

Abstract
Several dysmorphic syndromes affect the development of both the eye and the ear, but only a few are restricted to the eye and the external ear. We describe a developmental defect affecting the eye and the external ear in three members of a consanguineous family. This syndrome is characterized by ophthalmic anomalies (microcornea, microphthalmia, anterior-segment dysgenesis, cataract, coloboma of various parts of the eye, abnormalities of the retinal pigment epithelium, and rod-cone dystrophy) and a particular cleft ear lobule. Linkage analysis and mutation screening revealed in the first exon of the NKX5-3 gene a homozygous 26 nucleotide deletion, generating a truncating protein that lacked the complete homeodomain. Morpholino knockdown expression of the zebrafish nkx5-3 induced microphthalmia and disorganization of the developing retina, thus confirming that this gene represents an additional member implicated in axial patterning of the retina.
Main Text
Whereas the development of the eye is initiated by the bilateral evagination of the telencephalon followed by inductive events of the neural ectoderm on the budding optic vesicle, the ear derives from an ectodermal patch of cells that sinks into the mesenchyme to form the otic vesicle. In the eye, sequential expression of homeotic genes including, among others, members of the PAX, SIX, OTX, CHX10 (MIM 142993), and EMX families governs this morphogenetic program.1 The development of the ear is by no means a less complicated process: The sequential expression of patterning genes determines the cochlear versus vestibular fate of the otic anlagen. In vertebrates, expression of homeotic genes including Dlx3, mshC, mshD, Sox9, and BMP7 in the dorso-lateral region of the otic placode triggers the development of the vestibular system, and the expression in the ventro-medial region of Pax2, Dlx4, Fgf2, Fgf3, BMP4, and Notch is accompanied by the development of the auditory sensory organ.2
Here, we describe a consanguineous family originating from Switzerland in which three members (two males and one female) out of 14 sibs born to two consanguineous healthy couples sharing a common ancestor were diagnosed with, to our knowledge, a new oculo-auricular syndrome (Figure 1A). The proband (IX.2) first examined at 2 months of age presented congenital nystagmus, bilateral microcornea, posterior synechiae, and cataract in both eyes, colobomatous microphthalmia of the right eye (OD), and anterior segment dysgenesis consisting of incomplete coloboma of the iris, stromal iris cyst (OD), and irido-corneal adherences in the left eye (OS) (Figure 1B). Fundus examination revealed dysplastic macropapillae reminiscent of Morning Glory syndrome, macular hypoplasia, and peripheral infero-nasal chorioretinal coloboma (Figure 1C). The rapidly progressive cataract required surgery at 11 months of age. At 7 yr of age, ophthalmoscopy revealed the presence of circumferential abnormalities of the retinal pigment epithelium and chorioretinal atrophic lacunae at the equator. Full-field electroretinogram documented a rod-cone dystrophy. The ear showed lobule aplasia (Figure 1D), a narrow intertragic incisure, and an abnormal bridge connecting the crus of helix and the anthelix resulting in complete separation between the cymba and the conqua. The external acoustic meatus was narrow, and the ear drum was small but had a normal appearance on both sides. Audiogram and vestibular function were normal.
Figure 1.
