

Abstract
Branchio-oculo-facial syndrome (BOFS) is a rare autosomal-dominant cleft palate-craniofacial disorder with variable expressivity. The major features include cutaneous anomalies (cervical, infra- and/or supra-auricular defects, often with dermal thymus), ocular anomalies, characteristic facial appearance (malformed pinnae, oral clefts), and, less commonly, renal and ectodermal (dental and hair) anomalies. The molecular basis for this disorder is heretofore unknown. We detected a 3.2 Mb deletion by 500K SNP microarray in an affected mother and son with BOFS at chromosome 6p24.3. Candidate genes in this region were selected for sequencing on the basis of their expression patterns and involvement in developmental pathways associated with the clinical findings of BOFS. Four additional BOFS patients were found to have de novo missense mutations in the highly conserved exons 4 and 5 (basic region of the DNA binding domain) of the TFAP2A gene in the candidate deleted region. We conclude BOFS is caused by mutations involving TFAP2A. More patients need to be studied to determine possible genetic heterogeneity and to establish whether there are genotype-phenotype correlations.
Main Text
Branchio-oculo-facial syndrome (BOFS [MIM 113620]) is a distinctive rare disorder1 of the first and second pharyngeal arches that includes thinned, erythematous cutaneous defects in the cervical or infra- and/or supra-auricular region, ocular anomalies (microphthalmia or anophthalmia, cataract, coloboma, strabismus, ptosis), and nasolacrimal duct obstruction. The characteristic craniofacial features are dolichocephaly, malformed pinnae, thick nasal tip, up-slanted eyes, and cleft lip (CL) (often a lesser form described as a microform, “pseudocleft,” or abnormal philtrum) with or without cleft palate (CP). Other common findings are conductive hearing loss, ectodermal anomalies (small teeth, dysplastic nails, sparse, prematurely gray hair), ectopic dermal thymus, and scalp cysts. Less frequent findings are renal anomalies, growth restriction, upper lip pits, and mild mental retardation. Autosomal-dominant inheritance is well documented.1 Given the clinical overlap with branchio-oto-renal syndrome, Kaiser et al. used a candidate-gene approach to exclude most genes in the EYA-DACH-SIX-PAX pathway,2 although a shared 37.37 Mb haplotype at chromosome 6p21.31-p25.3 was found. We studied five families (European ancestry) in which probands demonstrated all three BOFS features (cervical skin defects, ocular anomalies, facial anomalies [five patients]) or two features and a first-degree affected relative (one patient). Institutional Review Board (IRB) Research informed consent and permission for publication of photos was signed by each participant and/or parent. Clinical features and pictures of each patient are shown in Table 1.
Table 1.
Molecular and Clinical Findings in BOFS Patients
| Patient | 1a | 1b | 2a | 3 | 4 | 5b |
|---|---|---|---|---|---|---|
| Picture |  |
 |
 |
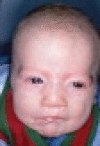 |
 |
 |
| DNA mutation | Del 6p24.3 region | Del 6p24.3 region | g. 10529 A → G | g. 10512 T → C | g. 10526 G → A | g. 12448 C → T |
| Exon | 4 | 4 | 4 | 5 | ||
| Protein consequence | R255G | L249P | R254G | G262E | ||
| De novo? | U | No | Yes | Yes | Yes | Yes |
| Sex | F | M | F | M | M | F |
| Family history | + | + | − | − | − | − |
| Age (years) | 25 | 1 | 18 | 2 | 14 | 17 |
| Branchial (Pharyngeal Arch) Anomalies | ||||||
| Cervical cutaneous anomaly | + R | + B | + B | + R | + B | + B |
| Dermal thymus | U | U | + | − | − | − |
| Ocular Anomalies | ||||||
| Coloboma | − | + R (iris, optic nerve, retina) | + R (iris) | − | + R | + B (iris, retina) |
| Microphthalmia | − | + R | + R | − | + R | + B |
| Nasolacrimal duct stenosis | − | − | + B | + B | + B | − |
| Facial Anomalies | ||||||
| Auricular malformation | + | + | + | + | + | + |
| Characteristic face | − | + mild | + | + | + | + |
| Chin dimple or cleft | − | − | Dimple | Dimple | − | Cleft |
| Cleft lip and palate | − | − (short tented philtrum) | CL and P, B | CL, B (minimicroform) | CL, B (microform) | CL, B |
| Facial nerve palsy | − | − | − | + Right | − | − |
| Preauricular pits | − | − | − | + | + B | + R |
| Additional Features | ||||||
| CHD | U | U | − | − | U | − |
| Dental anomalies | − | − | + | − | + | + |
