

Abstract
We present clinical, radiological, biochemical, and genetic findings on six patients from two consanguineous families that show EDS-like features and radiological findings of a mild skeletal dysplasia. The EDS-like findings comprise hyperelastic, thin, and bruisable skin, hypermobility of the small joints with a tendency to contractures, protuberant eyes with bluish sclerae, hands with finely wrinkled palms, atrophy of the thenar muscles, and tapering fingers. The skeletal dysplasia comprises platyspondyly with moderate short stature, osteopenia, and widened metaphyses. Patients have an increased ratio of total urinary pyridinolines, lysyl pyridinoline/hydroxylysyl pyridinoline (LP/HP), of ∼1 as opposed to ∼6 in EDS VI or ∼0.2 in controls. Lysyl and prolyl residues of collagens were underhydroxylated despite normal lysyl hydroxylase and prolyl 4-hydroxylase activities; underhydroxylation was a generalized process as shown by mass spectrometry of the α1(I)- and α2(I)-chain-derived peptides of collagen type I and involved at least collagen types I and II. A genome-wide SNP scan and sequence analyses identified in all patients a homozygous c.483_491 del9 SLC39A13 mutation that encodes for a membrane-bound zinc transporter SLC39A13. We hypothesize that an increased Zn2+ content inside the endoplasmic reticulum competes with Fe2+, a cofactor that is necessary for hydroxylation of lysyl and prolyl residues, and thus explains the biochemical findings. These data suggest an entity that we have designated “spondylocheiro dysplastic form of EDS (SCD-EDS)” to indicate a generalized skeletal dysplasia involving mainly the spine (spondylo) and striking clinical abnormalities of the hands (cheiro) in addition to the EDS-like features.
Introduction
The Ehlers-Danlos syndrome (EDS) is a heterogeneous group of heritable disorders of connective tissue characterized by articular hypermobility, skin hyperelasticity, and tissue fragility. It affects skin, ligaments, joints, blood vessels, and internal organs. The natural history and mode of inheritance differ between the six major types.1,2
Among them, the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VI [MIM 225400]) is characterized at birth by severe muscular hypotonia (requiring often invasive neuromuscular workup), kyphoscoliosis that is progressive, severe joint hypermobility and luxations, and marked skin hyperelasticity. In addition, there is fragility of the skin with abnormal scarring, osteopenia without a tendency to fractures, often a Marfanoid habitus and microcornea, and occasionally rupture of the arteries and the eye globe. The disorder is caused by a deficiency of the enzyme collagen lysyl hydroxylase (LH1; EC 1.14.11.4; procollagen-lysine,2-oxoglutarate 5-dioxygenase), which normally hydroxylates lysyl residues in -Xaa-Lys-Gly- sequences of the helical region of the collagen α chains to -Xaa-Hyl-Gly-. In bone and other tissue collagens, two Hyl residues, one in each of two amino telopeptides or two carboxyl telopeptides, together with one Hyl or one Lys residue of the triple helix form the trivalent pyridinium crosslinks hydroxylysyl pyridinoline (HP) and lysyl pyridinoline (LP), respectively. In addition, some helical Hyl residues of the α chains serve as attachment sites for carbohydrate units (either galactose or glucosyl-galactose). As a consequence, lysyl hydroxylase (LH1) deficiency results in underhydroxylation of lysyl residues, fewer glycosylated hydroxylysyl residues in collagens, and an abnormal crosslink formation with consequent mechanical instability of the affected tissues, as seen in these patients. As a result of underhydroxylation and underglycosylation, the collagen α chains produced by fibroblasts in culture display a faster electrophoretic mobility on SDS-PAGE gels. Furthermore, the enzyme deficiency gives rise to an abnormal urinary excretion pattern of LP and HP crosslinks. The ratio of urinary total LP to HP in patients with EDS VI is high (5.99 ± 0.99; range 4.30–8.10; n = 17) as compared with normal controls (0.19 ± 0.02; 0.12–0.25; n = 179)3 and is diagnostic for EDS VI.2,4,5 The diagnosis of EDS VI may then be confirmed by measuring the activity of the enzyme in cultured skin fibroblasts6 and/or directly by mutation analysis of PLOD1 (MIM 153454), which encodes the enzyme.7
Here, we report on a novel clinical entity overlapping with but distinguishable from EDS VI that is caused by a mutation in the zinc transporter gene SLC39A13 (MIM 608735). The clinical features of 6 patients from 2 consanguineous families with Ehlers-Danlos syndrome-like features, short stature, finger contractures, distinct radiological features, elevated ratios of LP to HP, underhydroxylated collagens in culture despite higher than normal and normal in vitro activities of lysyl hydroxylase and prolyl 4-hydroxylase, respectively, are described.
Subjects and Methods
The anthropometric, salient clinical and radiological features, and results of urinary pyridinolines of the six patients and the other members of the two families are summarized in Tables 1–4 and Figures 1–3. Routine blood and urine analyses were normal or not contributory. The karyotypes in P1/I and P3/II were normal. Family members participating in the study gave their informed consent.
Table 1.
Anthropometric Synopsis of the Members of the Two Families at Last Investigation
| Individuals (Age in Years); Sex | Height, cm (Centile)a | Sitting Height, cm (Centile)c | Arm Span, cm | Weight, kg (Centile)a | Head Circumference, cm (Centile)d |
|---|---|---|---|---|---|
| P1/I (14.5); m | 146 (8 cm < P3) | 76 (P3) | 140 | 51 (P10-25) | 56 (P75) |
| P2/I (8.5); m | 123 (P10) | 70 (P25) | 119 | 25 (P25) | 53 (P50) |
| F1/I (38); m | 172 (P25) | 101 (P97) | na | 93 (>P97) | 56 (P50) |
| M1/I (31); f | 158 (P25) | 92 (P90-97) | na | 73 (P97) | 56 (P75) |
| S1/I (12); f | 149 (P25) | 80 (P75) | na | 44 (P25-50) | 54 (P75) |
| P3/II (10.1); f | 121 (5 cm < P3) | 64 (4 cm < P3) | 108 | 20 (3 kg < P3) | 49 (P2) |
| P4/II (4.5); f | 95 (P3)b | 54 (1 cm < P3) | 86 | 15 (P25)b | 48 (P2)b |
| F2/II (43); m | 183 (P90) | 92 (P25-50) | 175 | 87 (P90-97) | 57 (P75) |
| M2/II (36); f | 155 (P25) | 82 (P3) | 150 | 51 (P10) | 52 (P2) |
| S2/II (13); m | 151 (P25) | 77 (P10-25) | 143 | 37 (P10) | 51 (P2) |
| S3/II (5.5); f | 113 (P75)b | 60 (P25) | 105 | 17 (P10)b | 47 (P2)b |
| P5/III (28); m | 152 (14 cm < P3) | 79 (8 cm < P3) | 156 | 63 (P10) | 57 (P50) |
| P6/III (12.5); f | 144 (P3) | 73 (P3) | 135 | 40 (P10) | 55 (P75) |
| F3/III (56); m | na (“normal”) | na (“normal”) | na (“normal”) | na (“normal”) | na (“normal”) |
| M3/III (48); f | 157 (P25) | 82 (P3) | 155 | na (“normal”) | 58 (P98) |
| S4/III (20.5); f | 162 (P50) | 81 (P3) | 153 | 55 (P50) | 55 (P50) |
| S5 (29); f | na (“normal”) | na | na | na (“normal”) | na (“normal”) |
Table 2.
Salient Clinical Findings in the Six Patients at Last Investigation
| P1/I | P2/I | P3/II | P4/II | P5/III | P6/III | ||
|---|---|---|---|---|---|---|---|
| Skin | |||||||
| hyperelastic | (+) | (+) | (+) | (+) | + | + | |
| velvety, smooth | − | − | + | + | + | + | |
| thin, translucent | + | + | + | (+) | + | + | |
| bruisable | + | − | + | + | + (infancy) | + | |
| scars atrophic, cigarette paper-like | + | + | + | + | − | + | |
| scars hypertrophic | + | nr | − | − | + (knee, ankle) | − | |
| Hands | |||||||
| palms wrinkled | + | + | + | + | (+) | + | |
| fingers tapering | + | + | + | (+) | + | + | |
| thenar hypotrophy | + | + | + | + | + | + | |
| Feet | |||||||
| flat feet | + | + | + | + | + | + | |
| Joints | |||||||
| hypermobility | nr | nr | + | + | (+) | (+) | |
| hypermobility of major joints | − | − | − | + | − | − | |
| hypermobility of minor joints (and subsequent contractures) | (+) | − | + (contractures) | + | + (contractures) | + (contractures) | |
| history of subluxations | − | − | + | + | + | − | |
| broadened metaphyseal regions (elbow, wrist, interphalanges) | − | − | + | + | + | + | |
| wrist sign | − | − | − | − | − | − | |
| thumb sign | − | − | − | − | − | − | |
| Skeletal | |||||||
| Marfanoid habitus | − | − | − | − | − | − | |
| chest deformity | − | − | − | − | − | − | |
| spine deformity | − | − | − | − | − | − | |
| Face and Eyes | |||||||
| down-slanting palpebral fissures | + | + | hypothelorism | − | (+) | − | |
| sclerae bluish | + | + | + | + | − (+ as a child) | (+) | |
| protuberant | + | + | (+) | (+) | (+) | (+) | |
| myopia | + | − | − | astigmatism | hyperopia | + | |
| cornea diameter (mm) | nr | 11 | 11 | 11 | nr | nr | |
| Oral | |||||||
| cleft palate/ bifid uvula | − | − | high palate | bifid uvula | − | − | |
| teeth (hypodontia), small | − | + | small, malocclusion | small, malocclusion | caries tendency | irregular ends of teeth | |
| retrognathia | − | − | − | − | − | − | |
| Cardiac Signs | |||||||
| heart murmur (MVP, AI) | − | − | − | − | − | − | |
The following symbols and abbreviations are used: nr, not recorded; +, present; (+), mildly present; and −, absent; findings marked by symbols printed in bold are depicted in Figure 2.
