

Abstract
Spondylothoracic dysostosis (STD), also known as Jarcho-Levin syndrome (JLS), is an autosomal-recessive disorder characterized by abnormal vertebral segmentation and defects affecting spine formation, with complete bilateral fusion of the ribs at the costovertebral junction producing a “crab-like” configuration of the thorax. The shortened spine and trunk can severely affect respiratory function during early childhood. The condition is prevalent in the Puerto Rican population, although it is a panethnic disorder. By sequencing a set of candidate genes involved in mouse segmentation, we identified a recessive E103X nonsense mutation in the mesoderm posterior 2 homolog (MESP2) gene in a patient, of Puerto Rican origin and from the Boston area, who had been diagnosed with STD/JLS. We then analyzed 12 Puerto Rican families with STD probands for the MESP2 E103X mutation. Ten patients were homozygous for the E103X mutation, three patients were compound heterozygous for a second nonsense mutation, E230X, or a missense mutation, L125V, which affects a conserved leucine residue within the bHLH region. Thus, all affected probands harbored the E103X mutation. Our findings suggest a founder-effect mutation in the MESP2 gene as a major cause of the classical Puerto Rican form of STD/JLS.
Main Text
Congenital vertebral-segmentation abnormalities in humans are present in a wide variety of rare but well-characterized disorders, as well as in many diverse and poorly understood phenotypic patterns.1 In some cases, patients with congenital scoliosis and chest-wall abnormalities present a major surgical challenge. In 1938, Jarcho and Levin described a Puerto Rican family whose two children presented with a shortened trunk, with abnormal segmentation throughout the vertebral column and irregularly aligned ribs, but with normal long bones and skull.2 Since then, the authors' names have frequently been used eponymously (and often inappropriately) for almost any form of costovertebral malformation.
Spondylothoracic dysostosis (STD) has been well characterized as an autosomal-recessive disorder with high prevalence in the Puerto Rican population, comprising 49% of the STD cases reported in the medical literature.3 The same phenotype has also been described in other patient populations.4–11 Patients with STD exhibit a short stature due to multiple defects in vertebral segmentation and spine formation, an increased antero-posterior (AP) thoracic diameter, and, radiologically, a characteristic “crab-like” appearance of the thoracic cage on AP projection. The ribs are fused posteriorly at the costovertebral junctions. The short spine and thoracic cage frequently cause respiratory insufficiency, with a mortality rate of 32% during early childhood.3
The characteristic, periodic vertebral arrangement of the spine is established during embryogenesis when the vertebral precursors, the somites, are rhythmically produced from the presomitic mesoderm in the embryo.12 This striking rhythmicity, observed in vertebrate model species, of the somite formation in the presomitic mesoderm results from the periodic activation of the Notch-, FGF-, and Wnt-signaling pathways by a molecular oscillator called the segmentation clock.12,13 The study of nonsyndromic, Mendelian forms of spondylocostal dysostosis (SCD; referred to as SCDO by OMIM) has resulted in an increased understanding of the causes of abnormal vertebral segmentation in humans. Three major SCD subtypes have been characterized thus far and include: (1) SCDO type 1 (SCDO1 [MIM 277300]), which appears to be the most common form and is due to a mutation of the delta-like 3 gene (DLL3 [MIM 602768]);14–16 (2) SCDO2 ([MIM 608681]), which is due to a mutation of the mesoderm posterior 2 homolog gene (MESP2 [MIM 605195]);17 and (3) SCDO3 ([MIM 609813]), which is due to a mutation of the LFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase gene (LFNG [MIM 602576]).18 All of these genes are components of the Notch-signaling pathway and are involved in the segmentation clock.1 Thus, in order to identify mutations that underlie congenital vertebral anomalies in humans, we adopted a candidate-gene approach and sequenced genes associated with somitogenesis in patients with congenital scoliosis.
We selected five genes, DLL3; LFNG; MESP2; hairy and enhancer of split 7 (HES7 [MIM 608059]); and presenilin 1 (PSEN1 [MIM 104311]), on the basis of their known association with SCD and/or their mouse mutant phenotypes.19–21 The coding frame of these five genes was first sequenced in a cohort of 31 patients from the Children's Hospital Boston, all of whom exhibited various degrees of congenital scoliosis with abnormal segmentation. The study was approved by the appropriate Institutional Review Boards (IRBs), and informed consent was obtained from all human subjects at the Children's Hospital Boston. Informed consent was also obtained from hospitals in Puerto Rico as described previously (see ref. 3).
Genomic DNA from patient blood samples was prepared with a PAXgene Blood DNA kit according to the manufacturer's protocol (PreAnalytiX, a QIAGEN/BD company). Polymerase chain reaction (PCR) was performed with 25 ng of genomic DNA, 0.6 μM primers, 0.5 units Biolace Taq polymerase (Bioline), 2.0 mM MgCl2, and standard PCR buffer. For products that were difficult to amplify, 0.6 units of failsafe enzyme (Epicenter) were used in place of the Biolace Taq polymerase and failsafe buffers H, J, and D were used for the PCR reaction (reaction details available upon request). The reactions were then amplified via the following thermocycler protocol: 5 min at 95°C, 40× (30 s at 95°C, 30 s at 60°C, and 60 s at 72°C), 10 min at 72°C. For some of the primer pairs, the annealing step was performed at 55°C or 65°C (Table 1). The PCR products were purified with ExcelaPure 96-well UF PCR Purification plates (Edge Biosystems) and sequenced with BigDye v3.1 chemistry on a 3730 DNA Analyzer (Applied Biosystems) with the primers described in Table 1 plus three additional primers to cover MESP2 exon 1 (5′-AAGATCGAGACGCTGCGCCT-3′, 5′-CACTGCAGACTCTCCTCGCT-3′, 5′-CAAGGGCAGGGGCAAGGACAG-3′); two additional primers to cover DLL3 exon 4 (5′-CCCTCCTTTGCCTGTCCTCG-3′, 5′-GGGTGAGTGGGTTGTGAGAGGG-3′), two additional primers to cover DLL3 exon 7 (5′-GCTGCTACGCCCACTTCTC-3′, 5′-TGGGAACTCACACCGCGCT-3′), and one additional primer to cover LFNG exon 8 (5′-ACGCACAGGCACGGCCCTAG-3′). Primer specificity was determined by blast and electronic PCR. Bioinformatic analysis and searches of pseudogene databases indicated no possibility of pseudogenes. Sequences of patient samples were compared to reference sequences in GenBank with SeqScape v2.5 software (Applied Biosystems).
