

Abstract
The possibility that organophosphorus (OP) compounds contribute to motor neuron disease (MND) is supported by association of paraoxonase 1 polymorphisms with amyotrophic lateral sclerosis (ALS) and the occurrence of MND in OP compound-induced delayed neuropathy (OPIDN), in which neuropathy target esterase (NTE) is inhibited by organophosphorylation. We evaluated a consanguineous kindred and a genetically unrelated nonconsanguineous kindred in which affected subjects exhibited progressive spastic paraplegia and distal muscle wasting. Affected subjects resembled those with OPIDN and those with Troyer Syndrome due to SPG20/spartin gene mutation (excluded by genetic linkage and SPG20/spartin sequence analysis). Genome-wide analysis suggested linkage to a 22 cM homozygous locus (D19S565 to D19S884, maximum multipoint LOD score 3.28) on chromosome 19p13 to which NTE had been mapped (GenBank AJ004832). NTE was a candidate because of its role in OPIDN and the similarity of our patients to those with OPIDN. Affected subjects in the consanguineous kindred were homozygous for disease-specific NTE mutation c.3034A→G that disrupted an interspecies conserved residue (M1012V) in NTE's catalytic domain. Affected subjects in the nonconsanguineous family were compound heterozygotes: one allele had c.2669G→A mutation, which disrupts an interspecies conserved residue in NTE's catalytic domain (R890H), and the other allele had an insertion (c.2946_2947insCAGC) causing frameshift and protein truncation (p.S982fs1019). Disease-specific, nonconserved NTE mutations in unrelated MND patients indicates NTE's importance in maintaining axonal integrity, raises the possibility that NTE pathway disturbances contribute to other MNDs including ALS, and supports the role of NTE abnormalities in axonopathy produced by neuropathic OP compounds.
Main Text
Exposure to neurotoxic organophosphorous (OP) compounds of sufficient magnitude and duration causes neurodegeneration in humans, livestock, and laboratory animals.1 OP compound exposures occur in industrial or agricultural environments and as the consequences of terrorism, accidents, suicide attempts, or chemical warfare. Recent attention has focused on OP neurotoxicity after the use of Sarin in the 1995 Tokyo subway terrorist incident2 and because of concerns about persistent neurological effects after exposures to neurotoxic OP agents in the Gulf War. During the Prohibition era in the United States, consumption of Jamaica ginger extract (“Ginger Jake”) adulterated with triorthocresyl phosphate (TOCP) led to OP compound-induced delayed neuropathy (OPIDN) in an estimated 50,000 Americans.3–5 Additional outbreaks of OPIDN causing paralysis of tens of thousands of individuals have occurred in Morocco, Fiji, and India due to consumption of cooking oil contaminated with lubricating oil (containing triorthocresyl phosphate).6–11
Neurologic syndromes following OP toxicity are highly variable and depend on such factors as the specific OP compound, dose and duration of exposure, species, age, and various physiologic factors at the time of OP exposure. Whereas acute OP toxicity usually produces acute cholinergic crisis,7 acute cholinergic crises are often absent in OPIDN. In contrast, OPIDN often begins with sensory impairment, ataxia, weakness, muscle fasciculation, and hyporeflexia and may progress to complete flaccid paralysis followed by progressive spastic paraplegia.4,7,12 Neuropathologic analyses of OPIDN have shown distal degeneration of the longest central- and peripheral-nervous-system axons.1,12 Distal axon degeneration is also the primary neuropathologic feature of hereditary spastic paraplegia (HSP) (reviewed in13). Some complicated forms of HSP,13 such as Troyer syndrome (MIM 275900), also exhibit lower-motor neuron involvement.14
OPIDN pathogenesis involves neuropathy target esterase (NTE), a neuronal membrane protein, either through direct OP-induced inhibition of NTE or through generation of OP-NTE neurotoxic complexes (“aged NTE”). NTE's functions are incompletely understood. NTE is capable of hydrolyzing several intrinsic membrane lipids and thus may be a factor in determining the composition of neuronal membranes.15 NTE's catalytic domain for esterase activity has been mapped to a 489 amino acid region between residues 727 and 1216.16 Participation of NTE in a cell-signaling pathway controlling interactions between neurons and accessory glial cells in the developing nervous system has also been proposed.17,18
We studied a consanguineous family of Ashkenazi Jewish ancestry (Figure 1A) and a nonconsanguineous family of northern European ancestry (Figure 1B) in which affected subjects developed childhood onset of insidiously progressive lower-extremity spastic weakness and progressive wasting of distal upper- and lower-extremity muscles. Electrophysiologic studies were consistent with a motor axonopathy affecting upper and lower extremities. Magnetic resonance imaging demonstrated spinal cord atrophy, particularly in the thoracic region. The affected phenotype in each family conformed both to OPIDN and to “Troyer syndrome,” an autosomal-recessive form of HSP associated with distal muscle wasting.14,19 Troyer syndrome patients variably exhibit additional neurologic and systemic abnormalities (delayed milestone acquisition, skeletal abnormalities, and cerebellar, extrapyramidal, and cognitive impairment). These features were not observed in our patients. The University of Michigan Institutional Review Board approved this study.