| Growth restriction | − | − | + (prenatal and postnatal) | − | + (prenatal) | + (prenatal and early childhood) |
| Hearing loss | − | + B, R > L | + B | + | + | + B |
| Kidney anomalies | − | − | − | − | U | + Right (multicystic dysplastic, VUR) |
| Lip pits | − | − | − | − | − | − |
| Nail anomalies | − | − | − | − | + | + |
| Prematurely gray hair | + | − | − | − | + | − |
| Scalp cysts | − | + | − | − | − | − |
| Sparse hair | − | − | − | − | − | + |
| New Features | ||||||
| Anxiety, hypoplasia left breast, precocious puberty | Depression, medulloblastoma | |||||
The following abbreviations are used: +, present; −, absent; B, bilateral; CHD, congenital heart defect; CL, cleft lip; CP, cleft palate; CL and P, cleft lip and palate; F, female; L, left; LDs, learning disabilities; M, male; R, right; U, unknown; and VUR, vesicoureteral reflux.
Genome-wide microarrays have proven useful in the identification of genetic regions that are either deleted or duplicated in specific malformation syndromes. These genomic alterations, even when found in a small percentage of cases, can significantly narrow the candidate region and allow successful discovery of the gene (e.g., CHD7 [MIM 608892] in CHARGE syndrome [MIM 214800]).4 We used the 500K SNP Affymetrix microarray to screen two sporadic BOFS patients and one affected mother and son pair for cryptic chromosomal aberrations. Genomic DNA was extracted with the AUTOPURE automated DNA extractor according to manufacturer's instructions (Gentra Systems, Minneapolis, MN). The 500K assay (Affymetrix, Santa Clara, CA) consists of two 250K arrays and was performed according to manufacturer's protocol. Copy-number analysis was performed with the Affymetrix GeneChip Genotyping Analysis software. This program combines the two 250K arrays into a single virtual chip that can be viewed for copy number. The arrays were compared against a reference set of 25 previously defined normal HapMap samples. A 3.2 Mb deletion was detected in the mother and son pair at chromosome 6p24.3 (Figure 1) in the previously implicated region harboring nine genes (based on UCSC Genome Browser Human March 2006 Assembly; Table 2). The two sporadic BOFS patients (BOFS pt. 2 and 3) had no copy-number alterations in this region. Five polymorphic dinucleotide repeat markers were selected (three flanking and two from within the detected deleted region) from the Genethon human linkage map HD-5 and MD-10 Prism Linkage Mapping Set v2.5 (Applied Biosystems, Foster City, CA.) and confirmed the chromosome 6 deletion (Figure 2A). We then employed Multiplex Ligation-dependent Probe Amplification (MLPA) to assess whether the Activating enhancer-binding protein 2 alpha (TFAP2A [MIM 107580]) gene was included in the deleted region. A specific MLPA kit was not available to confirm the copy-number changes detected by the 500K microarray. Two synthetic MLPA probes (from within exons 5 and 6 of the TFAP2A gene) were designed according to detailed methodology (Medical Research Council [MRC], Holland). The synthetic probes were added to an existing MLPA kit and assayed according to standard protocols (MRC, Holland). The results were analyzed with GENEMARKER (SoftGenetics, State College, PA) and confirmed the presence of a chromosome 6 deletion that included TFAP2A (Figure 2B). Larger chromosomal deletions including this region have been reported previously in patients with cleft lip and palate.5 Although neither the affected mother nor her son with BOFS in this report had overt cleft lip and palate (CL and P), the boy does have an abnormally short philtrum and bilateral notched vermilion-mucosa border, which are on the spectrum of microform CL in BOFS. Donnai et al. reported three members of a family with a balanced translocation t(6;9)(p23;q22.3) who had CL and P, malformed pinnae, nasolacrimal duct obstruction, up-slanting palpebral fissures, and premature graying, but without the other typical BOFS facial gestalt and cutaneous anomalies.5 Davies et al. described a boy with a larger deletion of 6p24-p25 and multiple congenital anomalies (no CL and P); although a photograph was not shown, the findings are not those of BOFS.6 It remains unknown whether the additional genes in the deleted region (Table 2) have impacted the phenotype of family 1. None of the genes have individually been associated with dominantly inherited or deletion phenotypes.