Table 1.
PCR Primers and Summary of Mutations Identified in Patient Samples
| Gene-Exon | Annealing Temp. | PCR Primers (5′→3′) |
Product Size (bp) | Nonsynonymous SNPs | |
|---|---|---|---|---|---|
| Forward | Reverse | ||||
| MESP2-Ex1 | 55 | GACACCTCTCTGCAACCTG | CCTGGAGTAGATAAGCTGGG | 1117 | A66G, E103X |
| MESP2-Ex2 | 60 | CCAGCCATACCATGGCAAC | CCAAGCTACAGGACTGATTC | 408 | P351H |
| DLL3-Ex1 | 60 | TCAGATATAAGGCTTGGAAGC | TCCCCAGGACCTTCAGGTCG | 208 | |
| DLL3-Ex2 | 55 | GCGTGGAAAGGGATGAATGC | TTTCGCTGGCAGGGTTAGGC | 465 | A115T |
| DLL3-Ex3 | 60 | CTCCATTCCTGAACTCTGGC | CACCAACCCCGTGTCTCACG | 208 | |
| DLL3-Ex4 | 55 | TCCGTATGCATCCATGTTCG | ATACATCCGCAGCAGTCAGC | 493 | L142Q, F172C |
| DLL3-Ex5 | 55 | GCCTCAGTTTCCCTATCTGT | GATCCCAAATCTCCAACCTAT | 749 | L218P |
| DLL3-Ex6 | 65 | CTTGAGACTGGACAAGGAGC | AACCCTGGACTGTCTGAGCC | 396 | |
| DLL3-Ex7 | 55 | CAGAGCTGGGAAACAGCGCG | CCAGCAGCGCAAGGCGTTCG | 695 | |
| DLL3-Ex8 | 60 | TTGGCCCGATTCCTTGATGC | AAAGAGAAGATGGCAGGTAGC | 435 | |
| Hes7-Ex1 | 60 | TTCTGGCTCCTGGAGTTCTGG | GCAGAATCTGGATGTCGAAGGG | 294 | |
| Hes7-Ex2 | 55 | GCCAACCAAGCTTGTGTCCC | AGCGGAGAGGGATCGAATGG | 340 | |
| Hes7-Ex3 | 55 | TTAACCTCGCCTCGGAGCAGAACC | ATTTGGTCGCGCGTTCTGTGGG | 367 | |
| Hes7-Ex4 | 55 | TCCCCAACACCTGCTCGCC | CTCCCTCTTTCCGTCCATCTGG | 699 | |
| LFNG-Ex1 | 55 | GGTGCTGCGCTGCTGGACTGC | ACTGGCGTCGCCCACAGATG | 666 | G38R |
| LFNG-Ex2 | 65 | AAACCAAGGCCCGGAGAAGG | TGAGTAAACTGCCCTCCCTGTCC | 319 | |
| LFNG-Ex3&4 | 65 | GGAAAAAGCTGCCTGAATGGG | ACTCCGGGTGTGCGCTCC | 632 | |
| LFNG-Ex5 | 65 | ATGGAGCGGGTCAGCGAGAA | AGAGTCCTGCCGGATGGTGC | 480 | |
| LFNG-Ex6 | 65 | AGATTCCCTCCACAGAGAGCCACG | CAGTGAAGAGAAGAGACGGCAGG | 581 | |
| LFNG-Ex7 | 60 | AGTTTGGGACCTTATTCCTGGG | ATGCCGCTTAGAGAGACCCTGC | 341 | V346M |
| LFNG-Ex8 | 65 | AAATGGGAGCTCAGCACCTGCC | AGAGGCACATAAGTGGCGCTGG | 351 | |
| PSEN1-Ex3 | 65 | TCTGGGAGCCTGCAAGTGAC | CTGTGTCCTCCAGCAATCAGC | 463 | |
| PSEN1-Ex4 | 65 | TGACGGGTCTGTTGTTAATCC | CCCCTCGCTCTCTCAACTG | 496 | |
| PSEN1-Ex5 | 65 | GAGTTGGGGAAAAGTGACTTA | TGAGCCTGGCATTACACA | 474 | |
| PSEN1-Ex6 | 55 | GGCGAAACCCTGTCTCTACTA | GGAGCAACAGAAGAATGTCTC | 511 | |
| PSEN1-Ex7 | 55 | GCCGTGATTGCACCACTTTAC | CATGCCCAGCCGAAATCT | 653 | |
| PSEN1-Ex8 | 60 | ACCCCAGTAACGATACACT | GAATCAACATCAGGTAGAAGA | 552 | |
| PSEN1-Ex9 | 60 | GGAAGACTGGCGATTTG | TGTATTTACTGGGCATTATCA | 329 | E318G |
| PSEN1-Ex10 | 60 | GGCCAGCTAGTTACAATG | CCAAATAAAAGTTACATGTGA | 407 | |
| PSEN1-Ex11 | 65 | AACACAGCTGAAGCCTAATTT | AGCTCCCAAGTGATTCTAATG | 453 | |
| PSEN1-Ex12 | 60 | TGCATAATGAACCCTATGAAA | ACAGTCCACTGCGATGAA | 584 | |
Ten nonsynonymous, single-nucleotide polymorphisms (SNPs) were identified in the patient samples, including nine missense mutations and one nonsense mutation (Table 1). Of the nine missense mutations, three were reported in the public SNP database (DLL3 F172C, dbSNP ID no. rs8107127; DLL3 L218P, dbSNP ID no. rs1110627 and PSEN1 E318G, dbSNP ID no. rs17125721) and six were previously unreported. It is unclear whether these missense mutations are simple polymorphisms or if they contribute to the disease phenotype in the patients. In a 12-year-old female of Puerto Rican origin with a severe form of STD (atypical, with scoliosis), a tethered spinal cord, and malrotation of the right kidney (Figure 1A), we identified a previously unreported homozygous nonsense mutation in the MESP2 gene. This patient harbored a single-base-pair substitution mutation (c.307G→T) in the basic helix-loop-helix (bHLH) domain of the MESP2 gene (base-pair numbering based on accession no. BC111413). The mutation resulted in the replacement of a glutamic acid codon (GAG) at position 103 with a stop codon (TAG) and the creation of an SpeI restriction-enzyme site (Figures 2A and 2B). The mutation, E103X (p.Glu103X), occurs in exon 1 in the middle of the bHLH domain and is likely to produce a truncated nonfunctional protein and to be susceptible to nonsense-mediated RNA decay (NMD)22 (Figure 2C).