Figure 1.
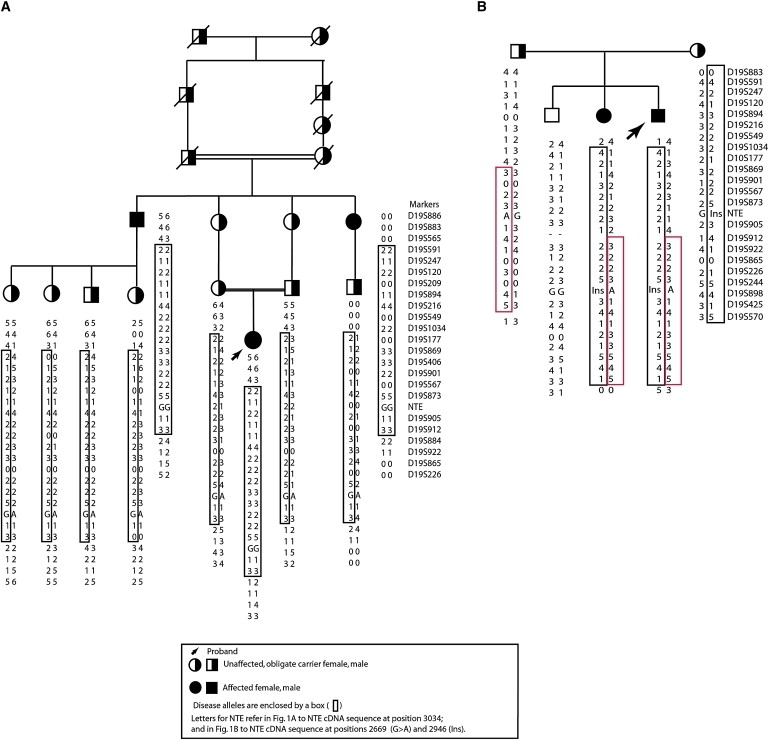
NTE-MND Kindreds
Analyzed portions of NTE-MND kindreds are shown together with haplotypes for linked and flanking microsatellite markers. Letters for marker NTE refer to the NTE sequence at cDNA position 3034 and cDNA position 2669 for the missense mutation and Ins for the insertion at position 2946.
(A) Analyzed portion of index NTE-MND kindred.
(B) Second NTE-MND kindred: affected subjects have compound heterozygous NTE mutations.
We analyzed the SPG20/spartin gene (MIM 607111) coding sequence, mutations in which cause Troyer syndrome (SPG20 HSP),14,19 and found no mutations in the index (consanguineous) family (data not shown), and we excluded the SPG2014 locus in the consanguineous family by genetic-linkage analysis (Table 1). Two-point linkage analyses for exclusion mapping were performed with the MLINK subroutine of the LINKAGE program20 with an autosomal-recessive model of disease inheritance and a disease allele frequency of 0.001. We assigned a genetic penetrance of 0.90 for LOD-score calculations.
Table 1.
Genetic-Linkage Analysis Excludes Chromosome 13q12.3
| Marker | LOD θ = 0 | 0.01 | 0.02 | 0.03 | 0.04 | 0.05 |
|---|---|---|---|---|---|---|
| D13S1841 | −3.4 | −1.23 | −0.94 | −0.77 | −0.66 | −0.57 |
| D13S1842 | −2.19 | −1.19 | −0.92 | −0.76 | −0.64 | −0.56 |
| D13S1851 | −3.4 | −1.23 | −0.94 | −0.77 | −0.66 | −0.57 |
| D13S1843 | −3.4 | −1.23 | −0.94 | −0.77 | −0.66 | −0.57 |
| D13S219 | 0.08 | 0.07 | 0.07 | 0.06 | 0.06 | 0.06 |
| D13S1844 | −2.19 | −1.19 | −0.92 | −0.76 | −0.64 | −0.56 |
Genetic-linkage analysis excludes SPG20 “Troyer syndrome” locus on chromosome 13q12.3.
We then initiated genome-wide linkage analysis (Figure 2) in the larger, consanguineous family with ten subjects (three living affected subjects, seven living unaffected subjects, no spouses of descendants) and in the second smaller, nonconsanguineous family. Marrying-in spouses were asymptomatic and had no evidence of similar neurologic disorders in their families. Maximum-likelihood estimates of marker allele frequencies were estimated from all 15 subjects in both families. The average number of alleles was 4 (range = 3–7), and the average observed heterozygosity of these markers in the families was 0.64 (range = 0.32–0.84).
Figure 2.