Table 3.
Salient Radiological Findings in the Six Patients
| Code | P1/I | P2/I | P3/II | P4/II | P5/III | P6/III | |
|---|---|---|---|---|---|---|---|
| Hand | |||||||
| CAa | 11.5 | 3.5 | 9.9 | 5.3 | 28 | 12.5 | |
| SAa | 11.5 | 3.0 | 8.9 | 5.0 | mature | 14 | |
| Broad ends of short tubular bones | + | + | + | + | + | + | |
| Flat epiphyses | + | + | − | + | na | + | |
| Short metacarpals and phalanges | + | − | − | − | + | − | |
| Osteopenia | − | − | − | + | − | − | |
| Knee | |||||||
| Shallow intercondylar fossa | + | − | − | − | + | − | |
| Spine | |||||||
| Platyspondyly | + | + | + | + | + | + | |
| Osteopenia | + | + | + | + | − | + | |
| Irregular endplates | + | + | − | − | + | − | |
| Pelvis | |||||||
| Small, broad ileum | + | ne | + | + | + | + | |
| Flat capital femoral epiphyses | + | ne | + | + | − | + | |
| Broad and short Femoral necks | + | ne | + | − | − | + | |
| Additional Regions | |||||||
| Chest | normal | normal | |||||
| Skull | normal | Wormian bones | |||||
| DXA total t score | −5.8 | +1.1 | −3.8 | ||||
Table 4.
Ratios of Total Urinary Pyridinolines (LP/HP) in Affected and Unaffected Family Members, Controls, and EDS VI Individuals
| Subjects | Age (y) | Single Values (LP/HP) | N | Mean | Range | 1 SD | Reference |
|---|---|---|---|---|---|---|---|
| P1/I | 11.5–14.5 | 0.76/0.67 (26.4.01); 0.73 (7.2.02); 0.91; 0.74 (10.3.04) | 4 | 0.76 | 0.67–0.91 | ||
| P2/I | 3.5–8.5 | 0.64/0.60 (26.4.01); 0.74 (7.2.02); 0.83; 0.77 (10.3.04); 0.82 (19.4.06) | 5 | 0.74 | 0.60–0.83 | ||
| P3/II | 7.5–10.1 | 0.89 (13.11.03); 1.07 (13.2.04); 1.04 (26.10.04); 0.93 (19.4.06); 0.92 (27.6.06) | 5 | 0.97 | 0.89–1.07 | ||
| P4/II | 1.9–4.5 | 0.79 (13.11.03); 0.98 (13.2.04); 0.91 (26.10.04); 0.99 (19.4.06); 0.90 (27.6.06) | 5 | 0.91 | 0.79–0.99 | ||
| P5/III | 26.8–28.3 | 0.79 (26.10.04); 0.73 (19.4.06) | 2 | 0.76 | |||
| P6/III | 11.0–12.5 | 1.25 (26.10.04); 1.13 (19.4.06) | 2 | 1.19 | |||
| F1/I | 38 | 0.26 (9.7.01); 0.22 (17.10.02) | 2 | 0.24 | |||
| M1/I | 31 | 0.34 (9.7.01); 0.30 (17.10.02); 0.35 (16.4.06) | 3 | 0.33 | 0.30–0.35 | ||
| F2/II | 43 | 0.31 (19.4.06) | 1 | 0.31 | |||
| M2/II | 36 | 0.324 (27.6.06) | 1 | 0.32 | |||
| F3/III | 36 | n.a. | — | — | |||
| M3/III | 48 | 0.39 (19.4.06) | 1 | 0.39 | |||
| S1/I | 10 | 0.27 (17.10.02) | 1 | 0.28 | |||
| S2/II | 13 | 0.27 (19.4.06) | 1 | 0.27 | |||
| S3/II | 5.5 | 0.20 (27.6.06) | 1 | 0.20 | |||
| S4/III | 3.5 | 0.19/0.19 (19.4.06) | 1 | 0.19 | |||
| EDS VI homozygotes | 10 (1–28) | 17 | 5.97 | 4.30–8.10 | 0.99 | 5 | |
| EDS VI obligate heterozygotes | 38 (26–54) | 7 | 0.33 | 0.30–0.37 | 0.035 | (B.S., unpublished data) | |
| Control children | 0–14 | 83 | 0.19 | 0.12–0.25 | 0.030 | 3 | |
| Control children | 0–1 | 15 | 0.17 | 0.12–0.21 | 0.03 | ||
| Control children | >1–3 | 14 | 0.19 | 0.15–0.25 | 0.04 | ||
| Control children | >3–5 | 15 | 0.19 | 0.13–0.25 | 0.04 | ||
| Control children | >5–8 | 12 | 0.21 | 0.15–0.25 | 0.03 | ||
| Control children | >8–11 | 14 | 0.18 | 0.12–0.24 | 0.03 | ||
| Control children | >11–14 | 13 | 0.19 | 0.16–0.23 | 0.02 | ||
| Control men | 56 (28–69) | 44 | 0.20 | 0.15–0.23 | 0.02 | 3 | |
| Control women | 41 (26–52) | 52 | 0.20 | 0.12–0.22 | 0.02 | 3 |
Note that biological replicates are indicated by the dates of urinary collection in parentheses; analytical duplicates are indicated by slashes; values in bold were obtained by an independent method,8 all others are obtained as described.5 The rows in italics refer to the data of patients and parents with EDS VI and highlight the difference to those of the patients described in the text. “n.a.” is short for not available.
Figure 1.

Pedigrees and Haplotype Analysis
Family I and family II are shown in (A) and (B), respectively. Affected individuals are depicted by black symbols. Genotypes for 39 microsatellite-marker loci spanning the linkage interval on chromosome 11p11.2-p13 are shown. The haplotypes are displayed by vertical bars. The disease-associated haplotype is denoted by a red box around the vertical bars. Recombinations in both families define a candidate region flanked by markers D11S1779 and D11S4191.
Figure 2.
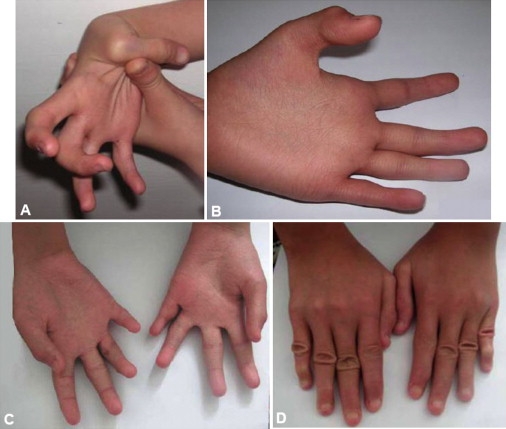
Hands with Characteristic Changes
(A) P3/II at age 10.1 years. The thumb is hypermobile and can easily be adducted to the forearm; the fingers are tapering and display flexion contractures.
(B) P3/II at age 10.1 years. The palm is excessively wrinkled, the thenar is hypotrophic, the fingers are tapering, and the endphalange of the thumb contracted.
(C) P6/III at age 12.5 years. The palms are wrinkled, the thenar and hypothenar muscles are hypotrophic, the fingers are tapering, and there is partial syndactyly of fingers 2 and 3, and 3 and 4.
(D) P6/III at age 12.5 years. The dorsal aspect of the hands show tapering fingers, broadened regions over the interphalangeal joints with abundant skin, and flexion contractures, especially of the fifth fingers.
Figure 3.

Radiographs of the Low Thoracic and Lumbar Spines, the Hands, the Pelvis, and the Knees
(A) P2/I at age 3.5 years. Note mild to moderate flattening, osteopenia, and irregular endplates of the vertebral bodies.
(B) P1/I at age 11.5 years. There are similar findings to (A) with flattening, irregular endplates and osteopenia of the vertebral bodies.
(C) P5/III at age 28 years. Again, platyspondyly, osteopenia, and irregular endplates of the vertebral bodies with depressions resembling notochordal remnants are seen.
(D) P1/I at age 11.5 years. Note the mildly short metacarpals and phalanges with widening of the ends and relative narrowing of the diaphyses. The epiphyses of the short tubular bones are flat. Osteopenia is not present.
(E) P2/I hand at age 3.5 years. Skeletal features of this younger child are similar but less pronounced as compared to (A); alterations of shape and flat epiphyses of the short tubular bones are noted.
(F) Pelvis of P3/II at age 10 years. Small ilia, mild flattening of the proximal epiphyses, and short and wide femoral necks are seen.
(G) Knee of P5/III at age 28 years. The knees show normal-sized epiphyses and a normal metaphyseal contour on the long tubular bones, but the intercondylar fossa of the distal femur is shallow in this adult patient.
Family I
The pedigree of family I is shown in Figure 1A. The proband (P1/I) was referred to one of us (BA) for evaluation at the age of 10 years because of short stature, walking pain, and a waddling gait.
P1/I was born spontaneously after an uneventful pregnancy at term. Birth weight was 2500 g (<3rd centile), length was 50 cm (10th–25th centile), and head circumference was not recorded. At birth, protruding eyes and reddish eye lids were noted. There was no muscular hypotonia. Psychomotor development was normal. Deciduous and permanent teeth erupted late.
At the age of 10 years, he was short (height 122 cm, 2 cm < 3rd centile; weight 26 kg, 10th centile; and head circumference 52 cm, 10th–25th centile). He had down-slanting palpebral fissures, protruding eyes, slightly bluish sclerae, and a normal cornea diameter. All the deciduous teeth were present, but one permanent tooth of the lower jaw was missing. His skin was thin and translucent with an easily visible venous pattern; the scars over the knees, sheens, and elbows had a cigarette-paper-like appearance. The hands looked prematurely aged. The fingers were slender and tapering, the palms had a thin, finely wrinkled and tight skin, and the thenar muscles were atrophic so that the thumbs could not be adducted.