Figure 1.
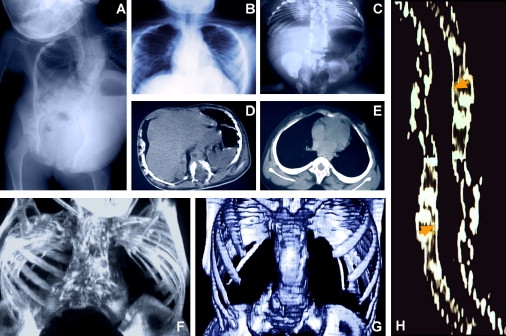
Rib and Vertebral Defects in Patients with Spondylothoracic Dysostosis
(A) Antero-posterior (AP) chest radiograph of patient 01-022 showing the shortened vertebral column characteristic of patients with spondylothoracic dysostosis (STD), also known as Jarcho-Levin syndrome (JLS). This patient is from the Boston area but is of Puerto Rican origin.
(B) AP chest radiograph demonstrating bilateral rib fusion at the costovertebral junction with the ribs' “crab-like” appearance as they fan out from their origins.
(C) Thoraco-abdominal radiograph characteristic of a patient with STD. The spine is short but straight (AP projection), and multiple segmentation abnormalities are evident. The pedicles of the vertebrae are prominent (“tramline sign”). The ribs are fused posteriorly at the costovertebral junctions and fan out, showing a “crab-like” appearance.
(D and E) Thoracic reconstructive CT scans. (D) Axial view of a thoracic vertebra shows both anterior and posterior cleft defects. (E) Vertebrae without clefts present an increased coronal diameter with a decreased sagittal diameter (sickle-shape vertebra).
(F) Patient with STD homozygous for the E103X mutation shows a thoracic spine measurement of 4.2 cm.
(G) Patient with STD heterozygous for the E103X mutation shows a spine measurement of 7.7 cm.
(H) Reconstructive image of the spinal canal of a patient with STD. Arrowheads indicate the measurement points at the thoracic and lumbar levels, demonstrating a spinal-canal width of 34.0 mm versus an expected 27.1 mm (patient genotype E103X/E103X).
Panels B–H are films from patients of Puerto Rican origin.
Figure 2.
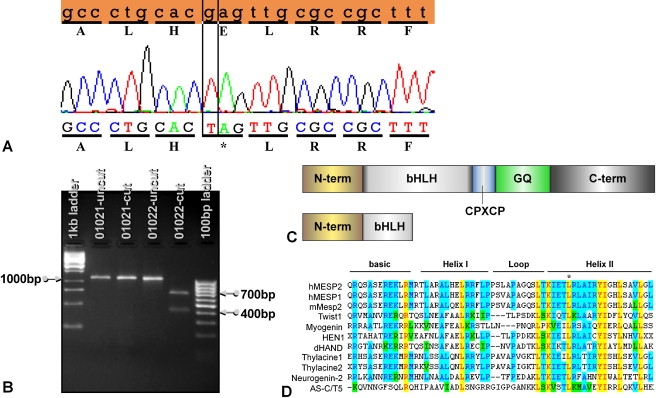
Detection of the E103X Mutation by DNA Sequencing and Restriction Fragment Length Polymorphism (RFLP) Analysis
(A) The amino acid and DNA sequences (top, orange) of MESP2 from GenBank are compared with the sequences from the patient with STD/JLS. The single-base-pair substitution mutation (c.307G→T) is indicated by a vertical box. The asterisk indicates the stop codon.
(B) The c.307G→T mutation results in the addition of an SpeI site, which can be detected as an RFLP. Sample no. 01021 is from a control patient who does not have the E103X mutation. Sample no. 01022 is from a patient who is homozygous for the E103X mutation.
(C) Comparison of MESP2 full-length protein versus truncated protein caused by the E103X mutation. The full-length MESP2 protein (top) has an N-terminal (N-term) domain followed by a basic helix-loop-helix domain (bHLH), a region rich in glycine and glutamine residues (GQ repeats), and a c-terminal (c-term) region. Between the bHLH domain and the GQ repeats is a conserved CPXCP motif. The predicted protein resulting from the E103X mutation (bottom) has a short, truncated bHLH domain and is missing the other domains.