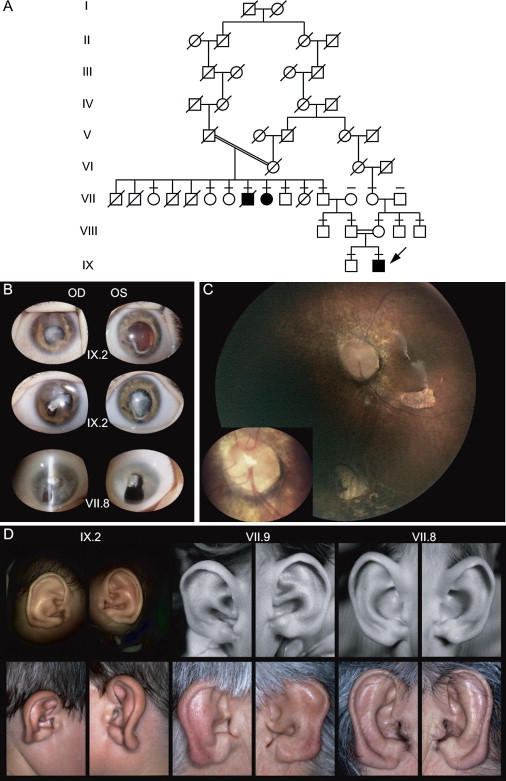
Family with Oculo-Auricular Syndrome
(A) Pedigree of consanguineous family with autosomal-recessive oculo-auricular syndrome. Affected individuals are indicated by black symbols, and the proband is indicated by an arrow. Individuals with line above symbol have been tested for mutation.
(B) Eyes of two affected patients. Top: Eyes of the proband (IX.2) showing an early cataract, coloboma, and microphthalmia of OD (15.5 mm); OS was borderline (16.5 mm). At age 7 yr, best corrected visual acuity was 0.05 in OD and 0.16 in OS; corneal thickness was 770 μm OD and 710 μm OS because of a diffuse cornea guttata. Middle: Eyes of the proband a few months later with advanced cataract. Bottom: Eyes of VII.8 presenting cataract and coloboma.
(C) Fundus photography of OS (Retcam II), showing the macular hypoplasia, inferior chorio-retinal coloboma, retinal dysplasia, and Morning Glory-like optic nerve head, with a magnified view of the latter (left corner).
(D) External ear of the three affected patients at two different ages. The three ear pinnas present a deformation of the lobule. Left: Patient IX.2. Middle: Patient VII.9. Right: Patient VII.8.
The two other patients were first examined 60 yr ago, when they were 3 (VII.9) and 6 (VII.8) yr old.3 Patient VII.8 had horizontal nystagmus, microphthalmia, sclerocornea, and vision limited to light perception. Patient VII.9 had nystagmus, microcornea (8 mm), microphthalmia, typical iris coloboma, cataract, and microphakia with vision of 1/60 in each eye (OU). The fundus was not visible in VII.9 but in VII.8, an inferior chorioretinal coloboma (OD), and lacunae (OS) were observed. Ear examination showed striking external-ear findings in both patients (Figure 1D). Painful complications led to the enucleation of OS in VII.9; the patient suffered partially calcified phtisis bulbi with total retinal detachment (OD). VII.8 conserved normal eye tension and vision through his inferior iris coloboma until his last visit, when he was 65 yr old, despite progression toward a total cataract with calcifications. There were neither additional dysmorphic signs nor abnormal systemic findings except for three maxillary dental rows in VII.8, as well as spina bifida occulta and moderate dyscrania with flattening of the cranial base and short mandibular rami in VII.9. In addition, orbito-cerebral computed tomography (CT) and magnetic resonance imagine (MRI) including the middle and inner ear performed in two of the three patients (VII.9 and IX.2) were within normal range.
Because the inheritance pattern was suggestive of a recessive disorder, we performed homozygosity mapping with dinucleotide markers from the ABI Prism Linkage Mapping Set 2.5. This study was approved by the Ethics Committee of the Faculty of Medicine of the University of Lausanne. A unique homozygous region on chromosome 4p16 and flanked by D4S2935 and D4S391 was identified. With MLINK, linkage analysis for these and additional markers provided a maximum LOD score of 3.37 at theta = 0 for D4S2906 (Figure 2A). Further single-nucleotide polymorphism (SNP) analysis reduced the homozygous interval to a region of less than 10 Mb between D4S2935 and D4S419 (Figure 2B). Because of the developmental defects present in the three patients, we concentrated our efforts on homeobox genes and genes expressed in the eye and sequenced the coding regions of all of them, but without identifying any base changes associated with pathogenesis. OMIM and a literature search indicated that HMX1 (MIM 142992) was another transcription factor that maps to the short arm of chromosome 4, although its precise location in the human genome had not been clearly established (build 36.1). HMX1, also known as NKX5-3, is expressed in the eye and ear.4 Using gene-specific primers (Table S1 available online), we amplified two exons from genomic DNA isolated from control individuals. The amplified gene encoded a putative protein that comprised 347 amino acids and contained the classical homeobox domain (HD) of all NKX5 members. However, it was significantly different from the published human HMX1, with 50% protein identity in the N-terminal part and only 24% identity at the C terminus (accession number M99587).4 Recently, this entry was modified to incorporate the new translation (accession number NM_018942.2, GI:116805349).
