 0.0163 | 0.0689 | |
| 5 (96 cM) | 0.5188 | 0.0252 | 0.2289 | 0.4638 | 0.0233 | 0.0607 | 0.6232 | 0.0673 | 0.0396 | 0.0183 | 0.0774 | |
| 5 (178 cM) | 0.5102 | 0.0249 | 0.3411 | 0.4568 | 0.0208 | 0.0194 | 0.5758 | 0.0526 | 0.0809 | 0.0211 | 0.0893 | |
| 7 (162 cM at D7S3070) | 0.5116 | 0.0253 | 0.3233 | 0.4500 | 0.0248 | 0.0228 | 0.5938 | 0.0660 | 0.0840 | 0.0167 | 0.0706 | |
| 8 (139 cM) | 0.5292 | 0.0271 | 0.1409 | 0.4817 | 0.0235 | 0.2195 | 0.6077 | 0.0644 | 0.0535 | 0.0444 | 0.1878 | |
| 11 (22 cM) | 0.5142 | 0.0253 | 0.2875 | 0.4628 | 0.0225 | 0.0508 | 0.6379 | 0.0715 | 0.0326 | 0.0190 | 0.0804 | |
| 12 (56 cM c12S1916) | 0.5152 | 0.2638 | 0.2817 | 0.4553 | 0.0250 | 0.0380 | 0.5588 | 0.0690 | 0.2007 | 0.0302 | 0.1277 | |
| 12 (72 cM) | 0.5045 | 0.0231 | 0.4216 | 0.4674 | 0.0210 | 0.0620 | 0.5940 | 0.0529 | 0.0439 | 0.0488 | 0.2064 | |
| 12 (88 cM) | 0.5238 | 0.0240 | 0.1617 | 0.4683 | 0.0208 | 0.0650 | 0.6148 | 0.0646 | 0.0438 | 0.0135 | 0.0571 | |
| 15 (20 cM at D15S165) | 0.5412 | 0.0168 | 0.0077 | 0.4722 | 0.0161 | 0.0439 | 0.5498 | 0.0409 | 0.1173 | 0.0009 | 0.0038 | |
| 16 (35 cM) | 0.5117 | 0.0232 | 0.3068 | 0.4634 | 0.0221 | 0.0498 | 0.5482 | 0.5074 | 0.2045 | 0.0418 | 0.1768 | |
| 19 (38 cM) | 0.5150 | 0.0241 | 0.2667 | 0.4704 | 0.0217 | 0.0876 | 0.6292 | 0.0517 | 0.0097 | 0.0254 | 0.1074 | |
| 20 (2 cM at D20S103) | 0.5137 | 0.0244 | 0.2874 | 0.4618 | 0.0211 | 0.0366 | 0.5469 | 0.0711 | 0.2574 | 0.0364 | 0.1540 | |
| 21 (13 cM at D21S1437) | 0.5542 | 0.0248 | 0.0152 | 0.4781 | 0.0237 | 0.1792 | 0.4726 | 0.0657 | 0.6596 | 0.0264 | 0.1117 | |
| Colon/Breast | ||||||||||||
| 1 (141 cM) | 0.5405 | 0.0390 | 0.1516 | 0.4181 | 0.0366 | 0.0142 | 0.5718 | 0.0927 | 0.2244 | 0.0067 | 0.0283 | |
| 2 (222 cM) | 0.6011 | 0.0327 | 0.0014 | 0.4874 | 0.0271 | 0.3219 | 0.4793 | 0.0508 | 0.6554 | 0.0150 | 0.0635 | |
| 8 (78 cM) | 0.5547 | 0.0397 | 0.0868 | 0.4543 | 0.0343 | 0.0937 | 0.5005 | 0.0710 | 0.4968 | 0.0354 | 0.1497 | |
| 10 (168 cM) | 0.5590 | 0.0325 | 0.0372 | 0.4560 | 0.0360 | 0.0775 | 0.4254 | 0.0591 | 0.8865 | 0.0366 | 0.1548 | |
| 13 (26 cM at D13S1493) | 0.5742 | 0.0398 | 0.0337 | 0.4307 | 0.0383 | 0.0388 | 0.5194 | 0.0727 | 0.3960 | 0.0070 | 0.0296 | |
| 17 (1 cM at D17s1308) | 0.5501 | 0.0340 | 0.0732 | 0.4567 | 0.0291 | 0.0710 | 0.5597 | 0.0671 | 0.1928 | 0.0086 | 0.0364 | |
| 18 (53 cM at D18s877) | 0.5279 | 0.0369 | 0.2259 | 0.4473 | 0.0360 | 0.0738 | 0.5825 | 0.0618 | 0.0991 | 0.0252 | 0.1066 | |
| 21 (12 cM at D21s1437) | 0.5860 | 0.0362 | 0.0102 | 0.4268 | 0.0274 | 0.0046 | 0.5258 | 0.0704 | 0.3589 | 0.0003 | 0.0013 | |
| 21 (59 cM at D21S446) | 0.5126 | 0.0328 | 0.3507 | 0.4232 | 0.0316 | 0.0087 | 0.5835 | 0.0500 | 0.0562 | 0.0086 | 0.0364 | |
| Multiple | ||||||||||||
| 1 (102 cM at D1S1665) | 0.5718 | 0.0250 | 0.0024 | 0.4082 | 0.0282 | 0.0007 | 0.4344 | 0.0688 | 0.8222 | 0.00007 | 0.00029 | |
| 1 (175 cM) | 0.5476 | 0.0258 | 0.0033 | 0.4422 | 0.0303 | 0.0295 | 0.4100 | 0.0921 | 0.8281 | 0.0170 | 0.0719 | |
| 5 (82 cM at D5S424) | 0.5322 | 0.0243 | 0.0938 | 0.4488 | 0.0281 | 0.0353 | 0.5812 | 0.0637 | 0.1103 | 0.0068 | 0.0288 | |
| 7 (163 cM at D7S3070) | 0.5050 | 0.0270 | 0.4208 | 0.4366 | 0.0300 | 0.0185 | 0.5393 | 0.0994 | 0.3487 | 0.0342 | 0.1447 | |
| 9 (84 cM) | 0.5336 | 0.0245 | 0.0859 | 0.4484 | 0.0275 | 0.0320 | 0.4466 | 0.0738 | 0.7587 | 0.0064 | 0.0271 | |
| 9 (110 cM) | 0.5224 | 0.0273 | 0.2070 | 0.4484 | 0.0258 | 0.0241 | 0.4322 | 0.0976 | 0.7511 | 0.0191 | 0.0808 | |
| 16 (105 cM) | 0.5227 | 0.0232 | 0.1649 | 0.4440 | 0.0246 | 0.0125 | 0.4526 | 0.0797 | 0.7197 | 0.0182 | 0.0770 | |
Entries in gray-shaded rows indicate where the H-E regression coefficient remained statistically significant after correction for multiple hypothesis testing.
Results
Here, we present the detailed results of the analyses of the complete set of 194 kindreds and of four phenotypic subgroups (oligopolyposis, young, colon/breast, and multiple cancer), along with summary results from our primary severe histopathology phenotype published previously.9 Of particular interest were regions for which both the H-E regression and the mean tests indicated “significant” linkage, defined as regions with a p value ≤ 0.016. This corresponded to a LOD > 1.0, reflecting a level of significance appropriate to control type II error. We report the most significant H-E regression p value for the region, along with the allele-sharing estimates, so that the reader can weigh the evidence resulting from both measures. Results from the H-E regression analyses have been plotted for all chromosomes and phenotypes (Figures 1 and 2).
Figure 1.