(D) Sequence comparison of the bHLH domain of MESP2 to other bHLH proteins. The asterisk indicates the L125V mutation in patient 0600905-01. Identical residues are highlighted in yellow, conserved residues are highlighted in blue, and blocks of similar residues are highlighted in green. Swiss-Prot accession numbers for the sequences are: human MESP2, Q0VG99; human MESP1, Q9BRJ9; mouse Mesp2, O08574; human Twist1, Q15672; human Myogenin, P15173; human HEN1, Q02575; mouse dHAND, Q9EPN2; Xenopus Thylacine1, O73623; Xenopus Thylacine2, O73624; mouse Neurogenin-2, P70447; and Drosophila AS-C/T5, P10083.
Because of the Puerto Rican origin of the patient and the high frequency of STD in the Puerto Rican population, we sequenced the MESP2 gene in a cohort of 14 Puerto Rican patients with STD. We first examined the MESP2 gene in ten Puerto Rican probands and family members previously reported.3 Eight of the ten probands were homozygous for the E103X mutation (Table 2). The unaffected parents were heterozygous carriers. An additional patient (0600905-01) was heterozygous for the E103X mutation and an L125V (c.373C→G, p.Leu125Val) missense MESP2 mutation (Table 2). The L125V mutation occurs in a conserved leucine residue in the bHLH domain (Figure 2D) and is predicted to be deleterious by the Sorting Intolerant From Tolerant (SIFT) prediction program23 but predicted to be neutral by the PMUT algorithm.24 The L125V mutation was not present in a panel of ethnically matched controls, suggesting that the L125V mutation is not a common polymorphism in the Puerto Rican population (n = 24, data not shown). The other patient (000408-05) was heterozygous for the E103X mutation, but a second MESP2 mutation was not identified, nor was there any mutation in the DLL3 or LFNG genes (data not shown). It is unclear how the E103X mutation contributes to the STD phenotype in this particular patient. The father of this patient was of Puerto Rican and Chinese ethnicity, which could explain the lack of a second MESP2 gene mutation. It is possible that this patient has a partial gene deletion or a mutation in the promoter or regulatory region, given that only coding exons and exon/intron junctions were sequenced. Alternatively, this patient might carry a mutation in an as-yet-unidentified gene. Two variants of the MESP2 gene—A66G and S220F—were identified in two parental carriers (Table 2) and were believed to be polymorphisms, given that these variants were not transmitted to their affected offspring.
Table 2.
MESP2 Mutations in Patients of Puerto Rican Origin
| Family No. | Relationship | Patient ID | Affected Status | E103stop Status | Other MESP2 Mutations |
|---|---|---|---|---|---|
| 1 | Proband | 990706-01 | AF | Hom | |
| 1 | Parent | 990706-03 | OC | Het | |
| 1 | Parent | 990706-04 | OC | Het | |
| 2 | Father | 980615-01 | OC | Het | |
| 2 | Mother | 980615-03 | OC | Het | |
| 2 | Proband | 980615-04 | AF | Hom | |
| 3 | Proband | 980524-01 | AF | Hom | |
| 3 | Father | 980524-03(A) | OC | Het | |
| 3 | Mother | 980504-04B | OC | Het | |
| 4 | Proband | 980712-01 | AF | Hom | |
| 4 | Parent | 980712-02 | OC | Het | A66G (Het) |
| 4 | Parent | 980712-03 | OC | Het | |
| 5 | Proband | 000308-01 | AF | Hom | |
| 5 | Parent | 000308-02 | OC | Het | |
| 6 | Proband | 980507-01 | AF | Hom | |
| 6 | Parent | 980725-02 | OC | Het | |
| 7 | Proband | 990617-01 | AF | Hom | |
| 7 | Parent | 990602-02 | OC | Het | |
| 8 | Proband | 000201-01 | AF | Hom | |
| 8 | Parent | 000201-03 | OC | Het | |
| 8 | Parent | 000201-02 | OC | Het | |
| 9 | Proband | 0600905-01 | AF | Het | L125V (Het) |
| 9 | Parent | 0600905-02 | OC | WT | L125V (Het), S220F (Het) |
| 9 | Parent | 0600905-03 | OC | Het | |
| 10 | Proband | 000408-05 | AF | Het | |
| 10 | Parent | 000408-07 | OC | WT | |
| 10 | Parent | 000427-02 | OC | Het |
AF denotes affected, OC denotes obligate carrier, Hom denotes homozygous, Het denotes heterozygous, and WT denotes wild-type.
The MESP2 gene was sequenced in two other affected families of Puerto Rican origin (Figures 3A and 3B), for whom radiographs were unavailable. The two probands of these families were homozygous for the E103X mutation. Unexpectedly, however, two affected third cousins of the proband in one of these families (Figure 3B) were compound heterozygotes for E103X and a second nonsense mutation, E230X. This mutation (c.688G→T) results in the replacement of glutamic acid at position 230 with a premature termination codon (p.Glu230X). Heterozygous carriers were unaffected.
Figure 3.