Follow-up examinations at ages 11.5, 13, and 14.5 years (Tables 1 and 2) showed that the standing height remained less than third centile. He had pain in the knees and occasionally in the hips. Chest and spine deformities and Marfanoid features were not present. With time his skin manifestations became more apparent with easy bruisability and slow wound healing resulting in thin cigarette-paper-like scars. For radiological findings, see below, Table 3, and Figures 3B and 3D.
A brother, P2/I, was born spontaneously at term after an uneventful pregnancy. Birth weight was 2490 g (<3rd centile), length 52 cm (50th centile), and head circumference 34 cm (10th–25th centile). No abnormalities were noted after birth. He walked independently at the age of 1 year. Clinical examination at the age of 2 years showed body measurements within the normal range (standing height 82 cm, weight 10 kg, head circumference 50 cm). He had down-slanting palpebral fissures and large, slightly protruding eyes with blue sclerae. His skin was normal. The hands showed tapering of the fingers and a fine texture of the skin of the palms. He had hypodontia with missing deciduous and permanent teeth of the lower jaw.
Follow-up examinations at ages 5, 6.5, and 8.5 years (Tables 1 and 2) showed that the standing height followed 10th centile. The skin of the palms was more wrinkled, and there was atrophy of the thenar. His skin was thin and translucent, with atrophic scars. He had occasional pain in both knees. For radiological findings, see below, Table 3, and Figures 3A and 3E.
The consanguineous family originates from North-Western Iraq. The parents were healthy, second cousins (Figure 1A). Both parents (F1/I and M1/I) and a daughter (S1/I) had normal skin, hands, joints, sclerae, and eyes and no spine or chest deformities.
Family II
The pedigree of family II is shown in Figure 1B. The proband, P3/II, was referred to one of us (NE) for evaluation at age 7.5 years because of short stature and finger contractures.
P3/II was born after an uncomplicated pregnancy at term with a birth weight of 2750 g (3rd centile). Perinatal history was uneventful, and psychomotor development was normal. Her family realized at age 1 year that her “wrists were weak” and “that she could not hold any objects with her hands and fingers.” At 7.5 years, she had a proportionally small stature with a standing height 109 cm (5 cm < 3rd centile), weight 15.6 kg (3 kg < 3rd centile), head 47 cm, arm span 99 cm, lower segment 52.5 cm, and upper/lower segment ratio of 1.08. There was hypotelorism, and the eyes were mildly protuberant with blue sclerae and a normal cornea diameter. There was a high palate, delayed teeth eruption, minimal umbilical hernia, and hypoplastic mammillae. Her skin was soft, velvety, and easily bruisable with scars. There was joint laxity especially of the small joints and elbows. There were atrophies of the thenar and hypothenar on both hands, and the right thumb was dislocated. On the hands there were some mild contracture-like deformities. The palms were wrinkled without real dermatoglyphics but with many thin lines. Flexion contractures were present on the first, third, and fifth fingers. There were bilateral pes planus and mild crura vara.
Follow-up at 10.1 years showed that the anthropometric measures (Table 1) remained similar. Her hands while shaking felt like “bags filled with little bones”; there was thumb instability on both sides, and the thumbs could easily touch the radius by flexion (Figure 2A). She was able to dislocate and reduce some phalangeal joints of other fingers. The fingers were long, spider-like, not straight, and tapering; there was also a dynamic swan neck deformity particularly of the fifth fingers and less so of the first and third fingers. Ligamentuous laxity was observed by pulling on the distal phalanges, giving a telescoping appearance. Thenar and hypothenar atrophy was obvious (Figure 2B), and to strengthen the grip, she used a stress ball. Her lower legs looked atrophic, and severe bilateral pes planovalgus needed support; swan-neck deformities were present on small toes. She had a physiotherapeutic program with weight bearing, swimming, and bicycling. The large joints (arms, hips, and knees) had a normal range of motion; the elbows were broadened. The skin was smooth, easily bruisable, slow healing, and somewhat hyperelastic, and the palms were wrinkled with abnormal dermatoglyphics (Figure 2B); the skin was particularly thin over the dorsal side of hands and feet. For radiological findings, see Table 3 and Figure 3F.
An affected sister of the proband, P4/II, was born at term with feet presentation and a weight of 3000 g; her perinatal history was uneventful and neuromotor development was normal.
At age 4.5 years, the eyes were mildly protuberant with blue sclerae and astigmatism. There was a bifid uvula, malocclusion of teeth, and bilaterally broadened elbows. The skin was smooth, velvety, and translucent particularly on legs and feet with easily visible veins and only a few atrophic scars and ecchymoses. Both hypermobile thumbs reached easily the radius. The palms were wrinkled, the thenar and hypothenar looked atrophic, and her thumb could easily be subluxated and replaced. There were bilateral severe pes planus and positional genua valga, and when the subject walked, it looked like “that there were not enough bones to hold the feet strong.” Arms, hips, and lower extremities had a full range of motion; she presented with bilateral genua recurvata and bilateral medial and lateral instability of knees, instability of ankles, bilateral flat feet that required physiotherapy and supramalleolar orthesis. Her skin was always fragile and easily bruisable with slow wound healing and few atrophic scars.
An affected cousin, P5/III, was admitted at age 28 years because of short stature and weakness of his fingers and toes.
P5/III was born at term with a weight of 3000 g and a length of 50 cm. He presented with finger contractures and bilateral club foot at birth, and these were treated by conservative means until the age of 7 years. His psychomotor development was normal. He had always complained about his short stature and weakness of his finger and toe tips; his fine motor skills of finger (and toe) tips were never as good as that of his peers but did not restrict his own routine day care. His fingers (particularly the fifth) were getting thinner and tapering with time on both sides. He had several dislocations of his fingers after moderate trauma. His mother described his skin as fragile, bruisable with atrophic scars after mild trauma during childhood but this improved with time.
At age 28 years, he had a short stature with a short trunk (Tables 1 and 2). The palpebral fissures were slightly down-slanting, the eyes were mildly protuberant with white sclerae—unlike in childhood when they were bluish—and there was moderate hyperopia (+3.25 Dpt). The skin was velvety, smooth, rather thin with mildly translucent appearance, and hyperelastic, with excessive skin folds over the broadened phalangeal joints. Both hands had muscular weakness with atrophy of the thenar, the hypothenar, and the interosseal muscles that made dorsiflexion difficult; there were no sensory deficits. The fingers were tapering with flexion contractures of the right third to fifth fingers and left fourth and fifth fingers that restricted the small-joint mobility in these fingers, and he could not bend his thumb. There was no hypermobility of elbows or genua recurvata, and flexion of the trunk was limited. Varicose veins over the dorsum of the feet were apparent. For radiological findings, see below, Table 3, and Figures 3C and 3G.
P6/III is the affected sister of P5/III and complained about the abnormalities of her skin and hands. She was born at term and had a normal perinatal history and a normal psychomotor development. Her skin had been always hyperelastic, thin, very bruisable, and fragile with slow wound healing. According to her mother, her fingers were less contracted at birth compared to those of her affected brother (P5/III), and although they improved with exercise, she lost some skills in her right hand during the years, such as buttoning and unbuttoning her clothes and carrying a cup.
At age 12.5 years, she was moderately short (Table 1). The teeth were malocclusive with irregular borders. The skin was velvety, smooth, thin, and translucent, with cigarette-paper-like atrophic scars; and over the hands and feet, the skin looked rather old with enlarged, easily visible vessels and was atrophic over the finger and toe tips. The skin was loose over hands and finger joints, and the palms were wrinkled (Figures 2C and 2D). The metaphyseal regions of elbow, wrist, and proximal interphalangeal joints were broadened. The fingers were tapering, with flexion contractures particularly on her left metacarpophalangeal and interphalangeal joints and swan-neck deformities, and the fascia and/or tendon were rather shortened between the thumb and the second, third, and fourth fingers, and there was some atrophy of the interosseous, thenar, and hypothenar muscles. There were bilateral flat feet. The eyes were moderately myopic, and the sclerae were bluish. For radiological findings, see below and Table 3.
The parents were consanguineous (Figure 1B), originating from the Southeastern part of Turkey, and had normal eyes and sclerae, no chest or spine deformities, and normal hands, joints, and skin. There was moderate familial microcephaly of M2/II, S2/II, S3/II, and P4/II (Table 1).
Radiological Findings
The radiological findings are summarized in Table 3. X-ray examinations of the hands, knees, and spine were performed in all patients and some healthy first-degree relatives (F2/II; S3/II; and M3/III). The pelvis was radiographed in all patients but P2/I, the chest was radiographed in P3/II and P4/II, and the skull was radiographed in P3/II and P6/III (Table 3).
All patients had mild to moderate flattening of vertebral bodies (platyspondyly). In addition, mild to moderate osteopenia was present in all but P5/III. Irregular endplates of the vertebral bodies were shown in P1/I and P5/III (Figures 3B and 3C, respectively); in P2/I, platyspondyly was already present at age 3.5 years (Figure 3A). The only adult patient of the series had endplate depressions resembling notochordal remnants (Figure 3C).
The hands were abnormal in all patients. Mild to moderate alteration of the shape of metacarpals and phalanges consisting of widening of the ends and relative narrowing of the diaphyses was present in all patients to a mild or moderate degree. The epiphyses of the short tubular bones, however, were flat in all patients (Figures 3D and 3E) except in P3/II. This latter feature distinguishes the present patients from progressive pseudorheumatoid arthropathy of childhood (MIM 208230), which otherwise bears similar radiographic features to our patients.
The ileum was small in all patients (P2/I was not examined). An abnormality of the proximal femurs consisting of mildly flat proximal femoral epiphyses and short and wide femoral necks was present in most patients (Figure 3F). The knees, however, showed normal-sized epiphyses and a normal metaphyseal contour of the long tubular bones. The intercondylar fossa of the distal femur was shallow in P1/I and P5/III (Figure 3G).