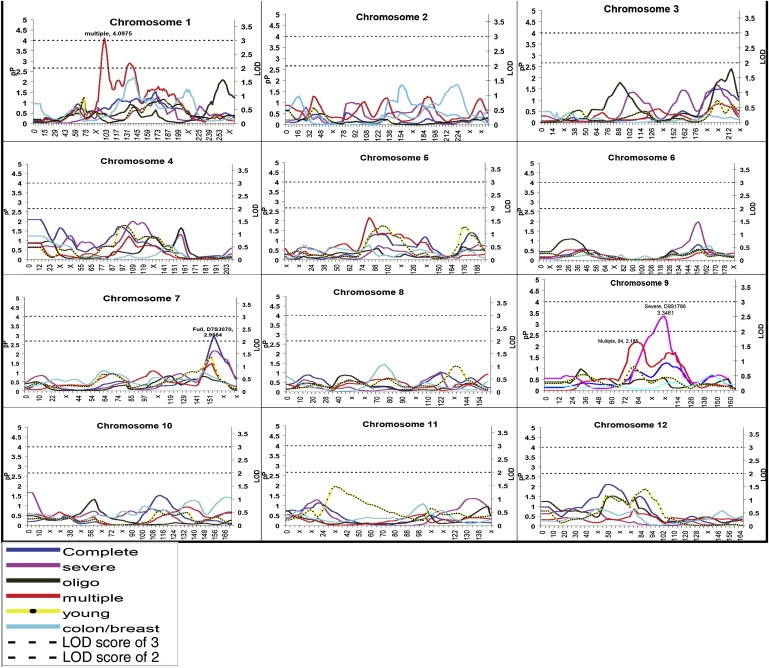
Plots for Chromosomes 1–12 for All Subgroups
Distance is plotted along the x axis and both −log10 (p value) denoted as pP and the LOD are plotted on the y axis for reference purposes. Reference lines at a LOD of 2 and 3 are provided, and significant data points are labeled with the phenotype location and pP value.
Figure 2.
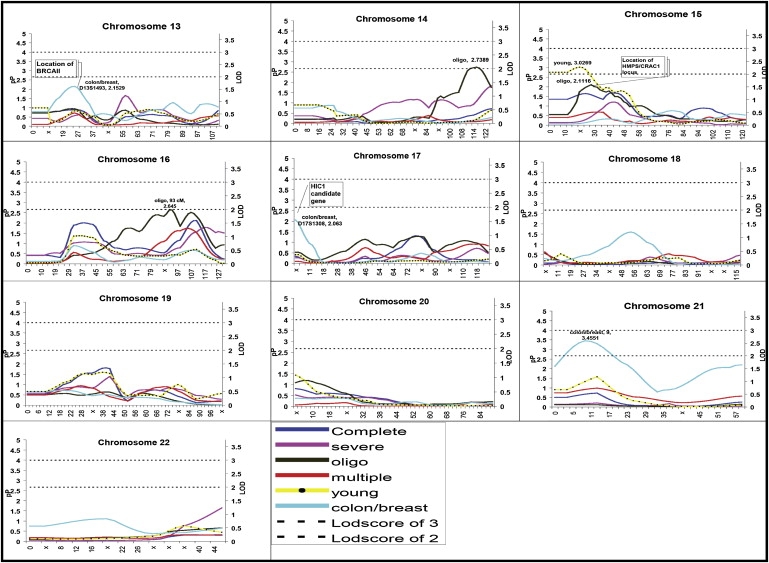
Plots for Chromosomes 13–22 for All Subgroups
Distance is plotted along the x axis and both −log10 (p value) denoted as pP and the LOD are plotted on the y axis for reference purposes. Reference lines at a LOD of 2 and 3 are provided, and significant data points are labeled with the phenotype location and pP value.
Complete Analysis
After permutation testing and correction for multiple hypothesis testing, significant evidence for linkage for the entire set of 194 kindreds was demonstrated for five regions on chromosomes 4, 7, 12, and 16 (Tables 2 and 3). The most significant signal was located at marker D7S3070 (163 cM; p = 0.0011) on chromosome 7, flanked by markers D7S2195 (155 cM) and D7S3058 (175 cM). This peak was also observed in the severe histopathology subgroup (Figure 1); it has not been previously identified with CRC or colon polyps. At D4S2366 (13 cM; p = 0.0080), the first marker on the chromosome demonstrated excess allele sharing among concordantly affected sib pairs ( = 05416; p = 0.0038). For chromosome 16, the peak at marker D16S3094 (111 cM) was flanked by markers D16S3096 (99 cM) and D16S539 (125 cM) and was observed in the oligopolyposis, severe-histopathology, and multiple-cancer subgroups.
Table 3.
Chromosomal Regions Suggestive of Linkage by the Mean Tests
| Group | Chromosome (cM) | Concordantly Affected Sib Pairs |
Discordantly Affected Sib Pairs |
Concordantly Unaffected Sib Pairs |
H-Ea p Value | Corrected H-E p Value (cP) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | ||||
| Full | ||||||||||||
| 1 (171 cM D1S1679) | 0.5291 | 0.0169 | 0.0433 | 0.4798 | 0.0171 | 0.1215 | 0.4949 | 0.0411 | 0.5486 | 0.0304 | 0.1286 | |
| 2 (94 cM D2S1394) | 0.5332 | 0.0174 | 0.0286 | 0.5240 | 0.0153 | 0.9406 | 0.4830 | 0.0379 | 0.6716 | 0.4653 | 1.0000 | |
| 4 (13 cM at D4S2366)b | 0.5416 | 0.0155 | 0.0038 | 0.4905 | 0.0147 | 0.2607 | 0.5324 | 0.0341 | 0.1727 | 0.0080 | 0.0338 | |
| 4 (99 cM) | 0.5391 | 0.0152 | 0.0053 | 0.4940 | 0.0144 | 0.3398 | 0.5094 | 0.0346 | 0.3925 | 0.0219 | 0.0926 | |
| 6 (152 cM) | 0.5323 | 0.0149 | 0.0156 | 0.5073 | 0.0139 | 0.7015 | 0.4925 | 0.0351 | 0.7835 | 0.2048 | 0.8663 | |
| 9 (95 cM D9S283) | 0.5306 | 0.0170 | 0.0362 | 0.5059 | 0.0165 | 0.6400 | 0.4731 | 0.0449 | 0.7404 | 0.2536 | 1.0000 | |
| 10 (128 cM) | 0.5277 | 0.0157 | 0.0396 | 0.5152 | 0.0156 | 0.8348 | 0.5426 | 0.0354 | 0.1167 | 0.2166 | 0.9162 | |
| 12 (94 cM) | 0.5393 | 0.0176 | 0.0133 | 0.4977 | 0.0159 | 0.4435 | 0.5097 | 0.0388 | 0.4016 | 0.2117 | 0.8955 | |
| 15 (43 cM D15S659)c | 0.5379 | 0.0169 | 0.0127 | 0.4884 | 0.0167 | 0.2450 | 0.5096 | 0.0368 | 0.3967 | 0.0213 | 0.0901 | |
| 16 (44 cM D16S403) | 0.5301 | 0.0172 | 0.0410 | 0.4700 | 0.0166 | 0.0365 | 0.4893 | 0.0405 | 0.6033 | 0.0166 | 0.0702 | |
| 16 (83 cM) | 0.5373 | 0.0139 | 0.0038 | 0.5078 | 0.0139 | 0.7134 | 0.4493 | 0.0302 | 0.9508 | 0.2540 | 1.0000 | |
| 18 (13 cM D18S976) | 0.5297 | 0.0162 | 0.0343 | 0.5294 | 0.0159 | 0.9672 | 0.4516 | 0.0339 | 0.9203 | 0.7583 | 1.0000 | |
| Oligo | ||||||||||||
| 3 (84 cM) | 0.5657 | 0.0351 | 0.0329 | 0.4482 | 0.0364 | 0.0823 | 0.4680 | 0.0674 | 0.6786 | 0.0326 | 0.1379 | |
| 3 (207 cM D3S2398) | 0.5812 | 0.0352 | 0.0120 | 0.4290 | 0.0381 | 0.0337 | 0.3690 | 0.0674 | 0.9644 | 0.0147 | 0.0622 | |
| 4 (13 cM) | 0.5751 | 0.0310 | 0.0090 | 0.5185 | 0.0358 | 0.6970 | 0.5595 | 0.0701 | 0.2046 | 0.1400 | 0.5922 | |
| 5 (54 cM) | 0.5697 | 0.0320 | 0.0164 | 0.5220 | 0.0361 | 0.7273 | 0.3937 | 0.0587 | 0.9549 | 0.4336 | 1.0000 | |
| 7 (108 cM D7S821) | 0.5782 | 0.0358 | 0.0161 | 0.5561 | 0.0396 | 0.9197 | 0.3516 | 0.0728 | 0.9700 | 0.7072 | 1.0000 | |
| 10 (4 cM D10S1435) | 0.5723 | 0.0336 | 0.0173 | 0.5447 | 0.0330 | 0.9100 | 0.5242 | 0.0849 | 0.3894 | 0.3277 | 1.0000 | |
| 11 (9 cM D11S2362) | 0.5744 | 0.0347 | 0.0181 | 0.5218 | 0.0377 | 0.7177 | 0.4623 | 0.0990 | 0.6456 | 0.3765 | 1.0000 | |
| 12 (64 cM) | 0.5586 | 0.0352 | 0.0502 | 0.4644 | 0.0328 | 0.1571 | 0.5538 | 0.0685 | 0.2224 | 0.0308 | 0.1303 | |
| 15 (43 cM D15S659) | 0.5707 | 0.0345 | 0.0219 | 0.4604 | 0.0402 | 0.1643 | 0.5831 | 0.0936 | 0.1941 | 0.0277 | 0.1172 | |