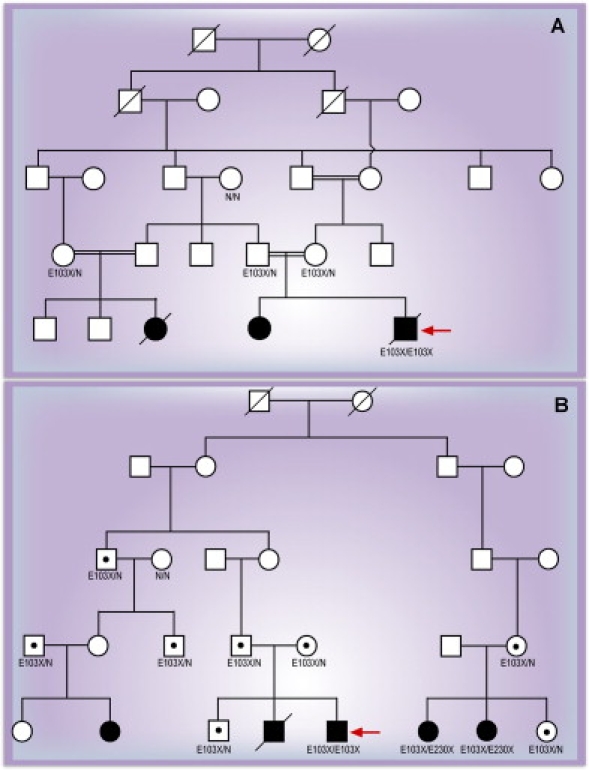
Confirmation of Homozygosity for the MESP2 E103X Mutation as the Cause of STD in Two Families of Puerto Rican Origin
(A) Confirmation of homozygosity for the MESP2 E103X mutation as the cause of STD in a family of Puerto Rican origin. Black circles and square represent affected individuals.
(B) Confirmation of homozygosity for the MESP2 E103X mutation as the cause of STD in a family of Puerto Rican origin and of heterozygosity for the E103X and E230X mutations. Black circles and squares represent affected individuals. Black dots represent unaffected carriers, and the red arrow in each panel indicates the proband.
To determine whether these mutations are responsible for the phenotype, transcriptional activities of MESP2 variants were analyzed with luciferase reporter assays. Previous studies in mouse revealed that Mesp2 exhibits synergy with Notch signaling to activate transcription from an Lfng reporter in cultured cells.25 We confirmed that the human MESP2 also retained similar activity on the mouse Lfng reporter, albeit weaker than the mouse Mesp2 protein (Figure 4A). For the luciferase reporter assays, a 2.5Kb EcoRI-NotI fragment of the mouse Lfng enhancer was cloned upstream of luciferase in the pGL4.10 vector (Promega, Madison, WI, USA). The reporter was transfected into NIH 3T3 cells with or without constructs expressing the Notch intracytoplasmic domain (NICD) fused to the venus variant of the yellow fluorescent protein (YFP) (50 ng) and one of the following: 3 × Flag-tagged mouse Mesp2 (50 ng), human MESP2 (50 ng), human MESP2 E103X (50 ng), human MESP2 500-503 dup (50 ng) or human MESP2 L125V (50 ng). In this assay the NICD-YFP expression construct acts as a constitutively active Notch receptor. pGL4.74 (Promega) was cotransfected as an internal control (10 ng) to normalize for differences in transfection efficiency. After 36 hr, luciferase activities were measured with a Dual Luciferase Assay kit (Promega), and the activities shown in Figure 4B represent average values, obtained from at least three independent experiments. We examined three different MESP2 mutant cDNA constructs harboring two mutations reported in this paper, E103X and L125V, as well as the 4-bp duplication (c.500–503 dupACCG) reported previously.17 The results indicate that all three mutations lack transcriptional activities in this assay (Figure 4B).
Figure 4.
Luciferase Reporter Assay Indicating that Human MESP2 Mutants Lack Transactivation Activities
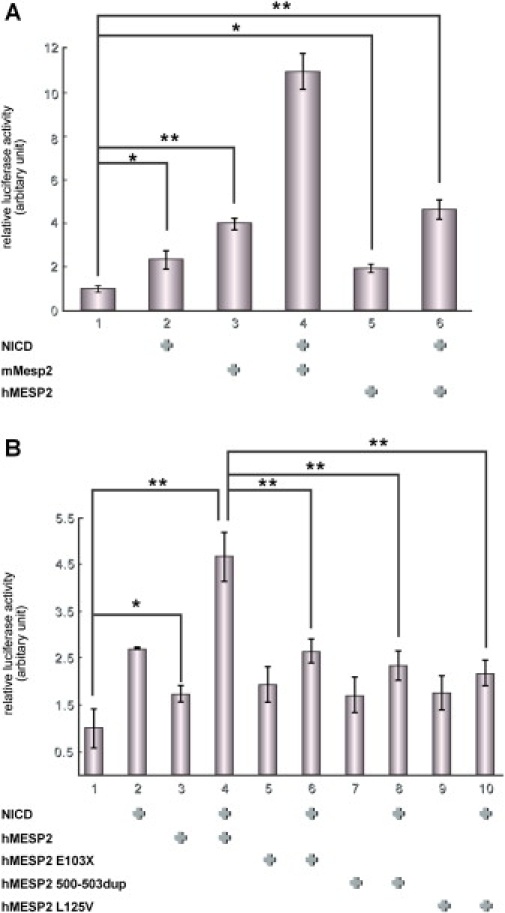
(A) The activity of MESP2 was compared between mouse (mMesp2) and human (hMESP2) with the use of a reporter construct containing luciferase driven by a mouse Lfng enhancer. Cotransfection of MESP2 with the notch intracellular domain (NICD) results in synergistic activation of the reporter (indicated by + at the bottom of each panel). Similar activation was observed in hMESP2 and NICD.
(B) The relative activities of hMESP2 and the variants. Three mutant MESP2 variants show reduced activities. The relative activities are expressed as means + SDs of three different experiments.
∗ indicates p < 0.05.
∗∗ indicates p < 0.01.
We compared the genotypes of ten patients with STD with their respective clinical phenotypes and natural histories. The two patients who died during early infancy were homozygous for the E103X mutation, but fewer clinical measurements and data were available for them. Clinical data are summarized in Table 3, and representative images of the skeletal defects are presented in Figures 1B–1H. Axial skeletal dimensions were compared to normal standards.26 Phenotypical data suggest that heterozygosity for the E103X mutation produces a milder phenotype in terms of the thoracic measurements (Figures 1F and 1G), although due to the small sample size the differences were not statistically significant.