The chest was examined in P3/II and P4/II, both with normal findings. The skull (Towne's projection) of P6/III showed innumerable, small Wormian bones of the lambdoid suture (not shown), whereas the skull of P3/II was normal.
X-rays of the examined hands, knees, and spine were normal in the first-degree relatives (F2/II, S3/II, and M3/III; not shown) who were clinically not affected and who had normal ratios of urinary crosslinks (Table 4).
Analysis of Pyridinium Crosslinks by HPLC
Spot urine samples and isolated peptide fractions from urine collections were acid hydrolyzed and analyzed by reverse phase HPLC in heptafluorobutyric acid as previously reported.5,8 The average intra-assay and inter-assay coefficients of variation for LP and HP were <8% and <6%, respectively.3
Collagen Peptides Isolation from Urine
Crosslinked telopeptides were isolated from urine with immunoaffinity columns with mAb 1H11, specific to crosslinked N-telopeptides of type I collagen (NTx) resulting from bone resorption, or with mAb 2B4, which binds to crosslinked C-telopeptides of type II collagen (CTx-II) resulting from growth-cartilage degradation. Antibody was coupled to CNBr-activated Sepharose 4B (Amersham Biosciences). For NTx isolation, the 1H11-column was equilibrated in 50 mM HEPES, pH 7.2, loaded with urine diluted with equilibrating buffer, and washed with 0.125 M NaCl, 0.025 mM HEPES, pH 7.2, and bound peptides were eluted with 1 M NaCl and 0.025 mM CAPS (N-cyclohexyl-3-aminopropanesulfonic acid), pH 10.4. For CTx-II isolation, the 2B4-column was equilibrated and loaded similarly, and the bound peptides were eluted with 0.1 M NaCl and 0.1 M glycine, pH 2.5.8
Dermal Fibroblast Cultures
Cells strains were established from skin biopsies from patients P1/I and P2/I and their father (F1/I) and mother (M1/I), as well as their unaffected sister (S1/I), and from patient P3/II and her mother (M3/II), by routine procedures. Cells were maintained under standard conditions, and radiolabeled collagen samples were prepared by digestion with pepsin, precipitated with ethanol, separated on a 5% SDS-polyacrylamide gel, and visualized by fluorography.9
Amino Acid Composition of Dermis
Skin biopsies were hydrolyzed with 6 N HCl in nitrogen at 110°C for 24 hr as described.10
Stability of Collagens
Thermal stability of triple helical collagens was assessed by the resistance to proteases at increasing temperatures.11
Collagen Purification and Mass Spectrometry
Cultures of exponentially growing fibroblasts from patients and controls were supplemented with 10% fetal-calf serum and 50 μg/ml ascorbic acid; the culture medium was serially collected 3 times a week and replaced by freshly prepared supplemented media; and the collected medium was stored at −20°C and pooled over a 2 week period. The combined medium was then acidified to 3% acetic acid and digested with pepsin (50 μg/ml) at 4°C for 24 hr. Collagens were precipitated with 1.0 M NaCl and run on SDS-PAGE either directly or after digesting a portion with CNBr in 70% formic acid. The α chains or individual CNBr peptides were cut from the gel and subjected to in-gel trypsin digestion. Electrospray MS was performed on the tryptic peptides with an LCQ Deca XP ion-trap mass spectrometer equipped with in-line liquid chromatography (LC) (ThermoFinnigan) that used a C8 capillary column (300 μm × 150 mm; Grace Vydac 208MS5.315) eluted at 4.5 μl per min. The LC mobile gradient was made from buffer A (0.1% formic acid plus 2% buffer B in MilliQ water) and buffer B (0.1% formic acid in 3:1 acetonitrile:n-propanol v/v). The sample stream was introduced into the mass spectrometer with an electrospray ionization source (ESI) under atmospheric pressure. The spray voltage was 3 kV and the inlet temperature 160°C.12
Lysyl Hydroxylase and Prolyl 4-Hydroxylase
Enzyme activities were measured in extracts of fibroblasts from two patients and a control (GM05659, Coriell Institute for Medical Research) as described.6 In brief, cells were grown to confluency in Dulbecco's modified Eagle's medium (Invitrogen-GIBCO) supplemented with 10% heat-inactivated fetal-calf serum (Sigma). Lysyl hydroxylase activity was measured with an L-[4,5-3H] lysine-labeled underhydroxylated procollagen substrate, and prolyl hydroxylase was determined with a similar underhydroxylated procollagen substrate labeled with L-[4-3H] proline. The enzyme activities were quantitated as released 3H2O in a minimum of four independent assays.
DNA and RNA Extraction
Genomic DNA was isolated from peripheral blood leukocytes or cultured fibroblasts with standard techniques. Total RNA was isolated from cultured dermal fibroblasts (2× T75 flasks) with an RNeasy Mini Kit (QIAGEN), in accordance with the manufacturer's protocols. Prior to RNA extraction, one of the two T75 fibroblast flasks was incubated for 6 hr with 1000 μg/ml cycloheximide in order for prevention of nonsense-mediated mRNA decay. Transcript quantification was performed by real-time quantitative PCR with the ABI Prism 7700 sequence detection system (Applied Biosystems) as previously described.7
Genome-wide Microsatellite-Marker Analysis
A genome-wide scan was performed on all individuals of family II with 218 highly polymorphic microsatellite markers distributed with an average spacing of 20 cM (ABI Prism Linkage Mapping Set 2.5, Applied Biosystems). Microsatellite analysis was carried out by standard semiautomated methods with an ABI 3100 Genetic Analyzer (Applied Biosystems). Parametric multipoint linkage analysis was performed by GENEHUNTER v2.1r5. Haplotypes were reconstructed with GENEHUNTER v2.1r5.
Genome-wide SNP Scan and Linkage Analysis
A genome-wide scan with SNP-arrays (Affymetrix GeneChip Human Mapping 10K array Xba 142 2.0) was performed on all individuals of family II in accordance with the manufacturer's instructions. The array interrogates 10,204 SNPs with a mean intermarker distance of 258 kb, equivalent to 0.36 cM. Genotypes were called by the GeneChip DNA Analysis Software (GDAS v2.0, Affymetrix). We assumed a fully penetrant autosomal-recessive trait locus with a disease-allele frequency of 0.0001 and a marker-allele frequency estimated from our sample. Multipoint parametric linkage analysis and haplotyping were carried out with GENEHUNTER v2.1r5 and ALLEGRO v. 2.0,13 respectively, through stepwise use of a sliding window with sets of 100 and 50 SNPs. Data handling was performed by EasyLinkage Plus v.5.05.14 The microsatellite-marker content of the linked ∼26.22 cM genomic region (deCode genetic map) between rs1350960 and rs870333 on chromosome 11p11.2-q13 (∼36 Mb) was examined with the NCBI Map Viewer (build 36.1). Genotyping of 39 highly polymorphic microsatellite markers spaced at intervals of ∼0.2–0.5 cM was carried out by PCR and fragment analysis of amplified products with ABI Prism 310 and ABI 3100 Genetic Analyzers (Applied Biosystems). Multipoint parametric linkage analysis and haplotyping were carried out with GENEHUNTER v2.1r5 and ALLEGRO v.2.0,13 under EasyLinkage Plus v.5.05. Haplotypes were displayed with HaploPainter v.029.5.15
Sequencing and Mutation Analysis of Candidate Genes
Positional candidate genes including SLC39A13 (NM_152264) within the ∼6.72 cM homozygous candidate region (58.73–64.96 cM according to the deCode genetic map, ∼16.9 Mb in size between D11S1779 and D11S4191) were prioritized for sequencing analyses on the basis of the known or putative functions of the encoded proteins, as well as on the basis of their expression profiling. Mutation screening of the candidate genes was performed by fluorescent bidirectional sequencing of genomic as well as cDNA-amplified PCR products on an ABI 3100 automated sequence detection system (Applied Biosystems). PCR conditions and primer sequences are available from the authors upon request.
Conservation Analysis
Multiple sequence alignment of the SLC39A13 (ZIP13) protein was performed with ClustalW and BOXSHADE programs, with sequences from human (Q96H72), orangutan (Q5R6I6), mouse (Q8BZH0), rat (Q2M1K6), bovine (A5D7H1), chicken (Q5ZI20), and zebrafish (Q8AW42).
Results
Because the clinical findings in patient 1 (P1/I) did not establish a diagnosis, a skin biopsy was taken for electron-microscopy study and fibroblast culture. Elastin was normal in amount and structure, and the collagen fibrils were normal in shape and diameter; specifically, no large fibrils with an irregular contour or round fibrils with an abnormally wide range of diameters were seen as in the classical form of EDS (EDS I/II [MIMs 130000 and 130010]).16 The collagens produced by his cultured skin fibroblasts showed on SDS-PAGE a slightly increased electrophoretic mobility of the α(I)-chains of collagen type I, seen also for P2/I, P3/II, and P5/III studied later. However, the faster migration of the collagen chains was not consistently observed (data not shown; see also the legend to Figure 4) and was less apparent than on SDS-PAGE of EDS VI collagen. In EDS VI, in addition to the α1(I)- and α2(I)-chains of collagen type I, the α1(V)-, the α2(V)-, and the α3(V)-chains of collagen type V and the crosslinked chains β11 and β12 of collagen type I display a faster electrophoretic mobility.2 Thus, we concluded that lysyl residues in collagens were underhydroxylated in these patients, albeit less than in individuals with EDS VI.
Figure 4.