| 16 (64 cM D16S3396) | 0.5712 | 0.0366 | 0.0278 | 0.4347 | 0.0351 | 0.0339 | 0.4433 | 0.0753 | 0.7682 | 0.0185 | 0.0783 | |
| 16 (89 cM) | 0.5962 | 0.0299 | 0.0009 | 0.4510 | 0.0371 | 0.0961 | 0.4909 | 0.0807 | 0.5437 | 0.0043 | 0.0182 | |
| 17 (45 cM D17S2196) | 0.5982 | 0.0355 | 0.0036 | 0.4847 | 0.0414 | 0.3568 | 0.4201 | 0.0582 | 0.9046 | 0.0722 | 0.3054 | |
| Young | ||||||||||||
| 2 (40 cM) | 0.5416 | 0.0236 | 0.0261 | 0.5146 | 0.0232 | 0.7350 | 0.5230 | 0.0681 | 0.3692 | 0.1907 | 0.8067 | |
| 4 (113 cM) | 0.5604 | 0.0231 | 0.0050 | 0.5177 | 0.0229 | 0.7800 | 0.5733 | 0.0658 | 0.1378 | 0.0807 | 0.3414 | |
| 7 (71 cM D7S1818) | 0.5397 | 0.0237 | 0.0480 | 0.5139 | 0.0244 | 0.7153 | 0.6195 | 0.0684 | 0.0465 | 0.1330 | 0.5626 | |
| 10 (130 cM) | 0.5509 | 0.0245 | 0.0196 | 0.5137 | 0.0234 | 0.7212 | 0.4509 | 0.5490 | 0.8100 | 0.2392 | 1.0000 | |
| 15 (20 cM) | 0.5405 | 0.0162 | 0.0067 | 0.4745 | 0.0158 | 0.0555 | 0.5547 | 0.0394 | 0.0884 | 0.0009 | 0.0038 | |
| 16 (93 cM) | 0.5406 | 0.0220 | 0.0338 | 0.5092 | 0.0217 | 0.6642 | 0.3946 | 0.0611 | 0.9513 | 0.6311 | 1.0000 | |
| 16 (110 cM D16S3091) | 0.5506 | 0.0241 | 0.0185 | 0.4980 | 0.0228 | 0.4652 | 0.3738 | 0.0602 | 0.9767 | 0.1955 | 0.8270 | |
| 18 (12 cM at D18S976) | 0.5418 | 0.0237 | 0.0219 | 0.5224 | 0.0238 | 0.8267 | 0.4918 | 0.0544 | 0.5586 | 0.2883 | 1.0000 | |
| 21 (13 cM at D21S1437) | 0.5542 | 0.0248 | 0.0152 | 0.4781 | 0.0237 | 0.1792 | 0.4726 | 0.0657 | 0.6596 | 0.0264 | 0.1117 | |
| Colon/Breast | ||||||||||||
| 1 (177 cM) | 0.5901 | 0.0372 | 0.0092 | 0.5071 | 0.0365 | 0.5768 | 0.4215 | 0.0762 | 0.8303 | 0.1882 | 0.7961 | |
| 2 (224 cM) | 0.6000 | 0.0318 | 0.0012 | 0.4875 | 0.0252 | 0.3119 | 0.4680 | 0.0515 | 0.7285 | 0.0152 | 0.0643 | |
| 3 (6 cM at D3S2287) | 0.5883 | 0.0403 | 0.0161 | 0.5425 | 0.0372 | 0.8709 | 0.4912 | 0.0726 | 0.5472 | 0.3272 | 1.0000 | |
| 7 (86 cM at D7S2204) | 0.6007 | 0.0435 | 0.0119 | 0.4969 | 0.0388 | 0.4686 | 0.3553 | 0.0736 | 0.9674 | 0.1968 | 0.8325 | |
| 7 (119 cM) | 0.5933 | 0.0315 | 0.0021 | 0.5220 | 0.0353 | 0.7323 | 0.4495 | 0.0744 | 0.7468 | 0.2306 | 0.9754 | |
| 10 (82 cM) | 0.5885 | 0.0393 | 0.0138 | 0.5205 | 0.3993 | 0.6957 | 0.4735 | 0.0755 | 0.6346 | 0.2269 | 0.9598 | |
| 10 (170 cM) | 0.5618 | 0.0336 | 0.0355 | 0.4520 | 0.0328 | 0.0744 | 0.4129 | 0.0656 | 0.8976 | 0.0384 | 0.1624 | |
| 11 (58 cM at D11S1344) | 0.5716 | 0.0390 | 0.0356 | 0.5108 | 0.0327 | 0.6298 | 0.3571 | 0.0619 | 0.9833 | 0.4218 | 1.0000 | |
| 13 (26 cM at D13S1493) | 0.5742 | 0.0398 | 0.0337 | 0.4307 | 0.0383 | 0.0388 | 0.5194 | 0.0727 | 0.3960 | 0.0070 | 0.0296 | |
| 17 (82 cM at D17S1290) | 0.5865 | 0.0400 | 0.0175 | 0.4676 | 0.0383 | 0.2009 | 0.4346 | 0.0756 | 0.8000 | 0.3271 | 1.0000 | |
| 19 (16 cM) | 0.5572 | 0.0313 | 0.0360 | 0.5312 | 0.0340 | 0.8190 | 0.6035 | 0.0548 | 0.0392 | 0.1940 | 0.8206 | |
| 19 (72 cM) | 0.5666 | 0.0349 | 0.0304 | 0.5465 | 0.0348 | 0.9069 | 0.4108 | 0.0777 | 0.8657 | 0.5986 | 1.0000 | |
| 21 (12 cM at D21S1437) | 0.5860 | 0.0362 | 0.0102 | 0.4268 | 0.0274 | 0.0046 | 0.5258 | 0.0704 | 0.3589 | 0.0003 | 0.0013 | |
| Multiple | ||||||||||||
| 1 (102 cM at D1S1665) | 0.5718 | 0.0250 | 0.0024 | 0.4082 | 0.0282 | 0.0007 | 0.4344 | 0.0688 | 0.8222 | 0.00007 | 0.0003 | |
| 1 (176 cM at D1S1677) | 0.5507 | 0.0258 | 0.0257 | 0.4433 | 0.0303 | 0.0324 | 0.4010 | 0.0942 | 0.8452 | 0.0234 | 0.0990 | |
| 2 (38 cM at D2S1360) | 0.5749 | 0.0276 | 0.0037 | 0.4900 | 0.0330 | 0.3844 | 0.4461 | 0.1105 | 0.6837 | 0.0518 | 0.2191 | |
| 2 (258 cM) | 0.5520 | 0.0237 | 0.0151 | 0.5251 | 0.0274 | 0.8187 | 0.6361 | 0.0816 | 0.0574 | 0.1591 | 0.6730 | |
| 4 (109 cM) | 0.5434 | 0.0257 | 0.0467 | 0.4862 | 0.0261 | 0.3008 | 0.5359 | 0.0882 | 0.3447 | 0.1318 | 0.5575 | |
| 5 (40 cM at D5S2848) | 0.5521 | 0.0282 | 0.0335 | 0.5356 | 0.0324 | 0.8633 | 0.4901 | 0.1108 | 0.5350 | 0.4103 | 1.0000 | |
| 7 (108 cM) | 0.5636 | 0.0259 | 0.0077 | 0.5152 | 0.0288 | 0.7005 | 0.4501 | 0.0709 | 0.7540 | 0.1691 | 0.7153 | |
| 8 (82 cM) | 0.5488 | 0.0285 | 0.0449 | 0.5039 | 0.0289 | 0.5530 | 0.4784 | 0.0865 | 0.5968 | 0.1904 | 0.8054 | |
| 10 (135 cM at D10S1237) | 0.5484 | 0.0270 | 0.0380 | 0.5064 | 0.0279 | 0.5914 | 0.5627 | 0.0589 | 0.1495 | 0.1174 | 0.4966 | |
| 12 (12 cM) | 0.5421 | 0.0240 | 0.0412 | 0.5361 | 0.0274 | 0.9049 | 0.5453 | 0.0741 | 0.2798 | 0.4321 | 1.0000 | |
| 12 (92 cM) | 0.5564 | 0.0272 | 0.0205 | 0.5238 | 0.0258 | 0.8211 | 0.4660 | 0.0916 | 0.6420 | 0.2885 | 1.0000 | |
H-E: Haseman-Elston regression method.