Figure 2.
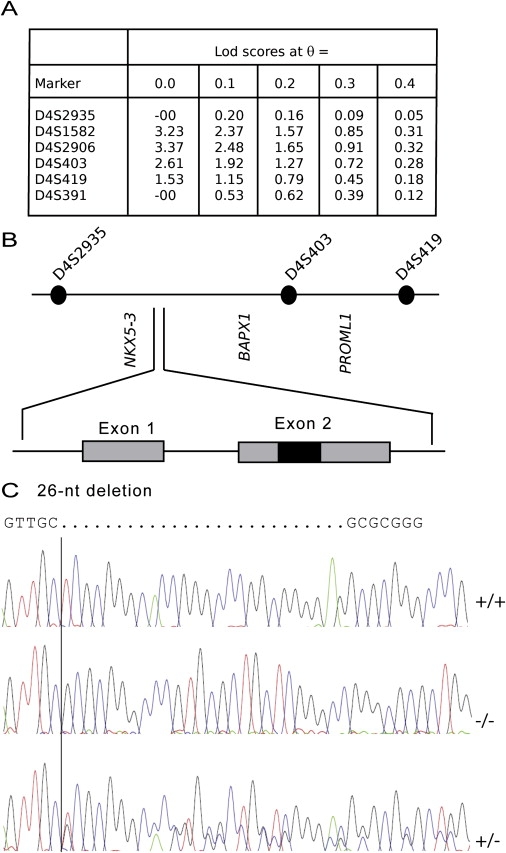
Linkage Analysis, Detailed Map of NKX5-3, and Mutation Analysis
(A) Linkage analysis with microsatellites from chromosome 4.
(B) The 11.9 Mb candidate region on chromosome 4p16.1 containing several candidate genes including NKX5-3 and genomic organization of NKX5-3. The coding region comprises two exons (shaded boxes); the homeodomain is designed by a black box.
(C) DNA sequence of NKX5-3 in a normal control (+/+), one heterozygous parent (VIII.2) (+/−), and the homozygous proband (IX.2) (−/−). The homozygous deletion was also found in patients VII.8 and VII.9.
Sequence analysis of this gene in the three affected patients showed a homozygous deletion of 26 nucleotides (c.215-240 del) resulting in a frameshift and the generation of a putative stop codon at amino acid position 112 (Figure 2C). This deletion abolished the translation of the homeobox. Both parents of patient IX.2 were heterozygous for the deletion, and no member of the family, other than the three patients, was homozygous for the deletion. This deletion was not found in more than 250 control individuals from the same ethnic group or from more than 500 patients with various eye diseases, including inherited rod-cone, cone-rod and macular dystrophies, coloboma, and other developmental defects (WAVE conditions described in Table S1).