Decreased Lysyl Hydroxylation of the Collagen α1(I) and α2(I) Chains Revealed by TMS
(A) SDS-PAGE resolution of collagen α1(I)- and α2(I)-chains of collagen type I from pepsin-treated culture medium. The α1(I) chains were excised from the gel and subjected to in-gel digestion with trypsin. Lanes 1–4 refer to samples obtained by fibroblasts from P1/I, P3/II, and controls 1 and 2. Please note that the electrophoretic mobility of the α(I) chains from the patients' samples is barely different from that of the controls.
(B) SDS-PAGE resolution of CNBr-peptides prepared from collagen type I from culture medium. Peptide α2(I)CB4 was excised from the gel and subjected to in-gel digestion by trypsin. Lanes 1, 2, and 3 were from patients P1/I, P3/II, and a control, respectively.
(C) Mass-spectral scans across the regions of the LCMS profiles from the α1(I) chains of one patient and a control that capture the post-translational variants of the tryptic peptide. The lowest panel shows an MS/MS spectrum from the 11492+ precursor ion, which establishes the identity of the peptide sequence including placement of hydroxyl groups on the three prolyl residues and the single lysyl residue. Similar MS/MS spectra showed that the 11412+ ion lacked the hydroxyl group (16 mass units) of the lysyl residue 684 in the two patients.
(D) Mass-spectral scans across regions of the LCMS profiles from peptide α2(I)CB4 of one patient and a control that capture posttranslational variants of the tryptic peptide shown. The lowest panel shows the MS/MS spectrum from the 12182+ precursor ion, which establishes the identity of the peptide including the placement of hydroxyl groups on three prolines and the single lysine. Similar MS/MS spectra showed that the 12102+ ion lacked the hydroxyl (16 mass units) on Lys219 in the two patients.
Previously, we have shown that underhydroxylation of collagen is reflected in the urine by the excretion of an abnormal LP/HP pattern and that a markedly increased LP/HP ratio (5.97 ± 0.99; range 4.30–8.10; n = 17) was diagnostic for EDS VI.2,4,5,8 Therefore, we measured the ratio of total urinary pyridinolines in the present patients and their family members. The ratio LP/HP was elevated in all six patients (mean 0.89 ± 0.18; range 0.67–1.25), remained so over an observation period of up to 5 years, and was independent of age (Table 4). The ratio was clearly lower in these patients than in individuals with EDS VI but markedly higher than in controls of all ages (mean 0.19 ± 0.02; range 0.12–0.25; n = 179),3 and there was no overlap of the ratios between the individuals with EDS VI, the present affected subjects, and the controls (Table 4). It is of interest that in seven obligate heterozygotes for EDS VI, the LP/HP ratio was indistinguishable (0.33 ± 0.03; 0.30–0.37) from those of five obligate heterozygotes of the present subjects (0.32 ± 0.05; 0.24–0.39) despite the fact that homozygotes for EDS VI had an LP/HP ratio far higher (∼6) than that of the affected subjects with a LP/HP ratio of ∼1.
The total amount of pyridinolines, i.e., lysyl pyridinoline plus hydroxylysyl pyridinoline expressed per creatinine was normal as compared to age-matched controls. This means that the formation of LP was increased at the expense of synthesized HP, as observed also in patients with EDS VI.4
Analysis of the immunopurified crosslinked N-telopeptide of collagen type I and of the crosslinked C-telopeptide of collagen type II from the urines of P1/I and P2/I revealed that both collagen types were underhydroxylated at the triple helical crosslinking sites. The underhydroxylation of collagen lysyl residues ranged between that of the controls and that of the subjects with EDS VI (Table 5).
Table 5.
Site-Specific Underhydroxylation of Crosslinking Lysyl Residues in Bone Collagen Type I and Cartilage Collagen Type II
|
LP/HP Molar Ratio |
|||
|---|---|---|---|
| Total Urine | NTx-I Peptides | CTx-II Peptides | |
| Control | 0.18 | 0.40 | <0.04 |
| P1/I | 0.91 | 3.00 | 0.30 |
| P2/I | 0.83 | 3.57 | 0.13 |
| EDS VI∗ | 5.26 | 20.0 | 0.50 |
The NTx-I LP/HP ratio is a surrogate index of the aggregate ratio of Lys/Hyl at K930 in α1(I) plus K933 in α2(I) chains of bone collagen type I, and the CTx-II LP/HP ratio is a surrogate index of the Lys/Hyl ratio at K87 in α1(II) chains of growth-plate cartilages. Underhydroxylation was assessed by HPLC analysis of the LP/HP pyridinoline ratio in urinary crosslinked peptides.
The patient with EDS VI was studied previously.8
As a next step, we measured hydroxylation of lysyl and prolyl residues in collagens produced in vivo and in culture. Acid hydrolysis of a skin biopsy of P5/III showed that lysyl residues were underhydroxylated, i.e., the Hyl-to-Lys ratio was 0.09 (5 controls: 0.14 ± 0.02) and the Hyl-to-Hyp ratio was 0.05 (controls: 0.10 ± 0.02); for comparison, the Hyl-to-Lys and Hyl-to-Hyp ratios in five patients with EDS VI were 0.008 ± 0.006 and 0.004 ± 0.002, respectively, and thus were more reduced than in P5/III.
Because hydroxylation of collagen prolyl residues confers stability to the triple helical molecules,17 we explored the possibility of lowered stability to proteases at increasing temperatures.11 Thermal stability of collagens produced by fibroblasts from P1/I and P3/II was normal for collagen types I, III, and V. Obviously, the general underhydroxylation of these collagens is moderate (see below) and not sufficient to lower their melting temperatures.
Mass-spectral analysis of a tryptic peptide from the α1(I)-chain of collagen type I (Figure 4A), composed of amino acid residues 658–684, confirmed that lysyl residues in collagen samples produced by fibroblasts of P1/I and P3/II were consistently underhydroxylated (Figure 4C). Specifically, hydroxylation of lysyl residue 684 in the peptide was 12% and 27% in P1/I and P3/II as opposed to 41% and 36% in the two controls. Similarly, in a tryptic peptide of a CNBr peptide from the α2(I)-chain of collagen type I (Figure 4B), composed of amino acid residues 193–219 (GEVGLP∗GLSGPVGPP∗GNP∗GANGLTGAK∗), hydroxylation of lysyl residue 219 was 46%, 48%, 74%, and 83% in P1/I, P3/II, and two controls, respectively (Figure 4D). Inspection of the mass-spectral results from the same in-gel digests in the controls and the subjects showed, in addition, partial but significant underhydroxylation at the single 3-Hyp site in the α1(I)-chain and underhydroxylation at a nearby 4-Hyp residue (Figure 5). Specifically, 3-hydroxylation of prolyl residue 986 was 88%, 93%, 100%, and 100%, and 4-hydroxylation of prolyl residue 981 was 67%, 78%, 93%, and 92% in P1/I, P3/II, and the two controls, respectively. These results indicate that lysyl underhydroxylation and prolyl 4-underhydroxylation occur along the whole molecule and are not confined to specific residues, and the single site of prolyl 3-hydroxylation is also underoccupied compared with control collagen.
Figure 5.
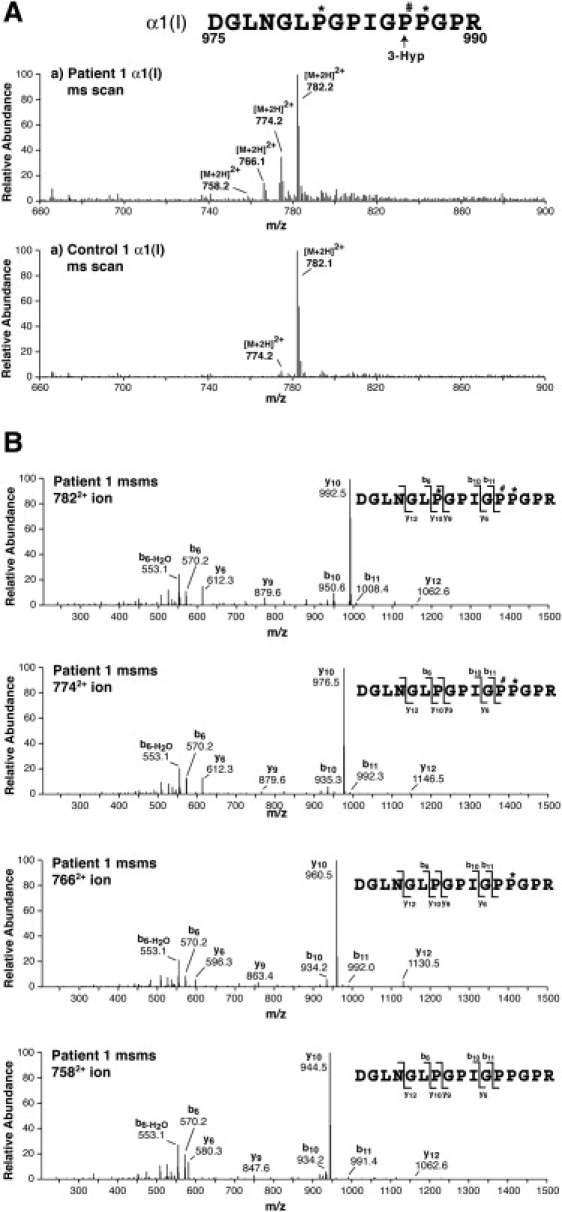
Decreased Prolyl 3-Hydroxylation and Prolyl 4-Hydroxylation Revealed by TMS
Mass spectra are shown for the tryptic peptide containing Pro986, the site of 3-Hyp formation in the α1(I) chain from one patient and one control fibroblast cultures run on SDS-PAGE (shown in Figure 4A).
(A) Mass-spectral scans encompassing all variants of the tryptic peptide detected in the LC profiles (∼28 min elution time). Four posttranslational variants are revealed in the patient's collagen.
(B) MS/MS spectra of the four peptide variants differing by 16 mass units identify the placement of hydroxyl groups. Underhydroxylation at the 3-Hyp position and the two 4-Hyp positions can be quantified from the identified structures and their relative abundance as shown in Figure 4.