Entries in gray-shaded rows indicate regions in which there was an allele-sharing pattern consistent with linkage and the H-E regression coefficient was significant after correction for multiple hypothesis testing.
Italicized entries indicate demonstrated linkage to the HMPS/CRAC1 locus.
Several of our significant linkage peaks (uncorrected) overlapped with previously identified candidate genes, including the hereditary mixed polyposis syndrome (HMPS/CRAC1 [MIM 601228]) locus on chromosome 15q15.3-q22.1 (H-E regression, p = 0.02; Table 2), and the region at 128 cM on chromosome 10 (Table 3), which overlapped both the BMPR1A locus, associated with HMPS38 and ANXA11, a putative low-penetrance gene for colorectal cancer.39
Severe Histopathology
The severe-histopathology phenotype was our primary analysis phenotype because we previously identified a chromosomal region on 9q22.2-31.2 as a putative susceptibility locus for colorectal neoplasia9 (CRCS1 [MIM 608812]). Linkage to this region has recently been confirmed in studies from Sweden and the UK.10,11 Results for this phenotype for the H-E regression are provided on the genome scan plots for ease of reference to our current findings and full-reporting purposes (Figure 1). We note that this is the original subgroup analyzed in our previous publication in which the full results for the mean tests and H-E regression can be found.9
Oligopolyposis or Multiple Polyps
After permutation testing and correction for multiple testing, there were five regions (chromosomes 1, 3, 14, 15, and 16) for which there was significant evidence for linkage (Tables 2 and 3). For chromosome 15 (Table 2), the identified linkage signal peaked at 28 cM (p = 0.008) between markers D15S165 (20 cM) and marker D15S1012 (36 cM). These markers overlap the HMPS/CRAC1 region associated with HMPS in families of Ashkenazi descent identified by Tomlinson and Jaeger,17,18 who reported a peak at marker D15S1007 (29.9 cM), flanked by markers D15S1031 (26 cM) and D15S118 (31.0 cM). The concordance between the two studies is illustrated in Figures 2 and 3. This region on chromosome 15q14 was identified in the complete set (p = 0.018), oligopolyposis (p = 0.003), and young (p = 0.0009) subgroups. We noted that the significance levels met and exceeded the thresholds required for significant replication of findings.40,41 Jaegar et al.17 reported a common haplotype that is shared in a large family of Ashkenazi descent and that is characterized by an uncommon and specific phenotype of multiple colorectal polyps (defined as greater than three polyps). To determine the effect of the families of Ashkenazi Jewish origin, we reanalyzed our linkage results after excluding the data from these families and found a subsequent increase in the linkage signal (i.e., decrease in the p value) was observed in all subgroups (Table 4 and Figure 3), indicating that in our sample, families of non-Jewish European ancestry were linked to this region. The p value was validated by 33,412 replicates in a Monte Carlo sample of the permutation distribution. To further refine the phenotype of families linking to this region, we identified and rank ordered the families linked to this region and examined the phenotype distributions in the top 12 families (see Table 5). We found three of the four highest ranked families met breast/colon criteria and two of these met young, breast/colon, and multiple polyp criteria.
Figure 3.
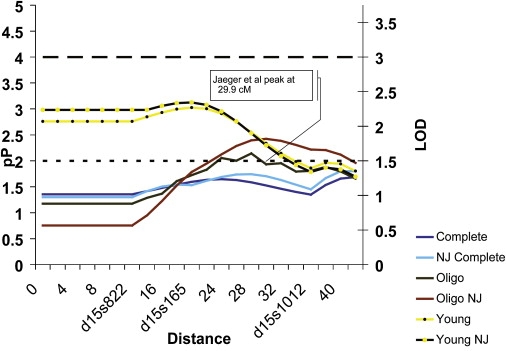
Overlap with HMPS/CRAC1 Locus
Distance is plotted along the x axis and both −log10 (p value) denoted as pP and the LOD are plotted on the y axis for reference purposes. Reference lines at a LOD of 2 and 3 are provided, and significant data points are labeled with the phenotype location and pP value. NJ indicates that families of Jewish ancestry have been removed from the analysis.
Table 4.
Haseman-Elston and Mean Tests Results for Chromosome 15 in the Complete Sample, Oligopolyposis, and Young Subgroups
| Sample | Distance (cM) | Concordantly Affected Sib Pairs |
Discordantly Affected Sib Pairs |
Concordantly Unaffected Sib Pairs |
H-E p Value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | |||
| Completea | (24 cM) | 0.5231 | 0.0113 | 0.0214 | 0.4911 | 0.0112 | 0.2152 | 0.5137 | 0.0263 | 0.3013 | 0.0225 |
| Complete—AJb | (28 cM) | 0.5312 | 0.0146 | 0.0166 | 0.4891 | 0.0138 | 0.2414 | 0.5133 | 0.0291 | 0.3248 | 0.0180 |
| Oligoc | (28 cM) | 0.5299 | 0.0279 | 0.1587 | 0.4614 | 0.0292 | 0.0990 | 0.6407 | 0.0661 | 0.0250 | 0.0091 |
| Oligo—AJd | (28 cM) | 0.5318 | 0.0357 | 0.1879 | 0.4558 | 0.0300 | 0.0928 | 0.6739 | 0.0741 | 0.0170 | 0.0037 |
| Younge | (20 cM at D15S165) | 0.5412 | 0.0168 | 0.0077 | 0.4722 | 0.0161 | 0.0439 | 0.5498 | 0.0409 | 0.1173 | 0.0009 |
| Young—AJf | (20 cM at D15S165) | 0.5435 | 0.0169 | 0.0055 | 0.4722 | 0.0163 | 0.0454 | 0.5498 | 0.0409 | 0.1173 | 0.0007 |
Complete set of families (n = 194).
Complete set without 13 families of Ashkenazi Jewish (AJ) descent (n = 181).
All families in oligopolyposis phenotypic subgroup (n = 32).
Oligopolyposis subgroup without three AJ families (n = 29).
All families in the Young subgroup (n = 74).
Young subgroup without four AJ families (n = 70).
Table 5.
Distribution of Phenotypes for the 13 Families Demonstrating Linkage to the HMPS/CRAC1 Locus
| Families with Both Affected and Unaffected Pairs (n) | Families with Multiple Polyps (>4 polyps) | Colon/Breast Families | Number of Individuals with CRC | Number of Adenomatous Polypsa | Average Number of Polyps per Affected Indivdiual | |
|---|---|---|---|---|---|---|
| Full Set | 12 of 13 | 8b | 3c | 8 | 80 | 3.2 |
Our polyp histology does not include hyperplastic polyps, and we cannot differentiate serrated adenomas.
Three of the four top-ranked families demonstrating linkage to HMPS/CRAC1 also meet criteria for breast/colon, and two of the families were classified as potential attenuated FAP when entering the study.
We note that two of these families also demonstrate linkage to BRCA2.
