

In the last decade the use of diamino N-heterocyclic carbenes (NHCs) A as ancillary ligands for transition-metal catalysts and as organic catalysts on their own has proven very fruitful.[1] The efficiency of NHCs is attributed to their strong σ-donor properties and sterically demanding structure.[1,2] Not only have NHCs yielded improved transition metal catalysts, but they have also lead to the isolation of unusual low-coordinate metal complexes,[2] which often play a key role in catalytic processes. Recently, we reported the synthesis of stable cyclic (alkyl)(amino)carbenes (CAACs) B[3] and demonstrated that CAACs can be even stronger σ donors than NHCs, and lead to highly efficient palladium catalysts for the α-arylation of ketones and aldehydes. Here we report that the peculiar steric and electronic properties of rigid CAAC ligands allow the preparation of low-coordinate metal complexes, hitherto not isolable with any other ligands.
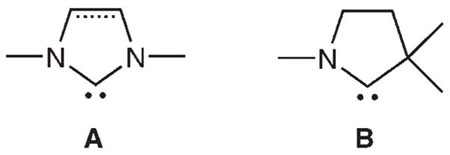
To further evaluate the electronic properties of rigid CAAC B1, we attempted to prepare the [RhCl(CO)2(B1)] complex, since the CO stretching frequencies of this type of rhodium complexes are recognized as an excellent measure of the σ-donor ability of the ligand L, and a large amount of data is available for comparison.[4] Reaction of carbene B1 with half an equivalent of [{RhCl(cod)2}2] (cod = 1,5-cyclooctadiene) afforded complex 1, which after purification by column chromatography on silica gel was obtained as orange crystals in 79% yield (Scheme 1). The structure of 1 was unambiguously determined by single-crystal X-ray diffraction.[5] Following the classical procedure, a solution of 1 in chloroform was treated with an excess of CO. Surprisingly, the 13 C NMR spectrum of the product (95% yield) showed only one CO signal, with a large coupling constant (JRh,C = 134 Hz), and not two signals with smaller J values (JRh,C = 33–82 Hz), as observed for [RhCl(CO)2(L)] complexes.[4] It is known that when bulky ligands L are present [RhCl(CO)2L] complexes can readily lose one CO ligand to give a chloro-bridged dimer.[6] However, single-crystal X-ray diffraction[5] revealed that even in the solid state complex 2 is formally a 14-electron [RhCl(CO)L] monomer (Figure 1).
Scheme 1.
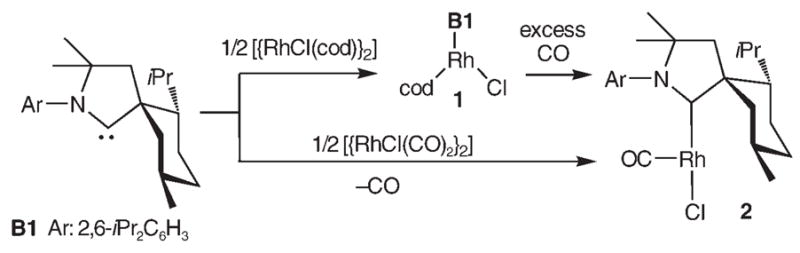
Synthesis of [RhCl(CO)(B1)] complex 2.
Figure 1.