Figure 1.
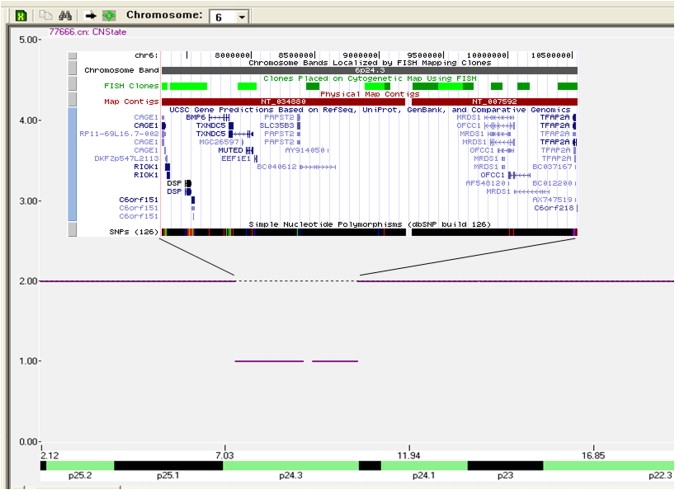
500K SNP Microarray in BOFS Family 1
A 3.2 Mb deletion of 704 SNPs at 6p24.3 is shown with the UCSC genes inserted above the deletion.
Table 2.
Genes Involved in Chromosome 6p24.3 Deletion in BOFS Family 1
| Gene Symbol | Gene Name | Gene Function | Expression |
|---|---|---|---|
| BMP6 | bone morphogenetic protein 6 preproprotein | Induces cartilage and bone formation | Multiple tissues |
| CAGE1 | cancer antigen1 | Unknown | Connective tissue; Prostate; Testis |
| DSP | desmoplankin | Involved in the organization of the desmosomal cadherin-plakoglobin complexes into discrete plasma-membrane domains and in the anchoring of intermediate filaments to the desmosomes | Ubiquitous |
| EEF1E1 | eukaryotic translation elongation factor 1 | Positive modulator of ATM response to DNA damage | Ubiquitous |
| OFCC1 | orofacial cleft 1 candidate 1 | Unknown | Testis |
| RIOK1 | RIO Kinase1 | Unknown | Ubiquitous |
| SLC35B3 | solute carrier family 35, member B3 | Probable sugar transporter (by similarity) | Ubiquitous |
| TFAP2A | transcription factor AP-2 alpha | See text | See text |
| TXNDC5 | thioredoxin domain containing 5 | Possesses thioredoxin activity reducing insulin disulfide bonds | Ubiquitous |
Figure 2.

Chromosome 6p24.3 Deletion in BOFS Family 1
(A) Microsatellite marker D6S309 showing absence of maternal allele (BOFS patient 1a) in her affected child (BOFS patient 1b) confirming the chromosome 6p deletion.
(B) MLPA confirmation of TFAP2A gene deletion with two different intragenic probes (exons 5 and 6).