In vertebrates, the NKX5 homeodomain family of genes comprises four members, NKX5-1, NKX5-2 (MIM 600647) NKX5-3—also known as HMX3, HMX2, and HMX1, respectively—and SOHO1.5,6 These genes probably arose from a tandem duplication followed by a chromosomal duplication leading to the formation of two clusters.7 One of these clusters contains NKX5-3 and SOHO1, the other NKX5-1 and NKX5-2. Whereas the latter cluster was found in many species, including on chromosome 7 of Mus musculus and on 10q26.3 in human, SOHO1 has only been reported in Gallus gallus (chicken), Oryzias latipes (medaka), and Takifugu rubripes (pufferfish), so far. Interestingly, in medaka, olnkx5-1 has further been duplicated into olnkx5-1.1 and olnkx5-1.2.7
In order to confirm that the identified sequence was NKX5-3, we tested its expression in human and mouse eyes and ears. We used tissue sections from embryonic mice at stages E10.5 to E18.5, sections from adult mice, and human fetal ocular and auricular tissue sections collected from embryos and fetuses. Morphologically normal human embryos and fetuses resulting from legal interruptions of pregnancies performed for medical reasons at five (n = 2), six (n = 2), and 20 (n = 2) weeks were legally provided by the departments of pathology of three French University Hospitals (Dr. P. Sarda, Montpellier; Dr. A.-L. Delezoide, Paris; and Dr. B. Gasser, Strasbourg) after approval by the Ethics Committee of the Necker-Enfants Malades Hospital (Paris, France). Hybridization was performed as followed: Two 60-mer oligonucleotide probes, located outside of the homeobox domain, were synthesized by Genset (France) and 3′ end-labeled with [alpha-35S]dATP (NEN) and terminal deoxyribonucleotidyl transferase (15 U/mL; Invitrogen-GIBCO) at a specific activity of ∼7 × 108 cpm/mg. The sequences of the human and mouse antisense NKX5-3 probes are listed in Table S1. The sense probes were used as controls.
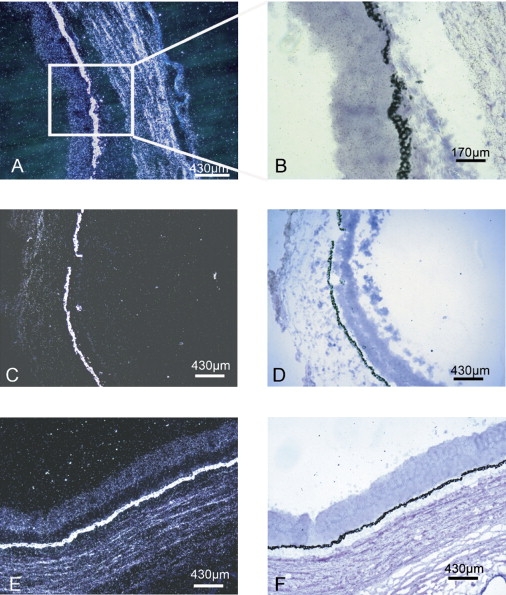
Mouse Nkx5-3 was expressed as early as E13.5 in the external ear, lens, and retina. In the latter, it showed clear polarized expression, whereas no signal was observed for the sense probe (Figure 3). A similar polarized expression was observed in the eye of 5- and 6-week-old human embryos with signals observed in the temporal and posterior retina (Figures 3 and 4). The pinna or auricule of 20-week-old fetuses did not display any NKX5-3 mRNA labeling in the developing cartilage per se. However, an intense hybridization signal was consistently detected in the perichondrium surrounding all the intrinsinc cartilages of the developing pinna. The developing auricular mesenchymatous cells, including those of the developing intrinsic auricular ligaments, contained significant levels of NKX5-3 transcripts (Figure 5). No signal was observed for the sense probe.
Figure 3.