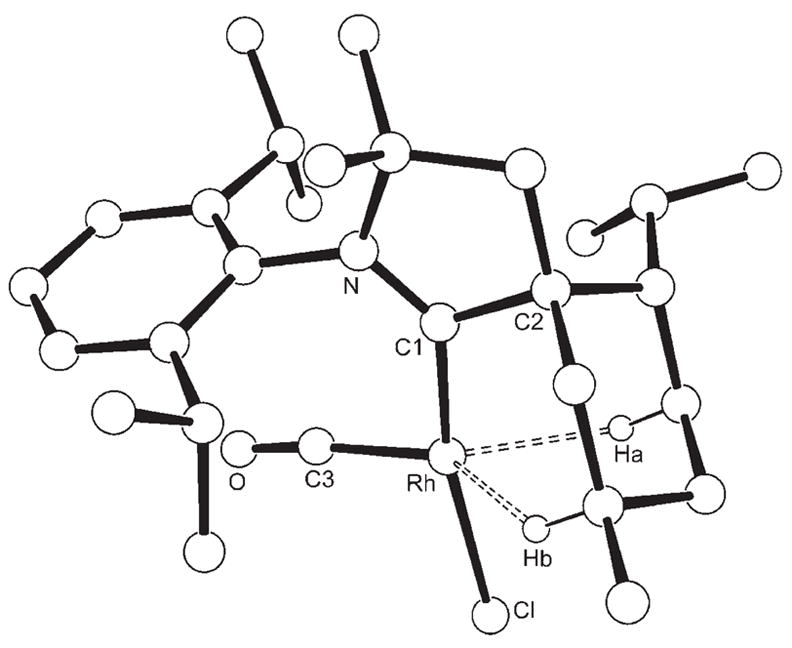
Molecular structure of 2 in the solid state. Selected bond lengths [Å] and angles [°]: N–C1 1.3174(14), C1–C2 1.5368(15), C1–Rh 1.9399(10) Rh–Cl 2.3740 (3), Rh–C3 1.7955(11), C3–O 1.1433(14), Rh–Ha 2.183(17), Rh–Hb 2.231(17); N-C1-C2 108.70(9), C1-Rh-C3 97.99(5), C3-Rh-Cl 94.55(4), C1-Rh-Cl 167.35(3), Rh-C3-O 173.46(10).
Interestingly, 2 can also be synthesized in 80% yield by reaction of one equivalent of carbene B1 with half an equivalent of [{RhCl(CO)2}2]. The extreme hindrance provided by the menthyl ring of CAAC B1, which is locked in the most sterically demanding conformation with respect to the metal center, explains in part why dimerization does not occur. Moreover, the short Rh–Ha (2.183 Å) and Rh–Hb (2.231 Å) distances suggest the existence of agostic interactions, which bring additional stabilization to the metal center. In solution, an agostic interaction is also apparent in the 1H NMR spectrum, which shows a broad multiplet (1H) at 0.08 ppm. Complex 2 is indefinitely stable at room temperature in air. Related [RhClL2] complexes, exemplified by the active species of Wilkinson3s catalyst [RhCl(PPh3)2], are known only as transient species.[7] They have only been generated in situ by ligand dissociation[8] or by hapticity changes;[9] otherwise they readily form chloro-bridged dimers, even when two very bulky ligands L are present.[6] Although other neutral T-shaped formally 14-electron RhI complexes have been isolated, they are still very rare, and none of them have a bridging-capable halide ligand like complex 2.[10,11]
The isolation of this low-coordinate RhI complex 2 made us confident that other low-ligated transition-metal complexes could be stabilized by rigid CAAC B1. After close scrutiny of the literature, it came to our attention that other ligands, particularly NHCs, have consistently failed to allow the isolation of cationic 14-electron [Pd(All)(L)]+ complexes (Scheme 2, All =allyl).[12] In 2003, Nolan et al.[12a] were able to abstract chloride from complex 3, but the cationic complex generated was either unstable and decomposed upon workup or isolated and structurally characterized as an acetonitrile adduct. In 2005, Porschke et al.[12b] reported that chloride abstraction with thallium salts from NHC complexes 3 led to ionic binuclear compounds, which result from combination of the transiently formed desired cationic species with the starting neutral complex. Most recently, Glorius et al.[12c] generated cationic complex 4, which, despite intramolecular stabilization by double-bond complexation, appeared to be rather unstable and was only characterized by 1H NMR spectroscopy.
Scheme 2.
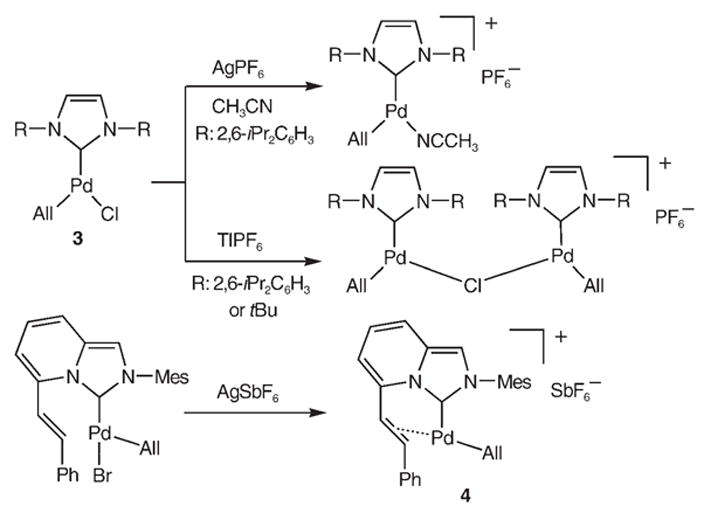
Previous attempts to prepare cationic 14-electron [Pd(All)(L)]+ complexes.