Candidate genes in the deleted region were selected for sequencing on the basis of their reported expression patterns and known involvement in developmental pathways associated with the clinical findings of BOFS (GeneCards, Weizmann Institute of Science, Israel). Our mutation-detection strategy was to polymerase chain reaction (PCR) amplify and sequence the coding exons and the intron-exon boundaries of each selected gene in the region. The method used to sequence the gene utilizes the ABI VariantSEQr Resequencing system (Applied Biosystems, Foster City, CA). Primer sequences for the generation of amplicons were derived from the NCBI Gene website. The mutation analysis was performed with the Mutation Surveyor Program (SoftGenetics, State College, PA). No mutations were found in BMP6 (MIM 112256), OFCC1, and SLC35B3 (MIM 610845). TFAP2A was among the first four genes sequenced in the remaining four sporadic BOFS patients. Deletions of the chromosomal region, which include this gene, have previously implicated TFAP2A as causing anterior ocular chamber anomalies.6 More recently, the TFAP2A gene has been shown to bind to a regulatory element of IRF6 (MIM 607199) involved in van der Woude syndrome (MIM 119300) (Rahimov et al. ASHG 57th meeting A87, 2007). This is intriguing because van der Woude syndrome is characterized by CL and P with lower lip pits, whereas only upper lip pits are seen in BOFS.
The AP-2 family of transcription factors bind to the DNA consensus sequence GCCNNNGGC and stimulate target-gene transcription, thus regulating gene expression during embryogenesis of the eye, ear, face, body wall, limbs, and neural tube.7–10 TFAP2A knockout mice exhibit abnormal neural-crest-derived facial structures.11,12 Specifically, this gene has been shown to regulate the development of the facial prominences, limb buds, cranial closure, and lens vesicle.7,13 Furthermore, Feng et al. have identified a conserved Tcfap2a intronic enhancer element required for expression in facial and limb-bud mesenchyme in mice.14
TFAP2A contains 437 amino acids and is a retinoic-acid-responsive gene. TFAP2A has a central basic DNA binding region, a carboxy terminus helix-span-helix motif that mediates dimerization, and an amino terminus that contains a transactivation domain.15
De novo missense mutations were found in exons 4 and 5 (basic region of the DNA binding domain) of the TFAP2A gene in the remaining four BOFS individuals (Table 1 and Figure 3; paternity proven). The mutated amino acids are highly conserved through the transparent sea squirt (Ciona Intestinalis; Figure 4). The mutations detected by sequencing all occurred at natural restriction-enzyme sites. Restriction digestion was performed with the appropriate enzyme (New England Biolabs, Beverly, MA) specific to each mutation. After the four sequencing mutations were confirmed, this method was used to screen 300 normal samples from individuals of similar ethnicity. The four sequencing mutations are not in the SNP database and were not found in more than 300 normal individuals. The L249P alteration (BOFS pt. 3) results in a predicted conformational space change with the substituted proline. The R254W (BOFS pt. 4) and R255G (BOFS pt. 2) alterations result in replacement of a charged polar side chain by a nonpolar side chain with a predicted conformational space change. The G262E alteration (BOFS pt. 5) results in a nonpolar side chain being replaced by a charged polar side chain. These four alterations are predicted to be probably damaging by PolyPhen and not tolerated by SIFT.
Figure 3.
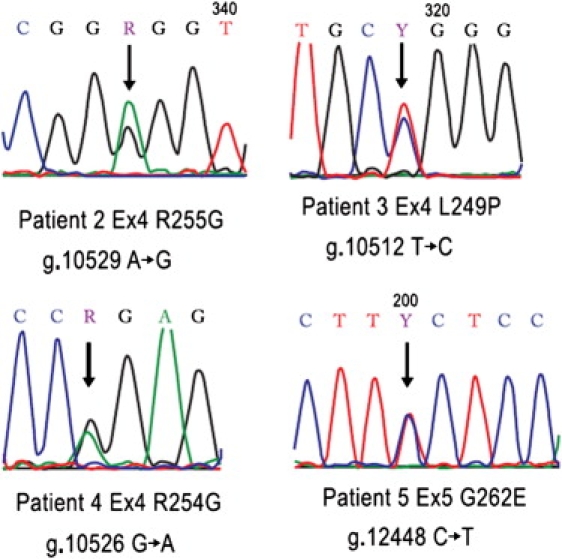
Sequence Chromatogram of TFAP2A Missense Mutations
Numbering is according to NCBI: NC_000006.