Additional regions of interest were located at ∼259 cM on chromosome 1 (H-E, p = 0.0078) and was flanked by markers D1S549 (240 cM) and D1S1609 (275 cM) (Table 2). This region includes EXO1, which maps to between 260 and 268 cM, depending on the reference genetic map (Marshfield or deCODE) used. EXO1, involved in DNA repair, is speculated to be involved in the development of CRC,39,42,43 and there is evidence to suggest that a recessive mode of inheritance for this locus.39 Because concordant sib pairs shared a greater proportion of two alleles IBD (0.39 for concordantly unaffected; 0.28 for concordantly affected) than the expected proportion (0.25), and because the proportion for discordant pairs was only 0.13 (p = 0.0001), these observations were consistent with linkage.44
For chromosome 3, the signal at marker D3S2418 (216 cM), flanked by markers D3S2427 (188 cM) and D3S1311 (226 cM), included the CRC candidate gene MFI2antigen,39 which is located at 224 cM. For chromosome 14, the signal at 116 cM (H-E, p = 0.0018) was flanked by markers c14S1937 (96 cM) and D14S1436 (126 cM). This region included the CRC candidate gene GOLGA5 (108 cM).39 For chromosome 16, the signal extended across a large region from D16S540 (58 cM) to D16S539 (125 cM) and peaked at ∼93 cM (H-E, p = 0.0026). Furthermore, there is no evidence for linkage to MutY-homolog (MYH [MIM 604933]), which is associated with a multiple colorectal adenoma phenotype, although the frequency is highest in patients with greater than ten polyps.45,46
Young
The most significant observation for both H-E regression and the mean tests and the only signal that survives multiple correction (young phenotype) was marker D15S165 (20 cM; p = 0.0009), located on chromosome 15 in the region of the HMPS/CRAC1 locus (Table 4). We tested the sensitivity of the analysis to the inclusion of data from the Ashkenazi families. When we excluded the data from these families, we observed a decrease in the p value, indicating that linkage to the region was generated by the data from families of non-Jewish European descent (i.e., excluding families of Jewish descent, Table 4). We found that six of the nine locations that had been identified because of excess allele sharing in concordantly affected sib pairs also showed excess sharing in the discordant sib pairs, highlighting the importance of evaluating data from discordant sib pairs (Table 3). For chromosomes 2, 4, and 7, all sib pairs have estimated allele sharing that exceeds 0.50, an observation that is not consistent with linkage (Table 3). This further highlights the need to weigh the evidence for linkage with the totality of evidence rather than place reliance upon excess sharing in affected sib pairs alone.
Colon/Breast
In the 33 colon/breast kindreds, there were several signals of considerable interest on chromosomes 1, 13, 17, and 21 (Table 2). For chromosome 1, the signal at 141 cM was flanked by markers D1S1588 (126 cM) and D1S534 (152 cM), in a region frequently altered in a number of solid tumors, including breast cancer47 and colon cancer;48 linkage to this region was also observed with the multiple-cancer phenotype. The signal at marker D13S1493, identified by both H-E regression and the mean tests, was within 5 cM of BRCA2; we did not see a signal in the region of BRCA1 (17q21) or CHEK2 (22q11-12). We observed linkage to marker D17S1308 (17p), in close proximity to the candidate gene Hypermethylated in Cancer 1 (HIC1 [MIM 603825]). There was evidence for linkage to much of chromosome 21, with two linkage peaks, one at D21S1437 (∼12 cM), flanked by markers D21S1432 (4 cM) and D21S1440 (37 cM), and the second peak at the last marker on the chromosome, D21S446 (59 cM).
One successful strategy to combat locus heterogeneity is to exclude families demonstrating linkage to known loci. To further evaluate our evidence for linkage to chromosomes 17 and 21, we identified 11 families linked to BRCA2 (Table 6), excluded these families from the analysis, and reevaluated the evidence for linkage to chromosomes 13, 17, and 21. As expected, linkage to D13S1493 (BRCA2) disappears (Table 7), as does the evidence for linkage at D21S1437. The signals for chromosomes 17 and for 21 at 57 cM remained. All evidence for linkage to D21S1437 was being provided by the same 11 families that demonstrate evidence for linkage to BRCA2. When we included markers D13S1493 (BRCA2) and D21S1437 in a multiple regression equation and tested for interaction (i.e., departure from additive effects), both markers remained statistically significant (for D13S1493, p = 0.015; and for D21S1437, p = 0.00019), with no evidence for interaction (epistasis) between the two markers. This leads to the conclusion that the two loci are segregating together in these 11 families but are either (1) acting in an additive fashion or (2) segregating together by chance alone. We note that two of the 11 families linked to BRCA2 also demonstrate evidence for linkage to HMPS/CRAC1.
Table 6.
Distribution of Phenotypes for the 33 Families in the Colon/Breast Subgroup
| Families with Affected and Unaffected Pairs | Families with Multiple Polyps (>4 polyps) | Breast Cancers | Number of Individuals with CRC | Number of Individuals with Adenomatous Polyps | Distribution of Ten Double Primaries | |
|---|---|---|---|---|---|---|
| Full Set | 24 | 5 (all MSI stable) | 16 (mom) 12 (sibling) 1 mother and daughter 2 sister-sister pairs | 13 | 67 | 3 Breast/Colon 2 Breast and HGD 5 Breast and polyps |
| 11 Families Linked to BRCA2 | 9 | 2 | 7 (mom) 4 (sibling, 1 bilateral breast diagnosed at age 49 and 53) | 5 | 18 | 2 Breast/Colon 2 Breast and polyps |
Table 7.
Colon/Breast Analysis after Excluding 11 Families Linked to the BRCA2 Locus on Chromosome 13
| Group | Chromosome (cM) | Concordantly Affected Sib Pairs |
Discordantly Affected Sib Pairs |
Concordantly Unaffected Sib Pairs |
H-E p Value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | Allele Sharing | SE | p Value | |||
| Colon/Breast | |||||||||||
| 13 (26 cM at D13S1493) | 0.4780 | 0.0407 | 0.7034 | 0.5672 | 0.0440 | 0.9328 | 0.4416 | 0.0759 | 0.7677 | 0.9574 | |
| 17 (1 cM at D17S1308) | 0.5512 | 0.0433 | 0.1223 | 0.4690 | 0.0355 | 0.1941 | 0.5514 | 0.0570 | 0.1824 | 0.0481 | |
| 21 (12 cM at D21S1437) | 0.5468 | 0.0488 | 0.1720 | 0.4929 | 0.0348 | 0.4202 | 0.4524 | 0.0858 | 0.7044 | 0.3145 | |
| 21 (57 cM) | 0.4865 | 0.0396 | 0.6317 | 0.4297 | 0.0314 | 0.0148 | 0.6184 | 0.0509 | 0.0201 | 0.0147 | |
Multiple Cancer
There were three peaks of interest in the multiple-cancer phenotype, on chromosomes 1, 5, and 9 (Tables 2 and 3). Statistically, the most significant linkage signal across all phenotypic subgroups was found at marker D1S1665 (102 cM; p = 0.00007) with the multiple-cancer phenotype. This signal was generated by a strong increase in allele sharing among concordantly affected sib pairs ( = 0.5718; p = 0.002) and a corresponding decrease in allele sharing among discordant sib pairs ( = 0.4082; p = 0.0007). These estimates indicated that being either affected or unaffected could be predicted by the allele sharing at D1S1665 and are consistent with a putative susceptibility locus. In the chromosome plots (Figure 1), there appeared to be three separate peaks: one at 102 cM, one at 137 cM, and a third at 175 cM. The second peak at 137 cM (Figure 1) was not reflected in the data in Tables 2 or 3 because both the allele-sharing estimates and the H-E regression values remained statistically significant for the entire region from 95 cM to 157 cM; the dip observed in the plot was relative. These observations were consistent with linked susceptibility loci. To further delineate these peaks, we used the weighted versions of the H-E regression (W2, W3, and W4), and these resulted in only a single peak, in the region of 137 cM (Figure 4). Weighted versions are asymptotically more powerful; however, with only 17 concordantly unaffected pairs in the multiple-cancer subgroup, the original H-E regression, which combined all concordant pairs, was probably more stable and accurate. Allele-sharing estimates for marker D1S1631 (second peak) demonstrate excess allele sharing for the concordantly unaffected pairs ( = 0.6767; p = 0.013), decreased sharing for the discordant pairs ( = 0.4228; p = 0.007), and a nonsignificant increase in the concordantly affected pairs ( = 0.5162; p = 0.282). All these findings are consistent with a protective allele. Our results for chromosome 1 are consistent with two linked loci, at 102 cM and at 137 cM, respectively.
Figure 4.
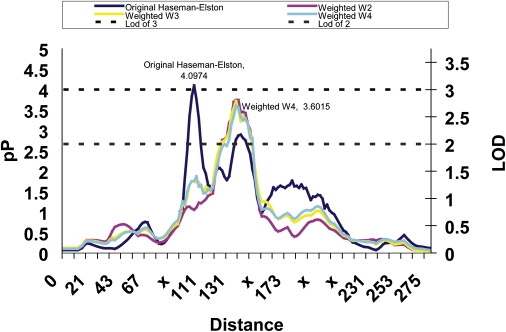
Mulitple Cancer Peak on Chromosome 1
Distance is plotted along the x axis, marker locations are indicated with an “x,” and both −log10 (p value) denoted as pP and the LOD are plotted on the y axis for reference purposes. Reference lines at a LOD of 2 and 3 are provided, and significant data points are labeled with the pP value.