In marked contrast, chloride abstraction from [PdCl-(All)(B1)] (5)[3] with a stoichiometric amount of AgBF4 produced the elusive cationic palladium complex 6 as yellow crystals (88% yield, Scheme 3). The structure of 6 was unambiguously established by single-crystal X-ray diffraction (Figure 2).[5] As expected, 6 has a T-shaped geometry with no interaction between the metal atom and tetrafluoroborate anion (shortest Pd–F distance 3.88 Å). Similar to the above-mentioned RhI complex 2, at least one of the axial H atoms of the menthyl ring provides a stabilizing agostic interaction (Pd–Ha 2.048, Pd–Hb 2.509 Å). This interaction is also apparent from the 1H NMR spectrum, which shows a broad multiplet (1H) at −0.17 ppm. This is the first example of a stable, formally 14-electron, PdII cation.[13]
Scheme 3.
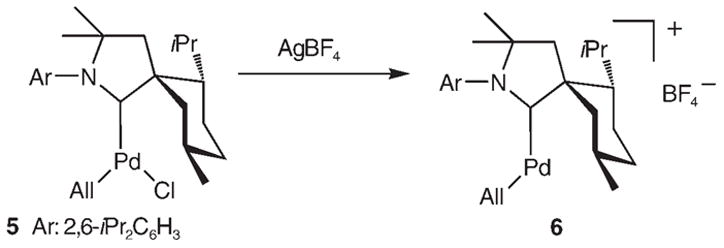
Synthesis of [Pd(All)(B1)]+ complex 6.
Figure 2.
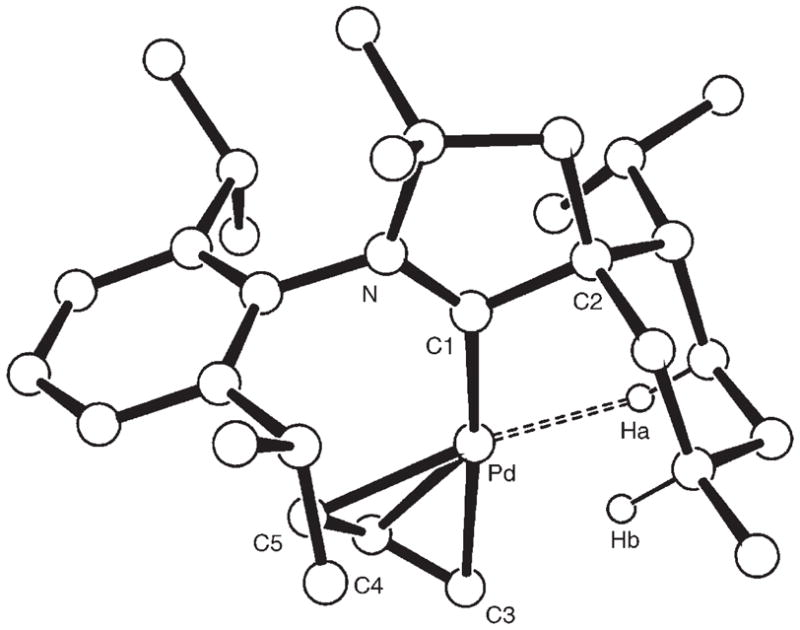
Structure of the cation of 6 in the solid state. Selected bond lengths [Å] and angles [°]: N–C1 1.293(3), C1–C2 1.519(3), C1–Pd 2.038(3), Pd–Ha 2.048, Pd–Hb 2.509, Pd–C3 2.272(13), Pd–C4 2.148(4), Pd–C5 2.126(9); N-C1-C2 109.4(2), N-C1-Pd 133.96(18), C2-C1-Pd 116.64(15).