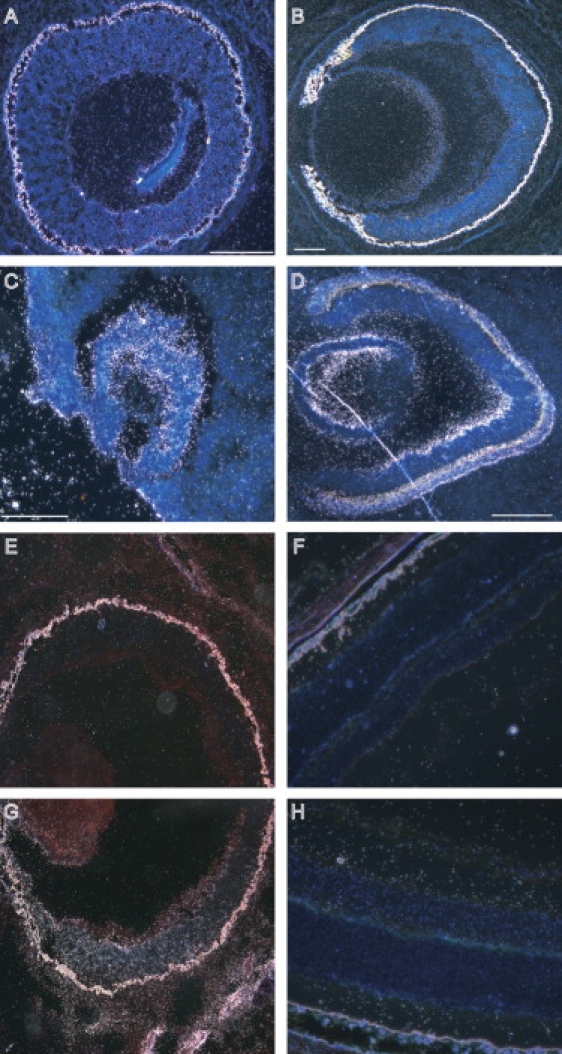
Expresssion of NKX5-3 in Eye
(A and B) Mouse embryo eyes at 13.5 and 18.5 days of gestation.
(C and D) Human embryo eyes at 5 and 6 weeks of gestation.
(E–H) Adult mouse retina.
High levels of transcripts were detected in the neural and pigment layers of retina (A). At 18.5 days, expression was found in the inner nuclear layer, the ganglion cell layer, and the lens (B). In human, NKX5-3 is expressed in the optic vesicle at 5 weeks of gestation (C) and at the ventral retina and anterior portion of the lens at 6 weeks (D). No expression was observed in the dorsal human retina (D). In the mouse retina, expression is observed at inner and ganglion cell layers of the ventral retina (G and H) but not at the dorsal retina (E and F).
Figure 4.
Expression of NKX5-3 in 20-Week-Old Human Eye
At this period, NKX5-3 displayed an asymmetric pattern of expression.
(A and B) Dark- and bright-field aspects of the labeled temporal retina, respectively.
(C and D) Dark- and bright-field aspects of the nasal retina, respectively, showing no expression.
(E and F) Dark- and bright-field aspects of the posterior retina, respectively, showing NKX5-3 mRNA signal in the developing inner nuclear layer, but not in the outer nuclear layer or in the developing ganglion cell layer.
Figure 5.
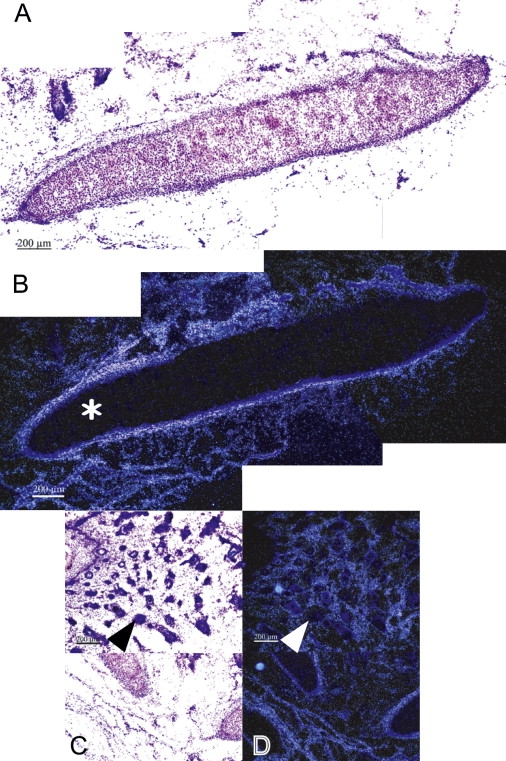
Expresssion of NKX5-3 in External Ear from 20-Week Old Human Fetus
(A) Bright-field aspect of a tissue section of the cartilage of the external ear.