Figure 4.

Evolutionary Conservation of the Amino Acids in the TFAP2A Gene Altered in BOFS Patients
Note the highly conserved amino acids from Homo sapiens through Ciona intestinalis in exons 4 and 5 of the TFAP2A gene.
Given the variable expressivity of BOFS, it will be necessary to study both classically affected cases having all three features as well as those with minor phenotypes. With strict inclusion criteria, some form of oral cleft was present in all BOFS patients in the largest review of 43 patients (54% microform CL).1 A deletion found in family 1 without CL and P suggests that additional genotype-phenotype studies are needed to determine whether this is a consistent observation and to determine the frequency of deletions in BOFS.
BOFS patient 5 is the first reported with medulloblastoma. TFAP2A has been shown to be involved in tumorigenesis with protein expression levels affecting cell transformation, tumor growth, metastasis, and survival.16–18 The tumor-suppressor activity of TFAP2A is mediated through a direct interaction with p53 (MIM 191170), altering its transcriptional activity and stability.19,20 In addition, TFAP2A suppresses the MYC (MIM 190080) oncogene.21 Both MYC and p53 are involved in medulloblastoma. Expression studies on medulloblastoma tumor tissue from patient 5 are planned.
As young teenagers, BOFS patients 2 and 5 had anxiety and depression, respectively. Although this may be related to the psychosocial context of having a craniofacial disorder, the AP-2 family may be involved in the regulation of the monoaminergic systems in the adult brain, resulting in neuropsychiatric disorders.22
We conclude that mutations involving TFAP2A result in BOFS. More patients are needed to investigate genetic heterogeneity. Binding partners as well as other members of the AP-2 family would be ideal candidates. The TFAP2A gene appears to be responsible for all aspects of the recognized phenotype. In family 1, the deletion phenotype may be milder (lacking classic CL and P) because of haploinsufficiency of the gene or contiguous modifier and/or enhancer genes. Hence, studies of additional BOFS patients are necessary to establish whether there are any genotype-phenotype correlations. Previous linkage studies have implicated the TFAP2A gene region in nonsyndromic CL and P.23–25 In a timely review of murine genetic models of CL and P, TFAP2A was one of several previously unexamined genes predicted to be a candidate gene for nonsyndromic CL and P.26 By demonstrating that TFAP2A plays an etiologic role in a CL and P syndrome, BOFS, its possible role in nonsyndromic CL and P, especially lesser forms of cleft lip, should be considered.
Acknowledgments
We thank the families who supported this project, as well as T. Erb and M. Kolthoff for assistance in examining patients. We also thank C. Baldwin and A. Milunsky for their critical review of this manuscript.
