

Main Text
In a recent issue of the American Journal of Human Genetics, Lin et al.1 showed that a change in linkage disequilibrium (LD) between a causal (B) and the observed (A) variants can lead to a reversal of genetic effect between two studies (a flip-flop). In this report, we show that a flip-flop can occur without a change in LD, even under simple noninteractive models. Further, we examine interactive models that allow for a flip-flop to take place under linkage equilibrium (LE). We provide specific conditions for the form of A and B interaction that permits a zero LD flip-flop, examine the behavior of allelic variance in the case of quantitative traits, and discuss potential implications of the findings for association mapping.
First, we consider a binary trait, , where indicates a condition, such as the presence of disease. As in Lin et al.,1 we will assume that the trait is influenced by two genetic variants, A:{A1,A2} (observed locus) and B:{B1,B2} (unobserved locus), and that there is no confounding. For simplicity, we disregard dominance effects, thereby allowing an essentially haploid treatment of the problem.2 The two-locus penetrance values are given by M and population frequencies of the haplotypes are given by P (see Table 1). This uses the notation for the entries of M. Among those with a particular allele, the expected proportion of individuals who will develop the condition is
| (1) |
and
| (2) |
Table 1.
Two-Locus Penetrance Values and Population Frequencies of the Haplotypes
| Haplotype | P | M | ||
|---|---|---|---|---|
| A1B1 | ||||
| A1B2 | ||||
| A2B1 | ||||
| A2B2 |
The relative risk is
| (3) |
The RR remains the same if M is multiplied by a constant. A flip-flop takes place whenever there is a change in sign of . For a given penetrance configuration, M, a flip-flop point is defined by a combination of haplotype frequencies such that (or equivalently, RR = 1).
Flip-Flop under a Constant LD
The simplest flip-flop case is the “proxy model,” in which the locus A has no functional significance but is related to the locus B only via LD. The penetrance array for the proxy model has the form M = {x, y, x, y}. In this case, it is necessary that the LD sign should change for a flip-flop to occur, as can be shown by writing the haplotype frequencies in terms of the LD coefficient (D) and solving for D. This calculation results in D = 0; thus, the proxy model most closely corresponds to the findings of Lin et al.1 Equations 1 and 2 show that for a given penetrance configuration, allelic effects are functions of haplotype frequencies. Thus, the driving force of flip-flip is a change in haplotype frequencies. There are multiple frequency configurations that can result in the same LD, and a flip-flop is not necessarily accompanied by a change in the LD. As an example, consider a simple noninteractive penetrance model of additive contributions by the two loci, , , , and , with haplotype effects given by . In this model, a flip-flop is possible without a change in LD. When P = {0.075, 0.01, 0.25, 0.665}, the association is positive; versus . A switch of population frequencies to P = {0.075, 0.25, 0.01, 0.665} leads to a flip-flop as follows: versus . In both cases, the LD is the same: rAB = 0.36, D′ = 0.83, D = 0.05, where the correlation rAB and D′ are the usual LD standardizations.3,4
Zero LD Flip-Flop
The population haplotype frequencies under LE are given by , where and . The penetrance values and the relative risk at the observed locus are given by
| (4) |
| (5) |
and
| (6) |
Note that these quantities do not depend on the frequencies of the observed locus, A. Whether any particular M is permitting a flip-flop under LE is determined by the solution of RR = 1 for (which defines the flip-flop point). This value is given by
| (7) |
For an effect reversal, this value has to be inside the (0, 1) interval. An equivalent condition is . The condition implies that the effect of A1 has to be reversed when the background of B is switched from B1 to B2. Two examples of such penetrance configurations are shown in Table 2, in which the values (δ1, δ2) of the same sign can be considered to be deviations from the “base values” (x, y). The M2, with some constraints on δ1, δ2, is an example of a “yin-yang” model, in which dissimilar haplotypes have similar susceptibilities. This model has been considered in several publications in the context of association mapping.5–8
Table 2.
Two Examples of Penetrance Configurations
| Haplotype | M1 | M2 |
|---|---|---|
| A1B1 | x | x |
| A1B2 | y | y |
| A2B1 | x + δ1 | y ± δ1 |
| A2B2 | y − δ2 | x ± δ2 |
The relations for the binary phenotype discussed above remain the same for the case when is quantitative. For example, the become the conditional expected values and . These are given by the same formulas as before. The “relative risk” becomes the ratio of the two allelic means. An additional quantity of interest in the case of a quantitative trait is the allele-specific variance:
| (8) |
where = Var , with a similar expression for A2. Assuming a common underlying variance, , and LE, the allele-specific variances become:
As a function of , the ratio of the variances under LE reaches the maximum or the minimum at . As a function of the joint effects of A and B, the variances and are unequal as long as . This condition excludes models in which A is a nonfunctional locus. Thus, under LE, requires that the A is not merely a proxy for the B but has a functional involvement. This argument assumes that there is no confounding; this is the same assumption that we would make when comparing allelic effects. Under LE, the ratio approaches 1 as approaches either 0 or 1. Figure 1 illustrates the behavior of the ratio of allelic means and allelic variances at the observed locus, under LE, as a function of the unobserved allele frequency . The variance contrast is strongest around the flip-flop point where the mean effect of A cannot be detected.
Figure 1.