(B) Dark-field aspect of the same tissue section showing the absence of labeling at this stage of the inner part of the outer auricular cartilage (∗), whereas the perichondrum is displaying an intense NKX5-3 mRNAs labeling.
(C) Bright-field aspect of a tissue section of the external ear showing the noncartilaginous mesenchyme from which derive various glands.
(D) Dark-field aspects of the same tissue sections showing NKX5-3 transcripts labeling of mesenchymatous cells surrounding cilia (arrow head) and developing sebaceous glands.
Nkx5-1, -2, and -3 are expressed very early in the developing central and peripheral nervous systems, and although they are all observed in sensory-organ-related structures, Nkx5-3 is mainly expressed in the retina and Nkx5-1 and Nkx5-2 in the inner ear.8 An Nkx5-1 mouse knockout model showed a circling behavior due to the fusion of the utricle and saccule, which created a common chamber with a contiguous endolymphatic space and a complete loss of the horizontal semicircular canal crista. Nkx5-3 has been less well studied. In mouse, it is mainly expressed in the second branchial arch, the retina, the lens, the dorsolateral mesenchyme of the craniofacial region, and also in the trigeminal, facial, acoustic, and vagal nerve ganglia, as well as in the dorsal root, the sympathetic nerve ganglia, and the external ear.6,8–10
On the basis of the evolutionary conservation of these genes, we cloned nkx5-3 and characterized its expression in Danio rerio (zebrafish). In zebrafish, the gene annotated as HMX1 in NCBI was in fact soho1, and we identified the true nkx5-3 1.7 kb upstream (Figure S1) (GenBank EU203551). Expression of nkx5-3 is localized to the naso-dorsal region of the eye and the ventral region of the otic placode as early as 24 hpf (Figure 6). No signal was observed for the sense probe.
Figure 6.
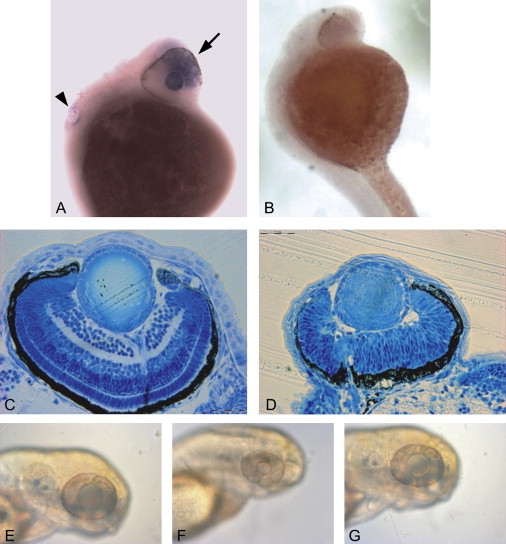
Expression of nkx5-3 in Zebrafish and Morpholino Experiments
(A and B) In situ whole-mount 1-day-old-embryo experiments with (A) antisense and (B) sense probes showing polarized expression of nkx5-3 in the eye (arrow) and in the ear (arrowhead). Up and left represent frontal and dorsal parts of animal.
(C and D) Toluidine-blue staining of transverse section in 3 dpf larvae (scale bar represents 0.02 mm). (C) shows a normal embryo, and (D) shows a morpholino-treated embryo from same stage (3 dpf) and with same magnification showing microphthalmia and incomplete retinal stratification.
(E–G) Photographs of the head of zebrafish at 3 dpf. (E) shows a normal embryo, (F) shows a morpholino-treated embryo, and (G) shows a morpholino-treated embryo rescued with nkx5-3 mRNA injection.