Table 3.
Summary of Clinical and Radiological Findings in Affected Subjects
| Observation or Measurement | Subjects and Findings |
|---|---|
| Genotypes | E103X/E103X (n = 8); E103X/L125V (n = 1); E103X/unknown (n = 1) |
| Age | Deceased (n = 2; 4 mos, 3 mos) |
| Preschool (n = 2; 2 yrs, 4 yrs) | |
| School-age (n = 4; avg. 12.2 yrs; range: 8–18) | |
| Adults (n = 2; 34 yrs, 35 yrs) | |
| Hospital treatment | < 1 yr: 3.7 admissions; avg. length 4.8 days (range: 2–18) |
| > 24 mos: 1.3 admissions per yr (range: 0–4) | |
| Abdomen/inguinal hernia | Protuberant, no visceromegaly; bilateral herniotomy (n = 8, i.e., all survivors) |
| Height | 1.15 percentile (range: 1–3) |
| Weight | 1.1 percentile (range: 1–3) |
| Upper/lower limbs | Within normal range (i.e., short stature due to truncal shortening) |
| BMI | 16.15 (range: 12–22.9) |
| Thoracic circumference: nipple line | 33rd percentile (range: 1st–68th) |
| Thoracic circumference: height ratio | 0.621 (range: 0.386–0.739) (expected 0.534 [range: 0.481-0.643]; p > 0.0001). No differences between children and adults, between survivors and nonsurvivors, or between genotypes. |
| Lung volumes compared with the normal range for age/gender | Average: 23%; range: 15.4–28% for E103X homozygotes (n = 6: survivors); 38–41% for E103X heterozygotes (n = 2) |
| Lung volumes compared with the normal range for height/age and thoracic spinal height/age (allowing for short stature) | Average: 40.5%; range: 16.2–101.3% |
| Spinal radiology | |
| Cervical | Clinically: neck rigid and extremely short |
| Atlas fused to occiput and axis fused with vertebrae—not possible to determine vertebral number | |
| Thoracic cage | Asymmetrically shortened, with posterior aspect more severely affected. Fusion of all ribs at costovertebral junctions (Figures 1A–1C); extent of rib fusion ranged from 30–60% of thoracic rib circumference. |
| Median vertebral number: 6 (range: 5–8), on the basis of normal pedicle shadows. Multiple segmentation and formation defects. Thoracic vertebrae showed anterior and posterior sagittal clefts (Figure 1D). | |
| Scoliosis rare—one subject (Cobb angle 21°) | |
| Median rib number: 10 per hemithorax (range: 7–12) | |
| Thoracic spinal height: 24.2% of normal (range: 19.2–33.3%). Subjects heterozygous for E103X at upper end of range (Figures 1F and 1G), i.e., milder than homozygotes. | |
| Antero-posterior chest diameter at T12 (sterno-vertebral distance) < 2.5th percentile. | |
| Lumbar | Median vertebral number: 5 (range: 4–6); multiple segmentation and formation defects. |
| Lumbar spinal height: 70.1% of normal (range: 45–116%) | |
| Overall appearance of lumbar vertebral body: normal width but decreased depth; vertebral canal: normal depth with a slightly increased width (Figure 1H). | |
| Sacrococcygeal | No apparent abnormality |
BMI denotes body-mass index.
We have shown that homozygosity for the MESP2 E103X mutation causes STD/JLS and is a common gene mutation in Puerto Rican patients, highly suggestive of a founder effect. Furthermore, we discovered two additional mutations—E230X and L125V—that are predicted to alter MESP2 function. The E230X mutation causes a premature stop codon in the c-terminal domain, and the L125V mutation destroys a conserved leucine residue in the bHLH domain. This suggests that multiple mutations in MESP2, including compound heterozygosity, can give rise to a wide range of vertebral phenotypes ranging from SCDO21,17 to STD. Whereas MESP2 is located on chromosome 15q26.1, initial homozygosity linkage-mapping studies using a pooling technique showed linkage to chromosome 2 in some of the patients included in this study.27 However, included in the initial pooling analysis was a subgroup of patients who, after review of their radiographs, did not meet the clinical criteria for the STD/JLS phenotype.
These different phenotypes associated with mutations in the same gene might be explained by the position and effect of the mutation. The E103X and E230X mutations identified in patients with STD are located within the first exon of the MESP2 gene, and the resulting mutant mRNA transcripts are predicted to be susceptible to NMD.22 Therefore, patients homozygous or compound heterozygous for these mutations are likely to have a reduced or absent MESP2 protein. However, the 4 bp duplication identified in a small consanguineous family with a milder form of SCD,17 which includes less-severe segmentation abnormalities and misaligned ribs than those observed in patients with SCDO1, occurs after the bHLH domain in exon 1 and causes a frameshift that results in a premature stop codon within the second and final exon of MESP2. Transcripts carrying this mutation would not be subject to NMD. In these patients, a truncated protein containing an intact bHLH domain, which could retain some function, is predicted. However, in the luciferase assay, the E103X, L125V, and 500–503 dup mutations all have a similar impact on MESP2 activity. Therefore, synergy with the Notch intracellular domain for control of the activation of specific targets is likely to be similarly altered in these mutants, but other functions are likely to be intact in the 500–503 dup mutation, thus potentially accounting for the milder phenotype observed.17
Our phenotypic data suggest that homozygosity for the E103X mutation gives rise to a more severe phenotype than does compound heterozygosity involving this mutation (according to thoracic measurements). Results from pulmonary-function tests of patients heterozygous for E103X should show a less-restrictive pattern than do results from those who are homozygous. In this study, thoracic CT scans had not been performed on the E103X homozygous patients who died; thus, comparison with homozygous E103X survivors was not possible.