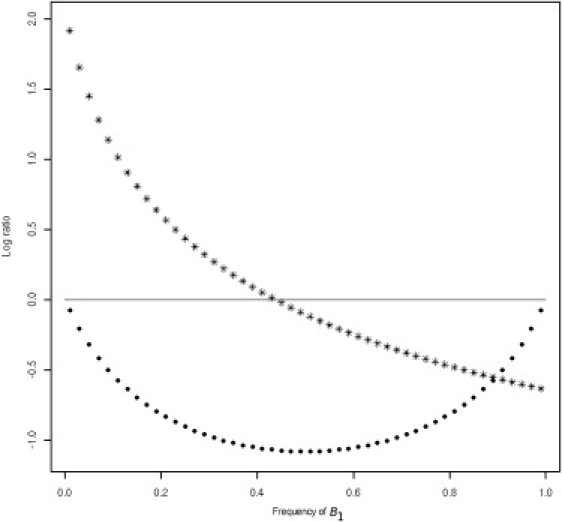
Values for and for a Zero-LD Model
Values for are indicated by the line of asterisks, and values for are indicated by the line of filled black dots. (M) = 10 × {0.5, 0.4, 0.95, 0.05} and
The flip-flop condition under LE between the studied and the unobserved variants has important implications outside of observational studies. Suppose that A1, A2 are levels of a factor under investigation, and these levels might be introduced in an interventional study on a random background of an unobserved B. A possible scenario is a study of efficacy of a drug A1 on a condition in the presence of genetic effects of the locus B. In an interventional study, a possible correlation between A and B is removed via randomization. Suppose an effect of A1 is claimed by a randomized study. Our analysis shows that in the presence of population heterogeneity with respect to the , there might be an effect reversal in a different study. Some studies could report that there is no effect of A at all, despite the importance of A at specific categories of B.
In the case of a quantitative trait, the allele-specific variances can be compared, in addition to the usual comparison of the means. In both types of comparisons, versus and versus , the unknown factor, B, can either be genetic or environmental. According to our analysis, in the absence of correlation between the A and the B, the allelic variances are unequal only in the presence of a functional involvement of the A. In this regard, interpretation of a comparison of the allelic variances is similar to interpretation that follows from the usual comparison of the allelic means. Rejection of the hypothesis leads to a similar claim as does the rejection of the hypothesis ; yet, the variance contrast might be substantial when the usual mean difference is undetectable. In either case, one can claim that the locus A is either directly associated with the trait or that it is a marker associated via correlation with an unobserved factor, B. When potential confounding due to population stratification is not an issue, the latter case leads to a standard claim that there is a nearby causal locus B correlated with the marker A via LD.
A practical question remains: How do we distinguish a genuine flip-flop from a statistical artifact? Our analysis shows that the underlying mechanism of a flip-flop is a change in the AB haplotype frequencies or, in the case of a zero-LD flip-flop, in the allele frequencies of B between populations. Examples can be constructed where both the allele frequency of the observed variant as well as the population prevalence of the trait (M · P) remain the same across populations, despite the flip-flop. Nevertheless, these are contrived situations that take place only at specific values of the four haplotype frequencies. Thus, a flip-flop is usually accompanied by a change in the population prevalence and in the case of a nonzero LD, by a change in the frequency of the observed variant as well. There would be a higher confidence that the flip-flop is genuine in those cases where studied populations are of distinct ancestry, with evidence of allele-frequency differences at many loci. In addition, we suggest that in the case of a quantitative trait, the allelic-variance contrast can be examined. This contrast can be informative even at the flip-flop point, where no allelic effect can be detected. If normality of the trait can be assumed, the variance contrast provides an independent evidence that the studied variant has a genetic involvement, either as a LD proxy for causal variation or as a part of a functional unit. A significant allelic-variance contrast in both samples that exhibit a flip-flop may serve as an additional evidence for a genuine genetic association. Statistical tests for comparison of allelic and haplotypic variances will be detailed in a subsequent paper.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. Luda Diatchenko, Norman Kaplan, Clare Weinberg, David Umbach, and Rongheng Lin provided stimulating discussion. We thank three anonymous reviewers for valuable comments.
References
- 1.Lin P.I., Vance J.M., Pericak-Vance M.A., Martin E.R. No gene is an island: The flip-flop phenomenon. Am. J. Hum. Genet. 2007;80:531–538. doi: 10.1086/512133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terwilliger J., Hiekkalinna T. An utter refutation of the ‘Fundamental Theorem of the HapMap’. Eur. J. Hum. Genet. 2006;14:426–437. doi: 10.1038/sj.ejhg.5201583. [DOI] [PubMed] [Google Scholar]
- 3.Weir B.S. Sinauer Associates; Sunderland, MA: 1996. Genetic Data Analysis II. [Google Scholar]
- 4.Lewontin R.C. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaykin D.V., Meng Z., Ehm M.G. Contrasting linkage-disequilibrium patterns between cases and controls as a novel association-mapping method. Am. J. Hum. Genet. 2006;78:737–746. doi: 10.1086/503710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J., Jin L., Xiong M. Test for interaction between two unlinked loci. Am. J. Hum. Genet. 2006;79:831–845. doi: 10.1086/508571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schrodi S.J., Garcia V.E., Rowland C., Jones H.B. Pairwise linkage disequilibrium under disease models. Eur. J. Hum. Genet. 2007;15:212–220. doi: 10.1038/sj.ejhg.5201731. [DOI] [PubMed] [Google Scholar]
- 8.Wang T., Zhu X., Elston R.C. Improving power in contrasting linkage-disequilibrium patterns between cases and controls. Am. J. Hum. Genet. 2007;80:911–920. doi: 10.1086/516794. [DOI] [PMC free article] [PubMed] [Google Scholar]