To further demonstrate the increased stabilization of metal centers provided by the rigid menthyl moiety of carbene B1, we attempted to prepare complexes analogous to 2 and 6, using the CAAC B2, featuring the unlocked and therefore flexible cyclohexyl ring.[3,14] Treatment of [{RhCl(CO)2}2] with two equivalents of carbene B2 afforded exclusively the classical dicarbonyl complex 7, while treatment of 8[3] with AgBF4 led to rapid formation of palladium black (Scheme 4). Clearly, in contrast to B1, the cyclohexane ring of CAAC B2 can flip to the less sterically demanding conformation, in which no H atoms are available for strong agostic interactions with the metal center.
Scheme 4.
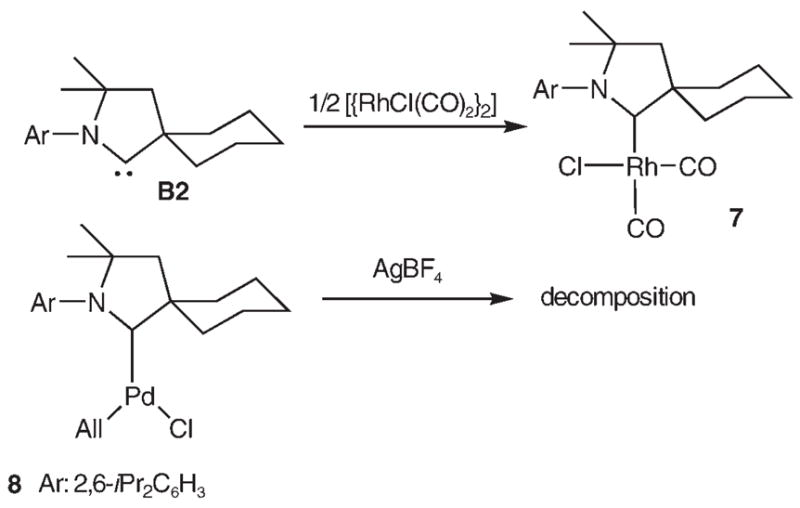
Synthesis of [RhCl(CO)2(B2)] complex 7, and attempted chloride abstraction from [PdCl(All)(B2)] complex 8.
The isolation of complexes 2 and 6 clearly demonstrates the advantages of rigid CAACs over classical ligands. By manipulating the quaternary carbon atom adjacent to the carbene center it is possible to design ligands with an electronically active wall of protection for the metal center. We are currently exploring the possibility that CAAC ligands will stabilize even lower coordinate metal complexes.
Experimental Section
All manipulations were performed under an inert atmosphere of argon by using standard Schlenk techniques. Dry, oxygen-free solvents were employed. 1H and 13C NMR spectra were recorded on Varian Inova 300, 500 and Bruker Avance 300 spectrometers.
1
A solution of carbene B1[3] (0.45 g, 1.18 mmol) in THF (10 mL) was added at −78°C to a stirred solution of [{RhCl(cod)2}2] (0.26 g, 0.53 mmol) in THF (5 mL). The solution was warmed to room temperature and stirred for 3 h. After evaporation of the solvent under vacuum, washing with hexane (15 mL), and purification by column chromatography on silica gel with diethyl ether as eluant, complex 1 was obtained as an orange powder (0.52 g, 79%). Recrystallization from diethyl ether/pentane at −20°C afforded 1 as orange crystals (m.p. 184–186°C). (CHCl3). 1H NMR (CDCl3, 25 °C, 300 MHz): δ=7.41–7.54 (m, 2H, Har), 7.17–7.20 (m, 1H, Har), 5.44 (m, 1H, CHcod), 4.65 (m, 1H, CHcod), 4.21 (m, 1H, CHcod), 3.06–3.23 (m, 5H), 2.68 (m, 2H), 2.51 (d, 1H, J = 12.9 Hz), 1.99–2.13 (m, 4H), 1.77 (d, 3H, CHCH3, J = 6.0 Hz), 1.45–1.71 (m, 10H), 1.29 (d, 3H, CHCH3, J = 6.6 Hz), 1.24 (d, 3H, CHCH3, J = 6.6), 1.20 (s, 6H), 1.11 (d, 3H, CHCH3, J = 6.3 Hz), 1.04 (d, 3H, CHCH3, J =6.3 Hz), 0.97 (d, 3H, CHCH3, J =6.6 Hz), 0.92 ppm (d, 3H, CHCH3, J = 6.3 Hz). 13C NMR (CDCl3, 25 °C, 75 MHz): δ=278.26 (d, J =44.5 Hz), 148.76, 146.13, 138.42, 128.94, 127.28, 124.30, 100.03 (d, J =6.0 Hz), 99.72 (d, J =5.3 Hz), 76.62, 72.44 (d, J =16.2 Hz), 70.69, 60.43 (d, J =14.5 Hz), 54.96, 51.34, 49.81, 36.42, 35.53, 33.69, 32.93, 31.63, 29.63, 29.34, 29.13, 29.03, 28.48, 27.97, 27.70, 26.71, 25.78, 25.59, 24.90, 22.35, 22.27 ppm.