Web Resources
The URLs for data presented herein are as follows:
GeneCards, http://www.genecards.org/
MRC Holland, http://www.mrc-holland.com/pages/indexpag.html
NCBI dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Lin A.E., Gorlin R.J., Lurie I.W., Brunner H.G., van der Burgt I., Naumchik I.V., Rumyantseva N.V., Stengel-Rutkowski S., Rosenbaum K., Meinecke P. Further delineation of the branchio-oculo-facial syndrome. Am. J. Med. Genet. 1995;56:42–59. doi: 10.1002/ajmg.1320560112. [DOI] [PubMed] [Google Scholar]
- 2.Kaiser R., Guillen Posteguillo E., Muller D., Just W. Exclusion of genes from the EYA-DACH-SIX-PAX pathway as candidates for branchio-oculo-facial syndrome (BOFS) Am. J. Med. Genet. 2007;143A:2185–2188. doi: 10.1002/ajmg.a.31875. [DOI] [PubMed] [Google Scholar]
- 3.Lin A.E., Losken H.W., Jaffe R., Biglan A.W. The branchio-oculo-facial syndrome. Cleft Palate Craniofac. J. 1991;28:96–102. doi: 10.1597/1545-1569_1991_028_0096_tbofs_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 4.Vissers L., van Ravenswaaij C., Admiraal R., Hurst J., de Vries B., Janssen I., van der Vliet W., Huys E., de Jong P., Hamel B. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 5.Donnai D., Heather L.J., Sinclair P., Thakker Y., Scambler P.J., Dixon M.J. Association of autosomal dominant cleft lip and palate and translocation 6p23;9p22.3. Clin. Dysmorphol. 1992;1:89–97. [PubMed] [Google Scholar]
- 6.Davies A.F., Mirza G., Flinter F., Ragoussis J. An interstitial deletion of 6p24-p25 proximal to the FKHL7 locus and including AP-2α that affects anterior eye chamber development. J. Med. Genet. 1999;36:708–710. [PMC free article] [PubMed] [Google Scholar]
- 7.Schorle H., Meier P., Buchert M., Jaenisch R., Mitchell P.J. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–238. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J., Hagopian-Donaldson S., Serbedzija G., Elsemore J., Plehn-Dujowich D., McMahon A.P., Flavell R.A., Williams T. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 1996;381:238–241. doi: 10.1038/381238a0. [DOI] [PubMed] [Google Scholar]
- 9.Ahituv N., Erven A., Fuchs H., Guy K., Ashery-Padan R., Williams T., de Angelis M.H., Avraham K.B., Steel K.P. An ENU-induced mutation in AP-2α leads to middle ear and ocular defects in Doarad mice. Mamm. Genome. 2004;15:424–432. doi: 10.1007/s00335-004-2334-z. [DOI] [PubMed] [Google Scholar]
- 10.Nelson D.K., Williams T. Frontonasal process-specific disruption of AP-2α results in postnatal midfacial hypoplasia, vascular anomalies, and nasal cavity defects. Dev. Biol. 2004;267:72–92. doi: 10.1016/j.ydbio.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 11.Morris-Kay G. Craniofacial defects in AP-2 null mutant mice. Bioessays. 1996;18:785–788. doi: 10.1002/bies.950181004. [DOI] [PubMed] [Google Scholar]
- 12.Nottoli T., Hagopian-Donaldson S., Zhang J., Perkins A., Williams T. Ap-2 null cells disrupt morphogenesis of the eye, face, and limbs in chimeric mice. Proc. Natl. Acad. Sci. USA. 1998;95:13714–13719. doi: 10.1073/pnas.95.23.13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West-Mays J.A., Zhang J., Nottoli T., Hagopian-Donaldson S., Libby D., Strissel K.J., Williams T. AP-2α transcription factor is required for early morphogenesis of the lens vesicle. Dev. Biol. 1999;206:46–62. doi: 10.1006/dbio.1998.9132. [DOI] [PubMed] [Google Scholar]