Two other signals of interest are located on chromosome 5 at D5S424, flanked by markers D5S2500 at 69 cM and D5S1725 at 98 cM, and on chromosome 9 at 84 cM, flanked by markers D9S1122 at 72 cM and D9S283 at 95 cM; both regions show increased allele sharing between concordantly affected pairs and a decrease in allele sharing for discordant pairs, a pattern that is consistent with linkage. Marker D5S424 is linked with bladder and ovarian cancer.49–51 The peak on chromosome 9 at 84 cM is 20 cM away from the 9q22.2 region linked to colorectal neoplasia in our previous paper.9
Discussion
Colon neoplasia is undoubtedly a heterogeneous disease, both in its genetic origins (allelic and locus) and in phenotypic presentation. Our strategy to combat genetic heterogeneity utilized clinical information, such as the size and number of polyps, histopathology, family history, age at onset, and other primary cancers, to identify families with similar clinical features, on the presumption that this would result in more genetic homogeneity and, hence, would increase the power to detect linkage signals.
Our results demonstrated that phenotypic classification could be used to identify linkage peaks associated with phenotypic subgroups. These linkage peaks had been masked in the analysis of the complete set of data, and many of the most interesting linkage signals overlapped with previously identified susceptibility genes (BRCA2 and HMPS/CRAC1) and candidate regions, suggesting that they are true linkage signals. Analyzing different phenotypic subgroups provides an opportunity to compare the evidence for linkage among phenotypic subgroups. We identified linkage at the level of genome-wide significance for a replication study to the HMPS/CRAC1 locus in the full (p = 0.018), oligopolyposis (p = 0.003), and young (p = 0.0007) subgroups. The HMPS/CRAC1 locus has previously only been reported with the uncommon and specific phenotype of HPMS, in families of Ashkenazi descent.17 Our analysis provides compelling new evidence linking this locus to families of European descent with a young age at onset (≤51) and/or multiple polyps, thereby confirming and generalizing the findings of Jaegar et al.,17 in a unique population of colon neoplasia families, by utilizing a study design with built-in “matched”-sibling controls and making the study robust to type I error.6,9 Furthermore, after correction for multiple phenotype testing and with permutation testing, these results remained statistically significant. Linkage to this locus is not identified in kindreds with the severe histopathology phenotype (see Figures 1 and 2), indicating that the HMPS/CRAC1 locus may be involved in the development of colorectal polyps but not necessarily involved with progression to CRC.
In the colon/breast subgroup, we identified 11 families that evidenced linkage to BRCA2 and to chromosome 21 at D21S1437. BRCA2 and D21S1437 are segregating together but acting independently. Further study of families segregating BRCA2 mutations, with and without colorectal cancer, will be useful in determining the importance of the linkage signal at D21S1437. After we had excluded data from families linked to BRCA2, the most statistically significant linkage peaks were at marker D17S1308 on chromosome 17 and at 57 cM on chromosome 21 (Table 5). D17S1308 is in the region of the tumor suppressor gene HIC1. HIC1 has not been previously associated with colon or breast cancer, but it is an attractive candidate gene. HIC1 is hypermethylated in cancer and is frequently silenced by epigenetic mechanisms.52 In murine models, Chen et al.53 demonstrated that disruption of HIC1 predisposes to gender-dependent tumors, with males being more likely to develop epithelial cancers and females being more likely to develop lipoid and mesenchymal cancers, thus making HIC1 an attractive candidate for gender-dependent cancers in humans.
More recently, it was shown that HIC1 binds to the SIRT1 promoter and reduces expression of SIRT1.54 SIRT1 is one of the SIR2 enzymes collectively called Sirtuins, which have been linked to cancer and diabetes.54 Both calorie restriction and fasting regulate the transcriptional regulation of SIRT1 through the HIC1:CtBP corepressor complex.54 Given the context of this candidate gene, examining lifetime dietary habits may be informative in determining whether “yo-yo” dieting trends and HIC1 expression are associated with breast and colon cancer. We found no evidence for linkage to BRCA1 or CHEK2.
The linkage peak identified for chromosome 7 in the complete analysis has not been previously associated with CRC/colon neoplasia, but the region has been associated with linkage to body mass index (BMI) in the Old Order Amish.55 BMI is a risk factor for CRC and colon neoplasia;56–59 it is speculated that circulating cytokines derived from adipose (fat) tissue may increase the risk for colon neoplasia. Several less prominent linkage signals overlapped with regions previously associated with colorectal cancer, including BMPR1A, ANXA11 (complete analysis), and EXO1, MFI2antigen, and GOLGA5 (oligopolyposis phenotype). Statistically, the most significant linkage signal was observed at D1S1665 (102 cM, p = 0.00007), with the multiple cancer phenotype; linkage to chromosome 1 was also observed with the breast/colon phenotype at 141 cM, (p = 0.0067). Chromosome 1 is altered in a number of cancer tumors, with 70%–100% of breast-cancer tumors demonstrating gains of 1q and 40% of colorectal cancers demonstrating losses of 1p.47,48 Our observations are consistent with linked loci (putative and/or protective) in this region.
We verified the histopathology of the adenomatous polyps and colon cancers; all other neoplasias were by self report only, which is a potential limitation of the study. However, there is high agreement between patient report and histology in cancer family history.60 In the future, we hope to both verify these cancers and extend the families.
This CNSS study is the first to report a comprehensive examination of multiple phenotypes derived from the familial clustering of colon cancer and colon neoplasia. Both accepted (oligopolyposis) and broad (breast/colon and multiple cancer) phenotypes were examined, in the context of a genome-wide linkage scan, thereby not only expanding our understanding of the phenotypes and of the ethnic groups that demonstrated linkage to the HMPS/CRAC1 locus but also identifying fresh candidate genes/regions, such as chromosome 1q (multiple cancer and breast/colon), HIC1 (breast/colon), and chromosome 21 (breast/colon). Because we stratified the complete set of 194 families into five phenotypic subgroups prior to performing any linkage analysis and corrected for the equivalence of 4.23 independent traits, this study demonstrated that the inclusion of unaffected individuals and the use of subphenotypes could increase analytical power in the search for colon neoplasia susceptibility genes.
Web Resources
The URLs for data presented herein are as follows:
Matrix Spectral Decomposition (matSpD), http://genepi.qimr.edu.au/general/daleN/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We gratefully acknowledge the individuals and families who generously participated in this study and the substantial assistance in reaching these individuals provided by the National Colon Cancer Research Alliance (NCCRA), the Entertainment Industry Foundation, the NBC “Today Show,” and Ms. Katie Couric. We additionally acknowledge the contribution of individual kindreds to this study by Dr. Henry Lynch (Creighton University, Omaha, NE) and Dr. Robert Bresalier (University of Texas M. D. Anderson Cancer Center, Houston). We also thank Christine Ticknor in the laboratory of Sanford D. Markowitz for technical support. Financial support for this work was provided by the following granting agencies: U.S. Public Health Service (PHS) Grants U01 CA82901 (to S.D.M.), P30CA43703 (to Case Western Reserve University/Ireland Comprehensive Cancer Center), K23 1K23CA81308 (to G.L.W.), P41 RR0365 and GM28356 (to R.C.E.), DAMD17-03-1-0289 (to D.D.), Michael Smith Foundation for Health Research Scholar awards (to D.D.), and CIHR IMPACT and IG/IPPH Postdoctoral Fellowships (to D.D.).