2
Carbon monoxide was bubbled (60 min) through a solution of complex 1 (0.58 g, 0.92 mmol) in chloroform (15 mL). After evaporation of the solvent under vacuum and washing with hexane (10 mL), complex 2 was obtained as an orange powder (0.48 g, 95%). Alternatively, a solution of carbene B1 (0.40 g, 1.04 mmol) in THF (5 mL) was added at −78°C to a stirred solution of [{RhCl(CO)2}2] (0.18 g, 0.47 mmol) in THF (5 mL). The solution was warmed to room temperature and stirred for 2 h. After evaporation of the solvent under vacuum and washing with hexane (20 mL), a solid residue was obtained (0.41 g, 80%). Recrystallization from chloroform by slow evaporation at room temperature afforded 2 as orange crystals (m.p. 251°C decomp). (CHCl3). 1H NMR (CDCl3, 25°C, 300 MHz): δ=7.45 (m, 1H, Har), 7.28 (m, 2H, Har), 2.83 (sept, 2H, CHCH3, J = 6.6 Hz), 2.60 (m, 1H), 2.47 (d, 1H, J = 13.5 Hz), 2.27 (m, 1H), 1.89–2.12 (m, 5H), 1.61 (d, 3H, CHCH3, J = 6.6 Hz), 1.24–1.44 (m, 20H), 1.13 (d, 3H, CHCH3, J = 5.7), 1.06 (d, 3H, CHCH3, J = 6.9), 0.08 ppm (m, 1H, Hagos). 13C NMR (CDCl3, 25 °C, 75 MHz): δ= 248.47 (d, J =48.8 Hz), 181.21 (d, J =134.3 Hz), 146.20, 145.72, 136.90, 130.28, 125.82, 125.70, 77.67, 71.85, 51.85, 50.39, 46.26, 40.60, 35.13, 34.78, 30.58, 30.38, 29.13, 27.11, 26.43, 26.26, 24.29, 24.16, 22.63, 20.19 ppm. IR (CH2Cl2): ν̃(CO) = 1989 cm−1.
6
A 1:1 mixture of carbene complex 5[3] (0.50 g, 0.9 mmol) and silver tetrafluoroborate was cooled to −40°C and 5 mL of fluorobenzene was added. The suspension was warmed to room temperature and stirred for 30 min. After filtration, evaporation of the solvent under vacuum, and washing with hexane (10 mL), a solid residue was obtained (0.48 g, 88%). Recrystallization from toluene/fluorobenzene at −20°C afforded 6 as yellow crystals (m.p. 157–159°C decomp). (C6H5F). 1H NMR (C6D5F, 25°C, 300 MHz): δ=6.78–7.15 (m, 3H, Har), 5.00 (m, 1H, Hallyl), 3.34–3.48 (m, 2H, Hallyl), 2.90 (m, 2H), 2.59 (m, 2H), 2.36–2.41 (m, 1H), 1.91–2.17 (m, 3H), 1.65–1.82 (m, 3H), 1.13–1.22 (m, 12H), 1.06 (d, 3H, CHCH3, J = 6.3 Hz), 1.05 (d, 3H, CHCH3, J = 6.9 Hz), 0.99 (d, 3H, CHCH3, J = 6.9 Hz), 0.98 (s, 3H, CH3), 0.88 (d, 3H, CHCH3, J = 6.9 Hz), 0.73 (d, 3H, CHCH3, J = 6.9 Hz), –0.17 ppm (m, 1H, Hagos). 13C NMR (C6D5F, 25 °C, 75 MHz): δ=251.46, 145.54, 145.33, 133.42, 130.92, 126.33, 126.17, 118.76, 81.45, 70.74, 51.49, 50.60, 45.86, 40.19, 35.20, 33.95, 29.87, 29.17, 28.93, 26.86, 26.16, 25.44, 23.88, 23.60, 22.94, 22.55, 18.58 ppm.