The in vitro activities of the enzymes lysyl hydroxylase and prolyl 4-hydroxylase were measured in fibroblasts of P1/I and P3/II in four independent assays. Lysyl hydroxylase activities were normal to higher-than-normal range, whereas prolyl 4-hydroxylase activities were within the normal range (Table 6). Because the in vitro assay was done under optimal substrate and cosubstrate concentrations, a Km variant for the binding of substrates and cosubstrates to the putative mutant enzyme was considered, and such a variant might reduce the enzyme activity in vivo under suboptimal conditions. Therefore, we explored the possibility of PLOD1 as a candidate gene. However, analyses of linkage, amounts of transcripts, and sequencing excluded it as a candidate; similarly, PLOD3 (MIM 603066) and PLOD2 (MIM 601865) were excluded.
Table 6.
Lysyl Hydroxylase and Prolyl 4-Hydroxylase Activities in Patients' Fibroblasts
| Lysyl Hydroxylasea | Prolyl Hydroxylasea | |
|---|---|---|
| P1/I | 142 ± 27; 105–166 | 103 ± 10; 95–115 |
| P3/II | 126 ± 16; 109–147 | 120 ± 22; 86–133 |
Activities of four independent assays are expressed as percentages of control values and given as mean ± SD and range.
The discrepancy of reduced hydroxylation of collagens produced in vivo or formed in intact cells and the normal lysyl hydroxylase and prolyl hydroxylase activities measured in cell homogenates in vitro prompted us to explore the hypothesis of in vivo reduced enzyme activities due to impaired cellular uptake of vitamin C, a cofactor of both enzymes. We hypothesized that SLC23A2 (MIM 603791) encoding the sodium-dependent vitamin C transporter 2 (SVCT2)18,19 would be defective in these patients. However, linkage and mutation analysis in the two families ruled out SVCT2 as a candidate. We likewise excluded genes involved in prolyl hydroxylation of collagen, P4HA1 (MIM 176710), P4HA2 (MIM 600608), P4HB (MIM 176790), P4HA3 (MIM 608987), and LEPRE1 (MIM 610339) by linkage analysis with microsatellite markers within and/or flanking the genes.
A 20 cM low-density genome scan was then performed on family II. A LOD score of 1.6 was obtained for a 20ptel chromosomal region. However, the only candidate gene SOX12 (MIM 601947) within the region was excluded by mutation analysis in F1/I, M2/II, and P3/II.
A genome-wide SNP scan with the Affymetrix GeneChip10 K SNPs array identified a region on chromosome 11p11.2-q13 flanked by the recombinant SNPs rs1350960 and rs870333 in family II defining a critical interval of ∼36 Mb. Fine-mapping with 39 highly polymorphic microsatellite markers within this region, and including all members of family I, reduced the critical region to a 16.9 Mb interval between markers D11S1779 and D11S4191, with a combined maximal LOD score of 5.3 (Figure 6). Within this region, several genes were regarded as relevant functional candidates because of their expression in connective tissues; however, sequencing at the genomic as well as at the cDNA level in M2/II, P3/II, M3/III, and P5/III detected only known polymorphic variants in 26 candidate genes except SLC39A13 (NM_152264.2), in which we detected a 9 bp in-frame deletion in exon 4, c.483_491 del9 (p.F162_164 del). All six patients were homozygous, and all parents as well as siblings S1/I, S2/II, and S4/III were heterozygous for this mutation, whereas S3/II was homozygous for the wild-type allele (Figure 7A). The mutation creates a Tail restriction-endonuclease recognition site (ACGTΔ) that was used for assessment of its frequency in 182 control individuals (51 with the same ethnic background); restriction digestion of 364 chromosomes showed no mutation.
Figure 6.
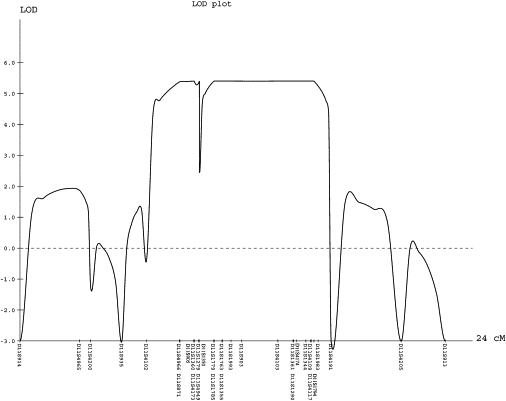
Results of Linkage Analysis
The parametric multipoint LOD score analysis for 39 microsatellite markers spanning the linkage interval on chromosome 11p11.2-q13 in families I and II is shown. Because of space constraints, only 31 markers are shown. Genetic distances, in cM, based on the deCODE map and the LOD scores are shown on the x axis and on the y axis, respectively.
Figure 7.
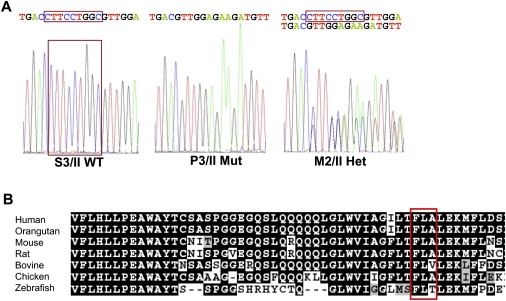
Mutation Identification and Comparative Analysis of the ZIP13 Protein
(A) Electropherograms indicating the wild-type (S3/II), the homozygous (P3/II), and the heterozygous (M2/II) mutation sequences. The 9 bp deletion in exon 4 of SLC39A13 (c.483_491 del9; p.F162_164 del) is boxed.
(B) ClustalW alignment and BOXSHADE analysis of the protein sequence of the transmembrane domain III of ZIP13 around the mutation (the deleted amino acids FLA are boxed). The protein sequence is highly conserved across vertebrates, from primates through to birds and primitive fish.
Discussion
The work-up of the initial proband with Ehlers-Danlos syndrome-like features and short stature revealed a pattern of urinary pyridinolines that was abnormal but less pronounced than in subjects with the kyphoscoliotic form of the Ehlers-Danlos syndrome (EDS VI). The subsequent study of five additional cases showed that they all had moderate underhydroxylation of collagen lysyl and prolyl residues despite normal lysyl hydroxylase and prolyl 4-hydroxylase activities in cell extracts and a clinical phenotype clearly distinct from EDS VI. These findings and a search of the literature indicated, to the best of our knowledge, that they all had a hitherto unrecognized clinical entity, which we named spondylocheiro dysplastic form of EDS (SCD-EDS) (see below).
SCD-EDS is inherited as an autosomal-recessive trait caused by mutations in SLC39A13 on chromosome 11p11.2. Clinically, it is characterized by the following features: (1) postnatal growth retardation, growth parallel below or along the lower centiles, moderate short stature; (2) hyperelastic, velvety, thin skin with an easily visible venous pattern and bruisability that leads to atrophic scars; (3) slender, tapering fingers, wrinkled palms, and considerable thenar (and hypothenar) atrophy; (4) hypermobility of the small joints, especially the fingers, which results later in contractures; (5) broadened metaphyseal regions of the elbows, wrists, knees, and interphalangeal joints; (6) protuberant eyes with bluish sclerae and normal cornea diameter; (7) primary platyspondyly, osteopenia of the axial skeleton, widening of the ends with relative narrowing of the diaphyses and flat epiphyses of metacarpals and phalanges, small ileum, mildly flat proximal femoral epiphyses, and short and wide femoral necks; and (8) a ratio of lysyl pyridinoline to hydroxylysyl pyridinoline (LP/HP) of ∼1.0.
There are clear differences and similarities between subjects with EDS VI and SCD-EDS. The phenotype of EDS VI differs from that of SCD-EDS by severe muscular hypotonia from birth, progressive kyphoscoliosis, severe hypermobility of small and large joints, absence of contractures of fingers, and normal height (in the absence of severe kyphoscoliosis), often by a Marfanoid habitus and microcornea, occasionally by ruptures of the eye globe and of large arteries, by the absence of platyspondyly,5,20–24 and by the high LP/HP ratio. On the other hand, individuals with SCD-EDS do have platyspondyly, wrinkled palms, tapering fingers, enlarged metaphyseal regions over the elbows, the wrists, and the knees, and the interphalangeal joints that are not observed in individuals with EDS VI. SCD-EDS shares common features with EDS VI such as smooth, thin, hyperelastic, and fragile skin with atrophic scars, hypermobile small joints, bluish sclerae, and osteopenia.
Platyspondyly is observed in SCD-EDS but not in EDS VI despite the complete lack of LH1 activity in the latter condition. Hydroxylysine content in bone collagen of the original EDS VI patients,25 however, was 40%–100% of the normal content, whereas in skin it was 5%–10%. The difference in lysyl hydroxylation of collagen in bone and skin is explained by the different activity ratios of lysyl hydroxylase to prolyl hydroxylase measured in bone cells and skin fibroblasts from these patients; their bone cells and fibroblasts had activity ratios of 46%–48% and 8% of normal activity ratios, respectively (Krane et al., 1980, Calcif. Tissue Int. 31, 57, abstract). These authors speculated on the presence of different isoenzymes with variable tissue expression and different substrate specificities that have been characterized recently.26 In SCD-EDS, the disturbed zinc homeostasis (see below) is expected to inhibit all iron-dependent collagen hydroxylases, including those in bone and growth-plate cartilages, and so may have more severe consequences on the vertebrae than isolated LH1 deficiency in EDS VI. Thus, the platyspondyly in SCD-EDS may be explained by the combined underhydroxylation of lysyl and prolyl residues in bone and cartilage, perhaps in combination with the normal muscular tone that thus may lead to platyspondyly; note that severe muscular hypotonia per se in genetic disorders similar to EDS VI and in acquired conditions result in tall vertebral bodies.