References
- 1.American Cancer Society . American Cancer Society; Atlanta: 2005. Colorectal Cancer Facts & Figures Special Edition. [Google Scholar]
- 2.Winawer S.J., Zauber A.G., Gerdes H., O'Brien M.J., Gottlieb L.S., Sternberg S.S., Bond J.H., Waye J.D., Schapiro M., Panish J.F. Risk of colorectal cancer in the families of patients with adenomatous polyps. National Polyp Study Workgroup. N. Engl. J. Med. 1996;334:82–87. doi: 10.1056/NEJM199601113340204. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein P., Holm N.V., Verkasalo P.K., Iliadou A., Kaprio J., Koskenvuo M., Pukkala E., Skytthe A., Hemminki K. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 4.Kemp Z., Thirlwell C., Sieber O., Silver A., Tomlinson I. An update on the genetics of colorectal cancer. Hum. Mol. Genet. 2004;13 Spec No 2:R177–R185. doi: 10.1093/hmg/ddh247. [DOI] [PubMed] [Google Scholar]
- 5.Bodmer W.F. Cancer genetics: Colorectal cancer as a model. J. Hum. Genet. 2006;51:391–396. doi: 10.1007/s10038-006-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elston R.C., Song D., Iyengar S.K. Mathematical assumptions versus biological reality: Myths in affected sib pair linkage analysis. Am. J. Hum. Genet. 2005;76:152–156. doi: 10.1086/426872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards J.H. Sib-pairs in multifactorial disorders: The sib-similarity problem. Clin. Genet. 2003;63:1–9. doi: 10.1034/j.1399-0004.2003.630101.x. [DOI] [PubMed] [Google Scholar]
- 8.Zollner S., Wen X., Hanchard N.A., Herbert M.A., Ober C., Pritchard J.K. Evidence for extensive transmission distortion in the human genome. Am. J. Hum. Genet. 2004;74:62–72. doi: 10.1086/381131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiesner G.L., Daley D., Lewis S., Ticknor C., Platzer P., Lutterbaugh J., Macmillen M., Baliner B., Willis J., Elston R.C. A subset of familial colorectal neoplasia kindreds linked to chromosome 9q22.2–31.2. Proc. Natl. Acad. Sci. USA. 2003;100:12961–12965. doi: 10.1073/pnas.2132286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skoglund J., Djureinovic T., Zhou X.L., Vandrovcova J., Renkonen E., Iselius L., Bisgaard M.L., Peltomaki P., Lindblom A. Linkage analysis in a large Swedish family supports the presence of a susceptibility locus for adenoma and colorectal cancer on chromosome 9q22.32–31.1. J. Med. Genet. 2006;43:e7. doi: 10.1136/jmg.2005.033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kemp Z.E., Carvajal-Carmona L.G., Barclay E., Gorman M., Martin L., Wood W., Rowan A., Donohue C., Spain S., Jaeger E. Evidence of linkage to chromosome 9q22.33 in colorectal cancer kindreds from the United kingdom. Cancer Res. 2006;66:5003–5006. doi: 10.1158/0008-5472.CAN-05-4074. [DOI] [PubMed] [Google Scholar]
- 12.Selby J.V., Friedman G.D., Quesenberry C.P., Weiss N.S. A case-control study of screening sigmoidoscopy and mortality from colorectal cancer. N. Engl. J. Med. 1992;326:653–657. doi: 10.1056/NEJM199203053261001. [DOI] [PubMed] [Google Scholar]
- 13.Rex D.K., Cummings O.W., Helper D.J., Nowak T.V., Mcgill J.M., Chiao G.Z., Kwo P.Y., Gottlieb K.T., Ikenberry S.O., Gress F.G. 5-year incidence of adenomas after negative colonoscopy in asymptomatic average-risk persons. Gastroenterology. 1996;111:1178–1181. doi: 10.1053/gast.1996.v111.pm8898630. [DOI] [PubMed] [Google Scholar]
- 14.Rex D.K., Johnson D.A., Lieberman D.A., Burt R.W., Sonnenberg A. Colorectal cancer prevention 2000: Screening recommendations of the American College of Gastroenterology. American College of Gastroenterology. Am. J. Gastroenterol. 2000;95:868–877. doi: 10.1111/j.1572-0241.2000.02059.x. [DOI] [PubMed] [Google Scholar]
- 15.Statistical Solutions Ltd. S.A.G.E. (2003) Statistical Analysis for Genetic Epidemiology, Release 4.3. Cork, Ireland.
- 16.Laken S., Petersen G., Gruber S., Oddoux C., Ostrer H., Giardiello F., Hamilton S., Hampel H., Markowitz A., Klimstra D. Familial colorectal cancer in Askenazim due to a hypermutable tract in APC. Nat. Genet. 1997;17:79–83. doi: 10.1038/ng0997-79. [DOI] [PubMed] [Google Scholar]
- 17.Jaeger E.E., Woodford-Richens K.L., Lockett M., Rowan A.J., Sawyer E.J., Heinimann K., Rozen P., Murday V.A., Whitelaw S.C., Ginsberg A. An ancestral Ashkenazi haplotype at the HMPS/CRAC1 locus on 15q13-q14 is associated with hereditary mixed polyposis syndrome. Am. J. Hum. Genet. 2003;72:1261–1267. doi: 10.1086/375144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomlinson I., Rahman N., Frayling I., Mangion J., Barfoot R., Hamoudi R., Seal S., Northover J., Thomas H.J., Neale K. Inherited susceptibility to colorectal adenomas and carcinomas: Evidence for a new predisposition gene on 15q14-q22. Gastroenterology. 1999;116:789–795. doi: 10.1016/s0016-5085(99)70061-2. [DOI] [PubMed] [Google Scholar]
- 19.Atkin W.S., Morson B.C., Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N. Engl. J. Med. 1992;326:658–662. doi: 10.1056/NEJM199203053261002. [DOI] [PubMed] [Google Scholar]
- 20.Otchy D.P., Ransohoff D.F., Wolff B.G., Weaver A., Ilstrup D., Carlson H., Rademacher D. Metachronous colon cancer in persons who have had a large adenomatous polyp. Am. J. Gastroenterol. 1996;91:448–454. [PubMed] [Google Scholar]
- 21.Thirlwell C., Howarth K.M., Segditsas S., Guerra G., Thomas H.J., Phillips R.K., Talbot I.C., Gorman M., Novelli M.R., Sieber O.M. Investigation of pathogenic mechanisms in multiple colorectal adenoma patients without germline APC or MYH/MUTYH mutations. Br. J. Cancer. 2007;96:1729–1734. doi: 10.1038/sj.bjc.6603789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slattery M.L., Kerber R.A. Family history of cancer and colon cancer risk: The Utah population database. J. Natl. Cancer Inst. 1994;86:1618–1626. doi: 10.1093/jnci/86.21.1618. [DOI] [PubMed] [Google Scholar]
- 23.Strul H., Kariv R., Leshno M., Halak A., Jakubowicz M., Santo M., Umansky M., Shirin H., Degani Y., Revivo M. The prevalence rate and anatomic location of colorectal adenoma and cancer detected by colonoscopy in average-risk individuals aged 40–80 years. Am. J. Gastroenterol. 2006;101:255–262. doi: 10.1111/j.1572-0241.2006.00430.x. [DOI] [PubMed] [Google Scholar]
- 24.Goldgar D.E., Easton D.F., Cannon-Albright L.A., Skolnick M.H. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J. Natl. Cancer Inst. 1994;86:1600–1608. doi: 10.1093/jnci/86.21.1600. [DOI] [PubMed] [Google Scholar]
- 25.Dong C., Hemminki K. Multiple primary cancers of the colon, breast and skin (melanoma) as models for polygenic cancers. Int. J. Cancer. 2001;92:883–887. doi: 10.1002/ijc.1261. [DOI] [PubMed] [Google Scholar]
- 26.Lipton L., Thomas H.J., Eeles R.A., Houlston R.S., Longmuir M., Davison R., Hodgson S.V., Murday V.A., Norbury C.G., Taylor C. Apparent Mendelian inheritance of breast and colorectal cancer: Chance, genetic heterogeneity or a new gene? Fam. Cancer. 2001;1:189–195. doi: 10.1023/a:1021101014264. [DOI] [PubMed] [Google Scholar]
- 27.Kadouri L., Hubert A., Rotenberg Y., Hamburger T., Sagi M., Nechushtan C., Abeliovich D., Peretz T. Cancer risks in carriers of the BRCA1/2 Ashkenazi founder mutations. J. Med. Genet. 2007;44:467–471. doi: 10.1136/jmg.2006.048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedenson B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed. 2005;7:60. [PMC free article] [PubMed] [Google Scholar]
- 29.Cybulski C., Gorski B., Huzarski T., Masojc B., Mierzejewski M., Debniak T., Teodorczyk U., Byrski T., Gronwald J., Matyjasik J. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004;75:1131–1135. doi: 10.1086/426403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liede A., Karlan B.Y., Narod S.A. Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: A review of the literature. J. Clin. Oncol. 2004;22:735–742. doi: 10.1200/JCO.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 31.Brose M.S., Rebbeck T.R., Calzone K.A., Stopfer J.E., Nathanson K.L., Weber B.L. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J. Natl. Cancer Inst. 2002;94:1365–1372. doi: 10.1093/jnci/94.18.1365. [DOI] [PubMed] [Google Scholar]