7
A solution of carbene B2 (0.56 g, 1.72 mmol) in THF (10 mL) was added at −78°C to a stirred solution of [{RhCl(CO)2}2] (0.30 g, 0.77 mmol) in THF (10 mL). The solution was warmed to room temperature and stirred for 3 h. After evaporation of the solvent under vacuum, washing with hexane (20 mL), and extraction with chloroform (20 mL), a gray solid residue was obtained (0.67 g, 84%). 1H NMR (CDCl3, 25 °C, 300 MHz): δ=7.42–7.47 (m, 1H, Har), 7.28–7.34 (m, 2H, Har), 3.02 (sept, 2H, CHCH3, J = 6.6 Hz), 2.58 (m, 2H, CH2), 2.09 (s, 2H, CH2), 1.39–1.90 (m, 8H, CH2), 1.34 (d, 6H, CHCH3, J =6.6 Hz), 1.33 (d, 6H, CHCH3, J = 6.6), 1.32 ppm (s, 6H, CH3). 13C NMR (CDCl3, 25 °C, 100.5 MHz): δ=259.82 (d, J =38.4 Hz), 185.95 (d, J =50.7 Hz), 184.61 (d, J =77.3 Hz), 146.41, 134.15, 129.56, 125.49, 81.81, 62.93, 45.27, 38.34, 30.47, 29.12, 28.78, 25.35, 24.94, 22.48 ppm. IR (CH2Cl2): ν̃(CO) = 1994, 2077 cm−1.
Footnotes
We are grateful to the NIH (R01 GM 68825) and RHODIA for financial support of this work.
References
- 1.Recent reviews: Peris E, Crabtree RH. Coord Chem Rev. 2004;248:2239.Crudden CM, Allen DP. Coord Chem Rev. 2004;248:2247.César V, Bellemin-Laponnaz S, Gade LH. Chem Soc Rev. 2004;33:619. doi: 10.1039/b406802p.Enders D, Balensiefer T. Acc Chem Res. 2004;37:534. doi: 10.1021/ar030050j.Kirmse W. Angew Chem. 2004;116:1799.Angew Chem Int Ed. 2004;43:1767.Alder RW, Blake ME, Chaker ME, Harvey JN, Paolini F, Schütz J. Angew Chem. 2004;116:6020. doi: 10.1002/anie.200400654.Angew Chem Int Ed. 2004;43:5896.Canac Y, Soleilhavoup M, Conejero S, Bertrand G. J Organomet Chem. 2004;689:3857.Perry MC, Burgess K. Tetrahedron: Asymmetry. 2003;14:951.Herrmann WA. Angew Chem. 2002;114:1342.Angew Chem Int Ed. 2002;41:1290.Jafarpour L, Nolan SP. Adv Organomet Chem. 2001;46:181.Enders D, Gielen H. J Organomet Chem. 2001;617–618:70.
- 2.Recent review: Scott NM, Nolan SP. Eur J Inorg Chem. 2005:1815.