Because the development of the eye can easily be followed in teleost, we examined the effect of a morpholino-based knockdown expression of nkx5-3 in zebrafish that potentially mimicked the loss-of-function human mutation. Antisense (MO) or control (ct-MO) oligonucleotide morpholinos (Genetools, Philomath, Oregon) targeting the intron 1-exon 2 splice site of nkx5-3 mRNA was used (Table S2). Five hundred micromolar morpholino solutions of either MO or ct-MO were injected into the eggs at the1–2-cell stage. The injected amount was ∼10 ng per zygote. At 3 dpf, inhibition of nkx5-3 induced a delay in the development of the eye. Microphthalmia was evident in 100% of the injected embryos, and a delayed or absent stratification of the retina was observed in 73% and 27% embryos, respectively (Figure 6), whereas the body length of the embryos was not affected. Injection with ct-MO had no effect on the size and stratification of the eye (Table S2). The phenotype observed in the morpholino experiments could be rescued by coinjection of nkx5-3 mRNA together with MO (Figure 6). The phenotype observed in the MO zebrafish does not completely reproduce the human syndrome. Further studies will show whether this is due to residual activity of nkx5-3 or to intrinsic variations between zebrafish and human eye development. The use of other animal models or the description of additional patients may help understanding these differences.
The polarized expression of the nkx5-3 in the eye resembling that found in other animal models6,7,11 together with the morpholino knockdown phenotype suggest a potential function of the homeobox in maintenance and differentiation of specific cell types in the eye. Many genes such as bone morphogenetic protein (BMP), sonic hedgehog (SHH), retinoic acid receptors (RAR), fibroblast growth factor 8 (FGF8), or Eph receptors are differentially expressed in the retina, where they contribute to cell differentiation and axon guidance.12–14 Is NKX5-3 part of such a program? In chicken, GH6, the ortholog of NKX5-3, is under the control of FOXG1 (MIM 164874), a gene that is implicated in the retinotectal topographical map along the anterioposterior axis that acts through a DNA-binding-independent mechanism and inhibits EPHA3 expression (MIM 179611), thus explaining the opposing pattern of expression between GH6 and EPHA3.15,16 Ephrins and ephrin receptors are involved in cell signaling. In the eye, EPHA3 is involved in targeting retinal axons to their correct topographic position in the tectum or the lateral geniculate nucleus. The loss-of-function mutation described here could inactivate the repressive action of NKX5-3 on EPHA3 and induce ectopic expression of EPHA3, thus modifying the retinal polarity16,17 and generating part of the phenotype observed in this family. Whether ephrin receptors are also involved in the developing lens remains to be seen. Alternatively, NKX5-3 could interact with unknown partners and indirectly modulate molecular pathways in the lens. This report confirmed that NKX5-3 represents another member of the genes implicated in axial patterning of the retina.
Acknowledgments
We thank the patients and family members for their participation in this study. D.F.S and F.L.M. were funded by the Swiss National Science Foundation, grant # 32.111948, and M.M.A. was funded by Retina France. We thank C. Agosti, T. Favez, V. Pittet, and M. Curchod for technical assistance.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/
National Center for Biotechnology Information (NCBI): http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Accession Numbers
The GenBank accession number for the zebrafish nkx5-3 sequence reported in this paper is EU203551.