The features observed in SCD-EDS, its disease locus, and/or its autosomal-recessive mode of inheritance distinguish it from skeletal dysplasias such as “SEMD with joint laxity (SEMD-JL) Beighton type” (MIM 271640), “spondyloepimetaphyseal dysplasia with multiple dislocations” (MIM 603546), and “spondylometaphyseal dysplasia with cone-rod dystrophy” (MIM 608940).27
The genetic defect in the two families with SCD-EDS is a homozygous 9 bp deletion in exon 4 of SLC39A13 (Figure 7A), which encodes for the zinc transporter SLC39A13 (ZIP13) that is widely expressed in most tissues. This protein is a member of the LIV-1 subfamily of ZIP zinc Transporters (LZT),28 a highly conserved group of eight transmembrane domain proteins known to transport zinc and/or other metal ions from the extracellular space or from the organellar lumen into the cytoplasm.29 They are mainly situated on the plasma membrane, but at least one member, SLC39A7 (HKE4 or ZIP7 [MIM 601416]), is located on the ER30 and on the Golgi31 membranes and transport zinc into the cytosol.30 Protein sequence alignments have shown that human SLC39A13 and SLC39A7 are phylogenetically closely related ZIP transporters32 that lack a highly conserved CPALLY motif, which immediately precedes the first transmembrane domain28 and characterizes all other LZT family members that are localized on the plasma membrane. Thus, in analogy to SLC39A7, the zinc transporter SLC39A13 may also reside on intracellular membranes.32 Indeed, our prediction analysis for subcellular localization of SLC39A13, based on its amino acid sequence with the online PSLDoc server, resulted in a 55% likelihood that SLC39A13 resides on the membrane of intracellular organelles, whereas the probability of it being localized on the plasma membrane of the cell was only 17%; for comparison, the probability of SLC39A7 to be located on these membranes is 60% and 17%, respectively. Although the exact localization of ZIP13 is not known so far, our biochemical findings (see below) strongly suggest it is on the ER membrane.
The mutation found in our patients causes a deletion of amino acid residues 162–164 (Phe-Leu-Ala) within the transmembrane domain III, which is highly conserved (Figure 7B). We suspect that the deletion disturbs the proper folding of the protein and thus impairs 3D conformation and function of the transporter.
Synthesis and posttranslational modifications of the nascent α chains of the various collagen types occur in the ER. Among the different modifications, hydroxylation of collagen lysyl and prolyl residues are due to highly conserved enzymatic processes that involve Fe2+, α-ketoglutarate, O2, ascorbate as cofactors, cosubstrates, and reducing agent.17 We suspect that an increased concentration of Zn2+ in the ER competes with Fe2+ for binding to lysyl hydroxylase, prolyl 4-hydroxylase, and prolyl 3-hydroxylase, thus impairing hydroxylation of lysyl and prolyl residues. Indeed, Zn2+ was found to be an effective competitive inhibitor with respect to Fe2+ for prolyl 4-hydroxylase33 and for lysyl hydroxylase.34 Proper hydroxylation of collagen lysyl residues is important for functional crosslinks as demonstrated by its deficiency in EDS VI, whereas 4-hydroxylation of collagen prolyl residues is important for the stability of the triple helix17 as shown in experimental scurvy.35
As a consequence of the defect, collagen hydroxylysyl residues (Hyl) are formed to a lesser extent in SCD-EDS than in controls as reflected by the abnormally high urinary LP/HP ratio and the lower Hyl contents of dermal collagens and of collagens formed by cultured fibroblasts. Underhydroxylation of collagen lysyl and prolyl residues observed in these individuals occurs along the entire molecule, is not confined to specific residues (although preferential effects dependent on sequence context are possible), and affects at least collagen types I and II (Table 5). The underhydroxylation of lysyl residues in SCD-EDS—although less pronounced than in EDS VI—suggests a relevant impairment of lysyl hydroxylase activity in SCD-EDS. However, its activity measured in fibroblast homogenates of SCD-EDS subjects is in the normal to higher-than-normal range. This is consistent with our hypothesis of Zn2+ induced inhibition of this and related hydroxylases. Homozygous EDS VI patients with a complete lack of LH1 still have ∼15%–20% of residual lysyl hydroxylase activity in cell extracts, an outcome has been attributed to the presence of its isoform LH3, and this results in a urinary LP/HP ratio of ∼6. In obligate heterozygotes for EDS VI, the estimated residual activity of 60%–65% (50% plus 15%–20%)2 leads to an LP/HP ratio of ∼0.3, which is clearly higher than in controls (∼0.2) but similar to that in obligate heterozygotes for SCD-EDS (Table 4). By extrapolating these results, we expect the lysyl hydroxylase activity in vivo to be considerably reduced in SCD-EDS. Moreover, it is possible that the slight but significant prolyl 3-underhydroxylation contributes to the phenotype. In osteogenesis, imperfecta type VII (OI type VII [MIM 610682]) caused by a homozygous mutation in CRTAP12 (MIM 605497) hydroxylation of prolyl residue 986 in the α1(I)-chain from cultured skin fibroblasts and from a bone biopsy was ∼70% of the control, compared with 90% in the patients' samples. It is unclear whether the deficiency in 3-Hyp in OI (moderate in OI type VII and nearly complete in OI type II [MIM 166210] and OI type III [MIM 259420]) is simply a marker or has pathological effects on matrix collagen. If the latter is true, it could contribute to the osteopenia in SCD-EDS. However, the pathogenesis may be more complex and may involve zinc as an intracellular second messenger36 and as an essential component of many proteins; thus, a disturbance of intracellular zinc homeostasis could affect many processes inside and outside the cell and add to the phenotype. Nevertheless, the common biochemical effect of mutations in PLOD1 and SLC39A13 on lysyl hydroxylation and the abnormal collagen crosslinks strongly implies that a similar defect in collagen structure is a key element of both phenotypes. Collagen assembly is complex, requiring for example collagen type V as an essential template for collagen type I deposition, so consequences of underhydroxylation in more collagen gene products than simply types I and II will need to be examined to understand fully the pathogenic mechanism. The observed underhydroxylation of collagen types I and II may explain the clinical findings in skin, joints, and the skeleton of our patients. It is an open question whether underhydroxylation of other collagen types is involved and whether this may be clinically relevant and responsible for subtle clinical features, i.e., as in the vascular form of EDS (EDS IV [MIM 130050], due to mutations in collagen type III), the classical form of EDS (EDS I/II, due to mutations in collagen type V), Bethlem myopathy ([MIM 158810] due to mutations in collagen type VI), epidermolysis bullosa dystrophica ([MIM 131750] due to mutations in collagen type VII), and various chondrodysplasias (due to mutations other than in collagen II such as in collagen types IX and XI) and as in other collagen disorders.
We have chosen the name “spondylocheiro dysplastic form of EDS (SCD-EDS)” to indicate a generalized skeletal dysplasia involving mainly the spine (spondylo) and striking clinical abnormalities of the hands (cheiro) in addition to the common EDS-like features.
As a diagnostic tool for SCD-EDS, the measurement of the urinary LP/HP ratio appears to be specific, reliable, and rapid; however, more subjects are needed for estimating the biochemical range of the LP/HP ratio as well as the clinical spectrum. The prevalence of this entity is not known. The common mutation in the two families may be explained by a founder effect in this geographic region. Indeed, an identical disease haplotype is indicated by the finding of identical alleles at the two SLC39A13-flanking satellite marker loci in our families. On the other hand, the disorder may have been underreported so far. With this publication, we hope to attract more cases of this entity in order to identify the spectrum of disease-causing mutations and the resulting clinical and biochemical variability.
Web Resources
The URLs for data presented herein are as follows:
EasyLinkage, http://www.uni-wuerzburg.de/nephrologie/molecular_genetics/molecular_genetics.htm
Entrez Gene, http://www.ncbi.nlm.nih.gov/sites/entrez
ExPASy, http://www.expasy.ch/sprot/
NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/ mapview/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
PSLDoc, protein subcellular localization prediction based on modified document classification methods, www.bio-cluster.iis.sinica.edu.tw/∼bioapp/PSLDoc
Acknowledgments
We are grateful to all patients and their family members for participating in this study. Our thanks go to Michael Raghunath (Munster) for sending material of the index patient; to Ingrid Hauser (Heidelberg) for the ultrastructural assessment of the skin biopsy; to Jörg Schaper (Dusseldorf) for initial radiological considerations; to Angelika Schwarze (Zurich), Udo Redweik (Zurich), and Christine Plüss (Basel) for expert technical assistance; to Konrad Oexle (Zurich), Ann Randolph (Zurich), and Frank Neuheiser (Zurich) for their help in the initial candidate linkage, genome-wide mapping and mutation analyses, respectively; to Linda Walker (Durham) for the enzyme assays; to Albert Schinzel (Zurich) for karyotyping; and to Andres Giedion, Patricie Paesold-Burda, and Matthias Baumgartner (Zurich) for useful discussions. This work was supported by grants from the Swiss National Science Foundation (grant Nr.3200B0-109370/1), the Wolfermann-Nägeli Stiftung to B.S., and NIH (AR 37318, AR 36794) to D.R.E.