- 14.Feng W., Huang J., Zhang J., Williams T. Identification and analysis of a conserved Tcfap2a intronic enhancer element required for expression in facial and limb bud mesenchyme. Mol. Cell. Biol. 2008;28:315–325. doi: 10.1128/MCB.01168-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckert D., Buhl S., Weber S., Jager R., Schorle H. The AP-2 family of transcription factors. Genome Biol. 2005;6:246. doi: 10.1186/gb-2005-6-13-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jean D., Gershenwald J.E., Huang S., Luca M., Hudson M.J., Tainsky M.A., Bar-Eli M. Loss of AP-2 results in up-regulation of MCAM/MUC18 and an increase in tumor growth and metastasis of human melanoma cells. J. Biol. Chem. 1998;273:16501–16508. doi: 10.1074/jbc.273.26.16501. [DOI] [PubMed] [Google Scholar]
- 17.Heimberger A.B., McGary E.C., Suki D., Ruiz M., Wang H., Fuller G.N., Bar-Eli M. Loss of the Ap-2alpha transcription factor is associated with the grade of human gliomas. Clin. Cancer Res. 2005;11:267–272. [PubMed] [Google Scholar]
- 18.Orso F., Fassetta M., Penna E., Solero A., De Filippo K., Sismondi P., De Bortoli M., Taverna D. The AP-2alpha transcription factor regulates tumor cell migration and apoptosis. Adv. Exp. Med. Biol. 2007;604:87–95. doi: 10.1007/978-0-387-69116-9_6. [DOI] [PubMed] [Google Scholar]
- 19.McPherson L.A., Loktev A.V., Weigel R.J. Tumor suppressor activity of AP2alpha mediated through a direct interaction with p53. J. Biol. Chem. 2002;277:45028–45033. doi: 10.1074/jbc.M208924200. [DOI] [PubMed] [Google Scholar]
- 20.Stabach P.R., Thiyagarajan M.M., Woodfield G.W., Weigel R.J. AP2alpha alters the transcriptional activity and stability of p53. Oncogene. 2006;25:2148–2159. doi: 10.1038/sj.onc.1209250. [DOI] [PubMed] [Google Scholar]
- 21.Gaubatz S., Imhof A., Dosch R., Werner O., Mitchell P., Buettner R., Eilers M. Transcriptional activation by Myc is under negative control by the transcription factor AP-2. EMBO J. 1995;14:1508–1519. doi: 10.1002/j.1460-2075.1995.tb07137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Damberg M. Transcription factor AP-2 and monoaminergic functions in the central nervous system. J. Neural Transm. 2005;112:1281–1296. doi: 10.1007/s00702-005-0325-1. [DOI] [PubMed] [Google Scholar]
- 23.Prescott N.J., Lees M.M., Winter R.M., Malcolm S. Identification of susceptibility loci for nonsyndromic cleft lip with or without cleft palate in a two stage genome scan of affected sib-pairs. Hum. Genet. 2000;106:345–350. doi: 10.1007/s004390051048. [DOI] [PubMed] [Google Scholar]
- 24.Schultz R.E., Cooper M.E., Daack-Hirsch S., Shi M., Nepomuncena B., Graff K.A., Obrien E.K., Obrien S.E., Marazita M.L., Murray J.C. Targeted scan of fifteen regions for nonsyndromic cleft lip and palate in Fillipino families. Am. J. Med. Genet. A. 2004;125:17–22. doi: 10.1002/ajmg.a.20424. [DOI] [PubMed] [Google Scholar]
- 25.Moreno L.M., Arcos-Burgos M., Marazita M.L., Krahn K., Maher B.S., Cooper M.E., Valencia-Ramirez C.R., Lidral A.C. Genetic analysis of candidate loci in non-syndromic cleft lip families from Antiquia-Colombia and Ohio. Am. J. Med. Genet. A. 2004;125:135–144. doi: 10.1002/ajmg.a.20425. [DOI] [PubMed] [Google Scholar]
- 26.Juriloff D.M., Harris M.J. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. A. Clin. Mol. Teratol. 2008;82:63–77. doi: 10.1002/bdra.20430. [DOI] [PubMed] [Google Scholar]
- 27.Lin A.E., Semina E.V., Daack-Hirsch S., Roeder E.R., Curry C.J., Rosenbaum K., Weaver D.D., Murray J.C. Exclusion of the branchio-oto-renal syndrome locus (EYA1) from patients with branchio-oculo-facial syndrome. Am. J. Med. Genet. 2000;91:387–390. doi: 10.1002/(sici)1096-8628(20000424)91:5<387::aid-ajmg13>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Lin A.E., Doherty R., Lea D. Branchio-oculo-facial and branchio-oto-renal syndromes are distinct entities. Clin. Genet. 1992;41:221–223. doi: 10.1111/j.1399-0004.1992.tb03667.x. [DOI] [PubMed] [Google Scholar]