References
- 1.Hever A.M., Williamson K.A., van Heyningen V. Developmental malformations of the eye: The role of PAX6, SOX2 and OTX2. Clin. Genet. 2006;69:459–470. doi: 10.1111/j.1399-0004.2006.00619.x. [DOI] [PubMed] [Google Scholar]
- 2.Represa J., Frenz D.A., Van De Water T.R. Genetic patterning of embryonic inner ear development. Acta Otolaryngol. 2000;120:5–10. doi: 10.1080/000164800760370756. [DOI] [PubMed] [Google Scholar]
- 3.Franceschetti A., Valerio M. Confin. Neurol. 1945;6:255–257. [PubMed] [Google Scholar]
- 4.Stadler H.S., Padanilam B.J., Buetow K., Murray J.C., Solursh M. Identification and genetic mapping of a homeobox gene to the 4p16.1 region of human chromosome 4. Proc. Natl. Acad. Sci. USA. 1992;89:11579–11583. doi: 10.1073/pnas.89.23.11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadler H.S., Murray J.C., Leysens N.J., Goodfellow P.J., Solursh M. Phylogenetic conservation and physical mapping of members of the H6 homeobox gene family. Mamm. Genome. 1995;6:383–388. doi: 10.1007/BF00355637. [DOI] [PubMed] [Google Scholar]
- 6.Yoshiura K., Leysens N.J., Reiter R.S., Murray J.C. Cloning, characterization, and mapping of the mouse homeobox gene Hmx1. Genomics. 1998;50:61–68. doi: 10.1006/geno.1998.5284. [DOI] [PubMed] [Google Scholar]
- 7.Adamska M., Wolff A., Kreusler M., Wittbrodt J., Braun T., Bober E. Five Nkx5 genes show differential expression patterns in anlagen of sensory organs in medaka: Insight into the evolution of the gene family. Dev. Genes Evol. 2001;211:338–349. doi: 10.1007/s004270100162. [DOI] [PubMed] [Google Scholar]
- 8.Wang W., Lo P., Frasch M., Lufkin T. Hmx: An evolutionary conserved homeobox gene family expressed in the developing nervous system in mice and Drosophila. Mech. Dev. 2000;99:123–137. doi: 10.1016/s0925-4773(00)00488-3. [DOI] [PubMed] [Google Scholar]
- 9.Wang W., Chan E.K., Baron S., Van de Water T., Lufkin T. Hmx2 homeobox gene control of murine vestibular morphogenesis. Development. 2001;128:5017–5029. doi: 10.1242/dev.128.24.5017. [DOI] [PubMed] [Google Scholar]
- 10.Wang W., Lufkin T. Hmx homeobox gene function in inner ear and nervous system cell-type specification and development. Exp. Cell Res. 2005;306:373–379. doi: 10.1016/j.yexcr.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Deitcher D.L., Fekete D.M., Cepko C.L. Asymmetric expression of a novel homeobox gene in vertebrate sensory organs. J. Neurosci. 1994;14:486–498. doi: 10.1523/JNEUROSCI.14-02-00486.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters M.A. Patterning the neural retina. Curr. Opin. Neurobiol. 2002;12:43–48. doi: 10.1016/s0959-4388(02)00288-x. [DOI] [PubMed] [Google Scholar]
- 13.Lupo G., Harris W.A., Lewis K.E. Mechanisms of ventral patterning in the vertebrate nervous system. Nat. Rev. Neurosci. 2006;7:103–114. doi: 10.1038/nrn1843. [DOI] [PubMed] [Google Scholar]
- 14.Picker A., Brand M. Fgf signals from a novel signaling center determine axial patterning of the prospective neural retina. Development. 2005;132:4951–4962. doi: 10.1242/dev.02071. [DOI] [PubMed] [Google Scholar]
- 15.Schulte D., Cepko C.L. Two homeobox genes define the domain of EphA3 expression in the developing chick retina. Development. 2000;127:5033–5045. doi: 10.1242/dev.127.23.5033. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi H., Shintani T., Sakuta H., Noda M. CBF1 controls the retinotectal topographical map along the anteroposterior axis through multiple mechanisms. Development. 2003;130:5203–5215. doi: 10.1242/dev.00724. [DOI] [PubMed] [Google Scholar]
- 17.Huberman A.D., Murray K.D., Warland D.K., Feldheim D.A., Chapman B. Ephrin-As mediate targeting of eye-specific projections to the lateral geniculate nucleus. Nat. Neurosci. 2005;8:1013–1021. doi: 10.1038/nn1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.