The discovery of three deleterious MESP2 mutations in a single population is unusual and could suggest a positive-selection pressure or heterozygous advantage as described in other disorders such as Cystic Fibrosis (CF [MIM 219700]) and Sickle Cell Anemia (MIM 603903).28,29 How frequent the MESP2 mutations are in the general Puerto Rican population and how heterozygosity for such mutations could be beneficial remain unclear. Future studies will focus on sequencing MESP2 in a larger sample size to determine allele frequency in the Puerto Rican population. Re-evaluation of mice heterozygous for Mesp2 knockout mutations might also provide clues to a possible mechanism for positive-selection pressure.
In the mouse embryo, Mesp2 expression is first observed in the presomitic mesoderm as a stripe which prefigures the future somite.19 These stripes provide the frame on which the periodic pattern of the vertebrae in the adult spine is built. Mesp2 expression subsequently refines and becomes localized in the future anterior somitic compartment. The rostrocaudal subdivision of somites is critical for the formation of vertebrae, which develop when the caudal half of one somite fuses to the rostral half of the next somite during a process called resegmentation.30 Genetic experiments in the mouse have demonstrated that Mesp2 plays a critical role in the somite-boundary formation and in the specification of the anterior somitic compartment.25,31 Knockout of the Mesp2 gene results in severe segmentation defects of the vertebral column, including proximal fusion of the ribs resembling that observed in patients with STD.19 Our studies suggest that MESP2 mutations also disrupt somitogenesis in humans, resulting in STD and SCD. Conservation of the role of MESP2 between mouse and humans provides further evidence in favor of the conservation of the segmentation clock and the associated segmentation machinery in mammals.
Acknowledgments
P.T., S.E., and E.Y. thank the British Scoliosis Research Foundation and Birth Defects Foundation for funding. P.T. is also grateful to Robert Soper, of New york, for referral of one of the families. We thank J. Michaud and members of the Pourquié Laboratory for critical reading and discussions, and we thank S. Esteban for artwork. Research was supported by the Stowers Institute for Medical Research. O.P. is a Howard Hughes Medical Institute Investigator.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
dbSNP database, http://www.ncbi.nlm.nih.gov/SNP/index.html
Sorting Intolerant From Tolerant (SIFT), http://blocks.fhcrc.org/sift/SIFT.html
UCSC In-Silico PCR, http://genome.ucsc.edu/cgi-bin/hgPcr
NCBI, GenBank and Blast, http://www.ncbi.nlm.nih.gov/ (For hMESP2 [#BC111413])
Ensembl Genome Browser, http://www.ensembl.org/index.html
UniProtKB/Swiss-Prot Protein Knowledge base, http://www.ebi.ac.uk/swissprot/
Accession Numbers
The GenBank accession numbers for the SNPs reported in this paper have been deposited in the dbSNP database under ss no. 99307026–99307034.
Note Added in Proof
Very recently, another nonsense mutation in exon 1 of MESP2, G81X, has been found in a patient with a typical STD phenotype as described in this paper (P.T., S.E., E.Y.). The affected individual is of European descent, from Norway, and homozygous for the mutation. We are grateful to Øivind Braaten and Stefan Kutzsche (Ullevål University Hospital, Oslo) for bringing this case to our attention.
References
- 1.Turnpenny P.D., Alman B., Cornier A.S., Giampietro P.F., Offiah A., Tassy O., Pourquie O., Kusumi K., Dunwoodie S. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev. Dyn. 2007;236:1456–1474. doi: 10.1002/dvdy.21182. [DOI] [PubMed] [Google Scholar]
- 2.Jarcho S., Levin P. Hereditary malformation of the vertebral bodies. Bull. Johns Hopkins Hosp. 1938;62:216–226. [Google Scholar]
- 3.Cornier A.S., Ramirez N., Arroyo S., Acevedo J., Garcia L., Carlo S., Korf B. Phenotype characterization and natural history of spondylothoracic dysplasia syndrome: a series of 27 new cases. Am. J. Med. Genet. 2004;128:120–126. doi: 10.1002/ajmg.a.30011. [DOI] [PubMed] [Google Scholar]
- 4.Lavy N.W., Palmer C.G., Merritt A.D. A syndrome of bizarre vertebral anomalies. J. Pediatr. 1966;69:1121–1125. doi: 10.1016/s0022-3476(66)80304-9. [DOI] [PubMed] [Google Scholar]
- 5.Moseley J.E., Bonforte R.J. Spondylothoracic dysplasia–a syndrome of congenital anomalies. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1969;106:166–169. doi: 10.2214/ajr.106.1.166. [DOI] [PubMed] [Google Scholar]
- 6.Pochaczevsky R., Ratner H., Perles D., Kassner G., Naysan P. Spondylothoracic dysplasia. Radiology. 1971;98:53–58. doi: 10.1148/98.1.53. [DOI] [PubMed] [Google Scholar]
- 7.Gellis, S.S., Feingold, M., and Pashayan, H.M. (1976). Picture of the month: the EEC syndrome. American journal of diseases of children (1960) 130, 653–654. [DOI] [PubMed]
- 8.Solomon L., Jimenez R.B., Reiner L. Spondylothoracic dysostosis: report of two cases and review of the literature. Arch. Pathol. Lab. Med. 1978;102:201–205. [PubMed] [Google Scholar]