- 32.Fasouliotis S.J., Schenker J.G. BRCA1 and BRCA2 gene mutations: Decision-making dilemmas concerning testing and management. Obstet. Gynecol. Surv. 2000;55:373–384. doi: 10.1097/00006254-200006000-00023. [DOI] [PubMed] [Google Scholar]
- 33.Lynch H.T., Harris R.E., Lynch P.M., Guirgis H.A., Lynch J.F., Bardawil W.A. Role of heredity in multiple primary cancer. Cancer. 1977;40:1849–1854. doi: 10.1002/1097-0142(197710)40:4+<1849::aid-cncr2820400813>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 34.Elston R.C., Buxbaum S., Jacobs K.B., Olson J.M. Haseman and Elston revisited. Genet. Epidemiol. 2000;19:1–17. doi: 10.1002/1098-2272(200007)19:1<1::AID-GEPI1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 35.Nyholt D.R. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am. J. Hum. Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sidak Z. On multivariate normal probabilities of rectangles: Their dependence on correlations. Ann. Math. Stat. 1968;39:1425–1434. [Google Scholar]
- 37.Sidak Z. On probabilities of rectangles in multivariate normal Student distributions: Their dependence on correlations. Ann. Math. Stat. 1971;41:169–175. [Google Scholar]
- 38.Cao X., Eu K.W., Kumarasinghe M.P., Li H.H., Loi C., Cheah P.Y. Mapping of hereditary mixed polyposis syndrome (HMPS) to chromosome 10q23 by genomewide high-density single nucleotide polymorphism (SNP) scan and identification of BMPR1A loss of function. J. Med. Genet. 2006;43:e13. doi: 10.1136/jmg.2005.034827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Webb E.L., Rudd M.F., Sellick G.S., El Galta R., Bethke L., Wood W., Fletcher O., Penegar S., Withey L., Qureshi M. Search for low penetrance alleles for colorectal cancer through a scan of 1467 non-synonymous SNPs in 2575 cases and 2707 controls with validation by kin-cohort analysis of 14 704 first-degree relatives. Hum. Mol. Genet. 2006;15:3263–3271. doi: 10.1093/hmg/ddl401. [DOI] [PubMed] [Google Scholar]
- 40.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: A unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 41.Lander E., Kruglyak L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 42.Liberti S.E., Rasmussen L.J. Is hEXO1 a cancer predisposing gene? Mol. Cancer Res. 2004;2:427–432. [PubMed] [Google Scholar]
- 43.Yamamoto H., Hanafusa H., Ouchida M., Yano M., Suzuki H., Murakami M., Aoe M., Shimizu N., Nakachi K., Shimizu K. Single nucleotide polymorphisms in the EXO1 gene and risk of colorectal cancer in a Japanese population. Carcinogenesis. 2005;26:411–416. doi: 10.1093/carcin/bgh335. [DOI] [PubMed] [Google Scholar]
- 44.Blackwelder W.C., Elston R.C. A comparison of sib-pair linkage tests for disease susceptibility loci. Genet. Epidemiol. 1985;2:85–97. doi: 10.1002/gepi.1370020109. [DOI] [PubMed] [Google Scholar]
- 45.Sieber O.M., Lipton L., Crabtree M., Heinimann K., Fidalgo P., Phillips R.K., Bisgaard M.L., Orntoft T.F., Aaltonen L.A., Hodgson S.V. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N. Engl. J. Med. 2003;348:791–799. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- 46.Bouguen G., Manfredi S., Blayau M., Dugast C., Buecher B., Bonneau D., Siproudhis L., David V., Bretagne J.F. Colorectal adenomatous polyposis associated with MYH mutations: Genotype and phenotype characteristics. Dis. Colon Rectum. 2007;50:1612–1617. doi: 10.1007/s10350-007-9027-0. [DOI] [PubMed] [Google Scholar]
- 47.Orsetti B., Nugoli M., Cervera N., Lasorsa L., Chuchana P., Rouge C., Ursule L., Nguyen C., Bibeau F., Rodriguez C. Genetic profiling of chromosome 1 in breast cancer: Mapping of regions of gains and losses and identification of candidate genes on 1q. Br. J. Cancer. 2006;95:1439–1447. doi: 10.1038/sj.bjc.6603433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Struski S., Doco-Fenzy M., Cornillet-Lefebvre P. Compilation of published comparative genomic hybridization studies. Cancer Genet. Cytogenet. 2002;135:63–90. doi: 10.1016/s0165-4608(01)00624-0. [DOI] [PubMed] [Google Scholar]
- 49.Kram A., Li L., Zhang R.D., Yoon D.S., Ro J.Y., Johnston D., Grossman H.B., Scherer S., Czerniak B. Mapping and genome sequence analysis of chromosome 5 regions involved in bladder cancer progression. Lab. Invest. 2001;81:1039–1048. doi: 10.1038/labinvest.3780315. [DOI] [PubMed] [Google Scholar]
- 50.Wang V.W., Bell D.A., Berkowitz R.S., Mok S.C. Whole genome amplification and high-throughput allelotyping identified five distinct deletion regions on chromosomes 5 and 6 in microdissected early-stage ovarian tumors. Cancer Res. 2001;61:4169–4174. [PubMed] [Google Scholar]
- 51.Tavassoli M., Steingrimsdottir H., Pierce E., Jiang X., Alagoz M., Farzaneh F., Campbell I.G. Loss of heterozygosity on chromosome 5q in ovarian cancer is frequently accompanied by TP53 mutation and identifies a tumour suppressor gene locus at 5q13.1–21. Br. J. Cancer. 1996;74:115–119. doi: 10.1038/bjc.1996.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Britschgi C., Rizzi M., Grob T.J., Tschan M.P., Hugli B., Reddy V.A., Andres A.C., Torbett B.E., Tobler A., Fey M.F. Identification of the p53 family-responsive element in the promoter region of the tumor suppressor gene hypermethylated in cancer 1. Oncogene. 2006;25:2030–2039. doi: 10.1038/sj.onc.1209240. [DOI] [PubMed] [Google Scholar]
- 53.Chen W.Y., Zeng X., Carter M.G., Morrell C.N., Chiu Yen R.W., Esteller M., Watkins D.N., Herman J.G., Mankowski J.L., Baylin S.B. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat. Genet. 2003;33:197–202. doi: 10.1038/ng1077. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Q., Wang S.Y., Fleuriel C., Leprince D., Rocheleau J.V., Piston D.W., Goodman R.H. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc. Natl. Acad. Sci. USA. 2007;104:829–833. doi: 10.1073/pnas.0610590104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Platte P., Papanicolaou G.J., Johnston J., Klein C.M., Doheny K.F., Pugh E.W., Roy-Gagnon M.H., Stunkard A.J., Francomano C.A., Wilson A.F. A study of linkage and association of body mass index in the Old Order Amish. Am. J. Med. Genet. C. Semin. Med. Genet. 2003;121:71–80. doi: 10.1002/ajmg.c.20005. [DOI] [PubMed] [Google Scholar]
- 56.Adams K.F., Leitzmann M.F., Albanes D., Kipnis V., Mouw T., Hollenbeck A., Schatzkin A. Body mass and colorectal cancer risk in the NIH-AARP cohort. Am. J. Epidemiol. 2007;166:36–45. doi: 10.1093/aje/kwm049. [DOI] [PubMed] [Google Scholar]
- 57.Kim S., Baron J.A., Mott L.A., Burke C.A., Church T.R., Mckeown-Eyssen G.E., Cole B.F., Haile R.W., Sandler R.S. Aspirin may be more effective in preventing colorectal adenomas in patients with higher BMI (United States) Cancer Causes Control. 2006;17:1299–1304. doi: 10.1007/s10552-006-0075-x. [DOI] [PubMed] [Google Scholar]
- 58.Ehrmann-Josko A., Sieminska J., Gornicka B., Ziarkiewicz-Wroblewska B., Ziolkowski B., Muszynski J. Impaired glucose metabolism in colorectal cancer. Scand. J. Gastroenterol. 2006;41:1079–1086. doi: 10.1080/00365520600587444. [DOI] [PubMed] [Google Scholar]
- 59.Larsen I.K., Grotmol T., Almendingen K., Hoff G. Lifestyle as a predictor for colonic neoplasia in asymptomatic individuals. BMC Gastroenterol. 2006;6:5. doi: 10.1186/1471-230X-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerber R.A., Slattery M.L. Comparison of self-reported and database-linked family history of cancer data in a case-control study. Am. J. Epidemiol. 1997;146:244–248. doi: 10.1093/oxfordjournals.aje.a009259. [DOI] [PubMed] [Google Scholar]