- 3.Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G. Angew Chem. 2005;117:5851. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:5705. [Google Scholar]
- 4.a) Alcarazo M, Roseblade SJ, Cowley AR, Fernandez R, Brown JM, Lassaletta JM. J Am Chem Soc. 2005;127:3290. doi: 10.1021/ja0423769. [DOI] [PubMed] [Google Scholar]; b) Martin D, Baceiredo A, Gornitzka H, Schoeller WW, Bertrand G. Angew Chem. 2005;117:1728. doi: 10.1002/anie.200462239. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:1700. [Google Scholar]; c) Canac Y, Conejero S, Donnadieu B, Schoeller WW, Bertrand G. J Am Chem Soc. 2005;127:7312. doi: 10.1021/ja051109f. [DOI] [PubMed] [Google Scholar]; d) Magill AM, Cavell KJ, Yates BF. J Am Chem Soc. 2004;126:8717. doi: 10.1021/ja038973x. [DOI] [PubMed] [Google Scholar]; e) Lavallo V, Mafhouz J, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. J Am Chem Soc. 2004;126:8670. doi: 10.1021/ja047503f. [DOI] [PubMed] [Google Scholar]; f) Mayr M, Wurst K, Ongania KH, Buchmeiser M. Chem Eur J. 2004;10:1256. doi: 10.1002/chem.200305437. [DOI] [PubMed] [Google Scholar]; g) Herrmann WA, öfele K, von Preysing D, Herdtweck E. J Organomet Chem. 2003;684:235. [Google Scholar]; h) Denk K, Sirsch P, Herrmann WA. J Organomet Chem. 2002;649:219. [Google Scholar]; i) Enders D, Gielen H, Runsink J, Breuer K, Brode S, Boehn K. Eur J Inorg Chem. 1998:913. [Google Scholar]; j) Herrmann WA, Elison M, Fischer J, Köcher C, Artus GRJ. Chem Eur J. 1996;2:772. [Google Scholar]; k) Coleman AW, Hitchcock PB, Lappert MF, Maskell RK, Müller JH. J Organomet Chem. 1985;296:173. [Google Scholar]
- 5.CCDC-278744 (1), -269951 (2), and -265857 (6) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 6.a) Canepa G, Brandt CD, Werner H. Organometallics. 2004;23:1140. [Google Scholar]; b) Canepa G, Brandt CD, Werner H. J Am Chem Soc. 2002;124:9666. doi: 10.1021/ja020560t. [DOI] [PubMed] [Google Scholar]
- 7.Collman JP, Hegedus LS, Norton JR, Finke RG, editors. Principles and Applications of Organotransition Metal Chemistry. University Science Books; Mill Valley, CA: 1987. [Google Scholar]
- 8.a) Ford PC, Netzel TL, Spillett CT, Pourreau DB. Pure Appl Chem. 1990;62:1091. [Google Scholar]; b) Maguire JA, Boese WT, Goldman ME, Goldman AS. Coord Chem Rev. 1990;97:179. [Google Scholar]; c) Vigalok A, Ben-David Y, Milstein D. Organometallics. 1996;15:1839. [Google Scholar]
- 9.a) Hofmann P, Meier C, Hiller W, Heckel M, Riede J, Schmidt MU. J Organomet Chem. 1995;490:51. [Google Scholar]; b) Werner H, Schäfer M, Nürnberg O, Wolf J. Chem Ber. 1994;127:27. [Google Scholar]; c) Schäfer M, Mahr N, Wolf J, Werner H. Angew Chem. 1993;105:1377. [Google Scholar]; Angew Chem Int Ed Engl. 1993;32:1315. [Google Scholar]; d) Price RT, Anderson RA, Muetterties EL. J Organomet Chem. 1984;267:407. [Google Scholar]; e) Fryzuk MD, McConville DH, Rettig SJ. J Organomet Chem. 1993;445:245. [Google Scholar]
- 10.a) Urtel H, Meier C, Eisenträger F, Rominger F, Joschek JP, Hofmann P. Angew Chem. 2001;113:803. doi: 10.1002/1521-3773(20010216)40:4<781::aid-anie7810>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2001;40:781. [Google Scholar]; b) Budzelaar PHM, Moonen NNP, de Gelder R, Smith JMM, Gal AW. Eur J Inorg Chem. 2000:753. [Google Scholar]
- 11.A cationic 14-electron RhI species has been isolated: Yared YW, Miles SL, Bau R, Reed CA. J Am Chem Soc. 1977;99:7076.
- 12.a) Viciu MS, Zinn FK, Stevens ED, Nolan SP. Organometallics. 2003;22:3175. [Google Scholar]; b) Ding Y, Goddard R, Pörschke KR. Organometallics. 2005;24:439. [Google Scholar]; c) Burstein C, Lehmann CW, Glorius F. Tetrahedron. 2005;61:6207. [Google Scholar]
- 13.Neutral, three-coordinate, T-shaped d8 palladium(II) complexes have recently been isolated: Yamashita M, Hartwig JF. J Am Chem Soc. 2004;126:5344. doi: 10.1021/ja0315107.
- 14.a) Altenhoff G, Goddard R, Lehmann CW, Glorius F. J Am Chem Soc. 2004;126:15195. doi: 10.1021/ja045349r. [DOI] [PubMed] [Google Scholar]; b) Altenhoff G, Goddard R, Lehmann CW, Glorius F. Angew Chem. 2003;115:3818. doi: 10.1002/anie.200351325. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:3690. [Google Scholar]