References
- 1.Beighton P., De Paepe A., Steinmann B., Tsipouras P., Wenstrup R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Steinmann B., Royce P.M., Superti-Furga A. The Ehlers-Danlos syndrome. In: Royce P.M., Steinmann B., editors. Connective Tissue and its Heritable Disorders. Second Edition. Wiley-Liss; New York: 2002. pp. 431–523. [Google Scholar]
- 3.Kraenzlin, M.E., Kraenzlin, C.A., Meier, C., Giunta, C., and Steinmann, B. Evaluation of an automated HPLC system for measurement of urinary collagen crosslinks: Effect of age, menopause and metabolic bone diseases. Clin. Chem., in press. [DOI] [PubMed]
- 4.Steinmann B., Eyre D.R., Shao P. Urinary pyridinoline cross-links in Ehlers-Danlos syndrome type VI. Am. J. Hum. Genet. 1995;57:1505–1508. [PMC free article] [PubMed] [Google Scholar]
- 5.Giunta C., Randolph A., Al-Gazali L., Brunner H.G., Kraenzlin M.E., Steinmann B. The Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VIA) Am. J. Med. Genet. 2005;133:158–164. doi: 10.1002/ajmg.a.30529. [DOI] [PubMed] [Google Scholar]
- 6.Murad S., Sivarajah A., Pinnell S.R. Serum stimulation of lysyl hydroxylase activity in cultured human skin fibroblasts. Connect. Tissue Res. 1985;13:181–186. doi: 10.3109/03008208509152396. [DOI] [PubMed] [Google Scholar]
- 7.Giunta C., Randolph A., Steinmann B. Mutation analysis of the PLOD1 gene: An efficient multistep approach to the molecular diagnosis of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA) Mol. Genet. Metab. 2005;86:269–276. doi: 10.1016/j.ymgme.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Eyre D., Shao P., Weis M.A., Steinmann B. The kyphoscoliotic type of Ehlers Danlos syndrome (type VI): Differential effects on the hydroxylation of lysine in collagens I and II revealed by analysis of cross-linked telopeptides from urine. Mol. Genet. Metab. 2002;76:211–216. doi: 10.1016/s1096-7192(02)00036-7. [DOI] [PubMed] [Google Scholar]
- 9.Steinmann B., Rao V.H., Vogel A., Bruckner P., Gitzelmann R., Byers P.H. Cysteine in the triple helical domain of one allelic product of the alpha 1(I) gene of type I collagen produces a lethal form of osteogenesis imperfecta. J. Biol. Chem. 1984;259:11129–11138. [PubMed] [Google Scholar]
- 10.Steinmann B., Gitzelmann R., Vogel A., Grant M.E., Harwood R., Sear C.H.J. Ehlers-Danlos syndrome in two siblings with deficient lysyl hydroxylase activity in cultured skin fibroblasts but only mild hydroxylysine deficit in skin. Helv. Paediatr. Acta. 1975;30:255–274. [PubMed] [Google Scholar]
- 11.Steinmann B., Nicholls A., Pope M.F. Clinical variability of osteogenesis imperfecta reflecting molecular heterogeneity: Cysteine substitutions in the α1(I) collagen chain producing lethal and mild forms. J. Biol. Chem. 1986;261:8958–8964. [PubMed] [Google Scholar]
- 12.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 13.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computor program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann K., Lindner T.H. easyLINKAGE-Plus - automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 15.Thiele H., Nürnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 16.Vogel A., Holbrook K.A., Steinmann B., Gitzelmann R., Byers P.H. Abnormal collagen fibril structure in the gravis form (type I) of the Ehlers-Danlos syndrome. Lab. Invest. 1979;40:201–206. [PubMed] [Google Scholar]
- 17.Kielty C.M., Grant M.E. The collagen family: Structure, assembly, and organization in the extracellular matrix. In: Royce P.M., Steinmann B., editors. Connective Tissue and its Heritable Disorders. Second Edition. Wiley-Liss; New York: 2002. pp. 159–221. [Google Scholar]
- 18.Rajan D.P., Huang W., Dutta B., Devoe L.D., Leibach F.H., Ganapathy V., Prasad P.D. Human placental sodium-dependent vitamin C transporter (SVCT2): Molecular cloning and transport function. Biochem. Biophys. Res. Commun. 1999;262:762–768. doi: 10.1006/bbrc.1999.1272. [DOI] [PubMed] [Google Scholar]
- 19.Clark A.G., Rohrbaugh A.L., Otterness I., Kraus V.B. The effects of ascorbic acid on cartilage metabolism in guinea pig articular cartilage explants. Matrix Biol. 2002;21:175–184. doi: 10.1016/s0945-053x(01)00193-7. [DOI] [PubMed] [Google Scholar]
- 20.Hyland J., Ala-Kokko L., Royce P., Steinmann B., Kivirikko K.I., Myllylä R. A homozygous stop codon in the lysyl hydroxylase gene in two siblings with Ehlers-Danlos syndrome type VI. Nat. Genet. 1992;2:228–231. doi: 10.1038/ng1192-228. [DOI] [PubMed] [Google Scholar]
- 21.Jarisch A., Giunta C., Zielen S., König R., Steinmann B. Sibs affected with both Ehlers-Danlos syndrome type VI and cystic fibrosis. Am. J. Med. Genet. 1998;78:455–460. doi: 10.1002/(sici)1096-8628(19980806)78:5<455::aid-ajmg11>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 22.Heim P., Raghunath M., Meiss L., Heise U., Myllylä R., Kohlschütter A., Steinmann B. Ehlers-Danlos syndrome type VI (EDS VI): Problems of diagnosis and management. Acta Paediatr. 1998;87:708–710. doi: 10.1080/080352598750014184. [DOI] [PubMed] [Google Scholar]
- 23.Yiş U., Dirik E., Chambaz C., Steinmann B., Giunta C. Differential diagnosis of muscular hypotonia in infants: The kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VI) Neuromuscul. Disord. 2008;18:210–214. doi: 10.1016/j.nmd.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Krane S.M. Genetic and acquired disorders of collagen deposition. In: Piez K.A., Reddi A.H., editors. Extracellular Matrix Biochemistry. Elsevier; New York: 1984. pp. 413–463. [Google Scholar]
- 25.Pinnell S.R., Krane S.M., Kenzora J.E., Glimcher M.J. A heritable disorder of connective tissue. Hydroxylysine-deficient collagen disease. N. Engl. J. Med. 1972;286:1013–1020. doi: 10.1056/NEJM197205112861901. [DOI] [PubMed] [Google Scholar]
- 26.Takaluoma K., Lantto J., Myllyharju J. Lysyl hydroxylase 2 is a specific telopeptide hydroxylase, while all three isoenzymes hydroxylate collagenous sequences. Matrix Biol. 2007;26:396–403. doi: 10.1016/j.matbio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Superti-Furga A., Unger S., the Nosology Group of the International Skeletal Dysplasia Society Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. 2007;143A:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 28.Taylor K.M., Nicholson R.I. The LZT proteins: The new LIV-1 subfamily of zinc transporters. Biochim. Biophys. Acta. 2003;1611:16–30. doi: 10.1016/s0005-2736(03)00048-8. [DOI] [PubMed] [Google Scholar]
- 29.Eide D.J. Zinc transporters and the cellular trafficking of zinc. Biochim. Biophys. Acta. 2006;1763:711–722. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Taylor K.M., Morgan H.E., Johnson A., Nicholson R.I. Structure-function analysis of HKE4, a member of the new LIV-1 subfamily of zinc transporters. Biochem. J. 2004;377:131–139. doi: 10.1042/BJ20031183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang L., Kirschke C.P., Zhang Y., Yu Y.Y. The ZIP7 gene (Slc39a7) encodes a zinc transporter involved in zinc homeostasis of the Golgi apparatus. J. Biol. Chem. 2005;280:15456–15463. doi: 10.1074/jbc.M412188200. [DOI] [PubMed] [Google Scholar]
- 32.Taylor K.M., Morgan H.E., Smart K., Zahari N., Pumford S., Ellis I.O., Robertson J.F.R., Nicholson R.I. The emerging role of the LIV-1 subfamily of zinc transporters in breast cancer. Mol. Med. 2007;13:396–406. doi: 10.2119/2007-00040.Taylor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuderman L., Myllylä R., Kivirikko K.I. Mechanism of the the prolyl hydroxylase reaction. 1. Role of cosubstrates. Eur. J. Biochem. 1977;80:341–348. doi: 10.1111/j.1432-1033.1977.tb11888.x. [DOI] [PubMed] [Google Scholar]
- 34.Puistola U., Turpeenniemi-Hujanen T.M., Myllylä R., Kivirikko K.I. Studies of the lysyl hydroxylase reaction. II. Inhibition kinetics and the reaction mechanism. Biochim. Biophys. Acta. 1980;611:51–60. doi: 10.1016/0005-2744(80)90041-8. [DOI] [PubMed] [Google Scholar]
- 35.Berg R.A., Steinmann B., Rennard S.I., Crystal R.G. Ascorbate deficiency results in decreased collagen production: Under-hydroxylation of proline leads to increased intracellular degradation. Arch. Biochem. Biophys. 1983;22:681–686. doi: 10.1016/0003-9861(83)90338-7. [DOI] [PubMed] [Google Scholar]
- 36.Yamasaki S., Sakata-Sogawa K., Hasegawa A., Suzuki T., Kabu K., Sato E., Kurosaki T., Yamashita S., Tokunaga M., Nishida K. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007;177:637–645. doi: 10.1083/jcb.200702081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neyzi O., Furman A., Bundak R., Gunoz H., Darendeliler F., Bas F. Growth references for Turkish children aged 6 to 18 years. Acta Paediatr. 2006;95:1635–1641. doi: 10.1080/08035250600652013. [DOI] [PubMed] [Google Scholar]
- 38.Neyzi O., Binyildiz P., Alp H. Growth centile charts from Turkish children (Türk çocuklarýnýn persentil büyüme eðrileri). Ist. Tip Fak. Mecm. 1978;41(Suppl. 74) [Google Scholar]
- 39.Prader A., Largo R.H., Molinari L., Issler C. Physical growth of Swiss children from birth to 20 years of age. Helv. Paediatr. Acta. 1988;43(Suppl. 52):1–125. [PubMed] [Google Scholar]
- 40.Nellhaus G. Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics. 1968;41:106–114. [PubMed] [Google Scholar]
- 41.Greulich W.W., Pyle S.I. Second Edition. Stanford University Press; Stanford, CA: 1993. Radiographic Atlas of Skeletal Development of the Hand and Wrist. [Google Scholar]