- 9.Tolmie J.L., Whittle M.J., McNay M.B., Gibson A.A., Connor J.M. Second trimester prenatal diagnosis of the Jarcho-Levin syndrome. Prenat. Diagn. 1987;7:129–134. doi: 10.1002/pd.1970070209. [DOI] [PubMed] [Google Scholar]
- 10.Schulman M., Gonzalez M.T., Bye M.R. Airway abnormalities in Jarcho-Levin syndrome: a report of two cases. J. Med. Genet. 1993;30:875–876. doi: 10.1136/jmg.30.10.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCall C.P., Hudgins L., Cloutier M., Greenstein R.M., Cassidy S.B. Jarcho-Levin syndrome: unusual survival in a classical case. Am. J. Med. Genet. 1994;49:328–332. doi: 10.1002/ajmg.1320490317. [DOI] [PubMed] [Google Scholar]
- 12.Dequeant, M.L., and Pourquie, O. (2008). Segmental patterning of the vertebrate axis. Nat. Rev. Genet. Published online 15 April 2008, doi:10.1038/nrg2320. [DOI] [PubMed]
- 13.Pourquie O. The segmentation clock: converting embryonic time into spatial pattern. Science. 2003;301:328–330. doi: 10.1126/science.1085887. [DOI] [PubMed] [Google Scholar]
- 14.Bulman M.P., Kusumi K., Frayling T.M., McKeown C., Garrett C., Lander E.S., Krumlauf R., Hattersley A.T., Ellard S., Turnpenny P.D. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 2000;24:438–441. doi: 10.1038/74307. [DOI] [PubMed] [Google Scholar]
- 15.Turnpenny P.D., Whittock N., Duncan J., Dunwoodie S., Kusumi K., Ellard S. Novel mutations in DLL3, a somitogenesis gene encoding a ligand for the Notch signalling pathway, cause a consistent pattern of abnormal vertebral segmentation in spondylocostal dysostosis. J. Med. Genet. 2003;40:333–339. doi: 10.1136/jmg.40.5.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonafe L., Giunta C., Gassner M., Steinmann B., Superti-Furga A. A cluster of autosomal recessive spondylocostal dysostosis caused by three newly identified DLL3 mutations segregating in a small village. Clin. Genet. 2003;64:28–35. doi: 10.1034/j.1399-0004.2003.00085.x. [DOI] [PubMed] [Google Scholar]
- 17.Whittock N.V., Sparrow D.B., Wouters M.A., Sillence D., Ellard S., Dunwoodie S.L., Turnpenny P.D. Mutated MESP2 causes spondylocostal dysostosis in humans. Am. J. Hum. Genet. 2004;74:1249–1254. doi: 10.1086/421053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sparrow D.B., Chapman G., Wouters M.A., Whittock N.V., Ellard S., Fatkin D., Turnpenny P.D., Kusumi K., Sillence D., Dunwoodie S.L. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am. J. Hum. Genet. 2006;78:28–37. doi: 10.1086/498879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saga Y., Hata N., Koseki H., Taketo M.M. Mesp2: a novel mouse gene expressed in the presegmented mesoderm and essential for segmentation initiation. Genes Dev. 1997;11:1827–1839. doi: 10.1101/gad.11.14.1827. [DOI] [PubMed] [Google Scholar]
- 20.Bessho Y., Sakata R., Komatsu S., Shiota K., Yamada S., Kageyama R. Dynamic expression and essential functions of Hes7 in somite segmentation. Genes Dev. 2001;15:2642–2647. doi: 10.1101/gad.930601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koizumi K., Nakajima M., Yuasa S., Saga Y., Sakai T., Kuriyama T., Shirasawa T., Koseki H. The role of presenilin 1 during somite segmentation. Development. 2001;128:1391–1402. doi: 10.1242/dev.128.8.1391. [DOI] [PubMed] [Google Scholar]
- 22.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 23.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrer-Costa C., Gelpi J.L., Zamakola L., Parraga I., de la Cruz X., Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto M., Takahashi Y., Endo M., Saga Y. The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity. Nature. 2005;435:354–359. doi: 10.1038/nature03591. [DOI] [PubMed] [Google Scholar]
- 26.Dimeglio A., Bonnel F. Growth of the Spine. In: AJ R., M C., C D.R., editors. The Pediatric Spine. Springer-Verlag; New York: 1989. pp. 39–83. [Google Scholar]
- 27.Santiago-Cornier A., Ramirez N., Franceschini V., Acevedo J., Roman H., Rosado E., Garcia L., Torres J. Mapping of Spondylothoracic dysplasia (Jarcho-Levin syndrome) to chromosome 2q32.1 in Puerto-Rican population. Am. J. Hum. Genet. 2001;69:514. [Google Scholar]
- 28.van de Vosse E., Ali S., de Visser A.W., Surjadi C., Widjaja S., Vollaard A.M., van Dissel J.T. Susceptibility to typhoid fever is associated with a polymorphism in the cystic fibrosis transmembrane conductance regulator (CFTR) Hum. Genet. 2005;118:138–140. doi: 10.1007/s00439-005-0005-0. [DOI] [PubMed] [Google Scholar]
- 29.Williams T.N., Mwangi T.W., Roberts D.J., Alexander N.D., Weatherall D.J., Wambua S., Kortok M., Snow R.W., Marsh K. An immune basis for malaria protection by the sickle cell trait. PLoS Med. 2005;2:e128. doi: 10.1371/journal.pmed.0020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirsinger E., Jouve C., Dubrulle J., Pourquie O. Somite formation and patterning. Int. Rev. Cytol. 2000;198:1–65. doi: 10.1016/s0074-7696(00)98002-1. [DOI] [PubMed] [Google Scholar]
- 31.Saga Y., Takeda H. The making of the somite: molecular events in vertebrate segmentation. Nat. Rev. Genet. 2001;2:835–845. doi: 10.1038/35098552. [DOI] [PubMed] [Google Scholar]