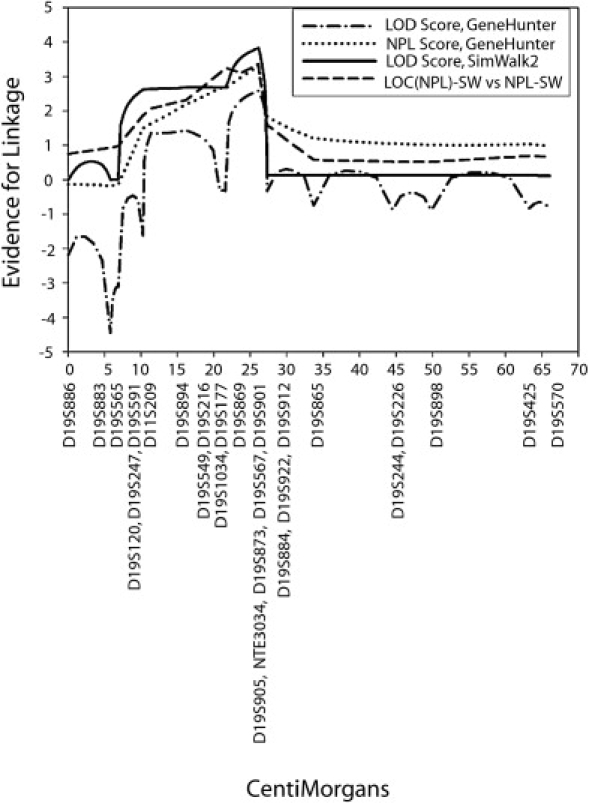
Multipoint Linkage Analysis of Chromosome 19 Markers
Multipoint analyses of markers on chromosome 19 show significant linkage regardless of platform (GeneHunter versus SimWalk2) and analysis assumptions (parametric LOD-score analysis versus nonparametric linkage [NPL]).
We analyzed 400 polymorphic microsatellite markers spaced ∼10 cM apart (ABI MD-10 Linkage Mapping Set), of which 164 markers heterozygous for at least one affected individual excluded 41% of the genome. Six of these markers were homozygous for all affected individuals in the consanguineous family. We analyzed adjacent markers for each homozygous marker. Only markers adjacent to D19S209 yielded an extended linked haplotype that spanned 22 cM between D19S565 and D19S884 on chromosome 19p13 (Figure 1A). In the smaller, nonconsanguineous family, these same markers also resulted in haplotype sharing in affected subjects consistent with genetic linkage of this region (Figure 1B).
The positive chromosome 19 region was analyzed with multipoint methods to incorporate information from all 27 informative markers. The GeneHunter program21 was used to obtain exact multipoint LOD scores and multipoint nonparametric linkage (NPL) scores. The NPL score is based on allele sharing identical by descent (IBD) and does not assume a transmission model. The NPL analysis allows an assessment of the robustness of the result to model misspecification. Because of the size and complexity of the largest pedigree, it had to be trimmed for analysis in GeneHunter. Therefore, analyses were also performed with SimWalk2.22–24 SimWalk2 uses Markov chain Monte Carlo (MCMC) and simulated annealing methods to perform multipoint analyses, allowing computation on large, complex pedigrees. Although the method is not exact, all pedigree members could be included in the analysis. We performed parametric LOD score and NPL analyses in SimWalk2. For each analysis, we set the “parallel runs” flag to generate two simulated annealing runs of the data. The runs produced a maximum score at the same location with the same scores within rounding error. p values given by SimWalk2 were calculated with 10,000 simulations. For nonparametric analyses, p values were used to generate the reported scores (score = −log10 [p value]).
Multipoint LOD score and nonparametric analyses of the two families clearly mirrored the shared haplotype region (Figure 2). With all methods (model-based parametric analysis or nonparametric analysis with either GeneHunter or SimWalk2) used, the maximum LOD score occurred near marker D19S869. Maximum multipoint parametric LOD scores were 2.58 with GeneHunter and 3.82 with SimWalk2. The maximum NPL scores were 3.30 with GeneHunter and 3.36 with SimWalk2. The p value associated with the GeneHunter statistics is 0.002; the simulation-based p value for the SimWalk2 statistics is 0.0004. The index family contributed most to these scores (3.28 and 3.09 for the parametric and NPL SimWalk2 analyses, respectively, and 2.07 and 3.07 for the GeneHunter parametric and NPL analyses, respectively). It is important to note that the second small, nonconsanguineous family supported this peak, with scores close to the maximum expected for an affected-sibling-pair family.
To further characterize the region on chromosome 19 and to fully characterize all shared chromosomal regions, we carried out high-density SNP typing with the Affymetrix 250K NspI chips on all family members for whom DNA was available: 12 subjects in the consanguineous family (including all three affected individuals and two additional unaffected subjects ascertained after the initial microsatellite linkage analysis had been performed) and five subjects in the nonconsanguineous family. We analyzed the SNP data by looking for regions of autozygosity in the entire genome with the Find Autozygous Regions program in the GeneSpring GT analysis package (Agilent). The results of this analysis are shown in Figure 3; they clearly show a single major autozygosity region on chromosome 19 in the three affected individuals. This 4.7 megabase region is defined by the map location from 2784431 to 7541689 bp (NCBI build 35). This region corresponds to the same region identified by the microsatellite markers. The region includes the interesting candidate gene neuropathy target esterase (NTE) (GenBank AJ004832), whose location on chromosome 19 is from 7505075—7532647 bp (UCSC Genome Browser on NCBI build 35).
Figure 3.
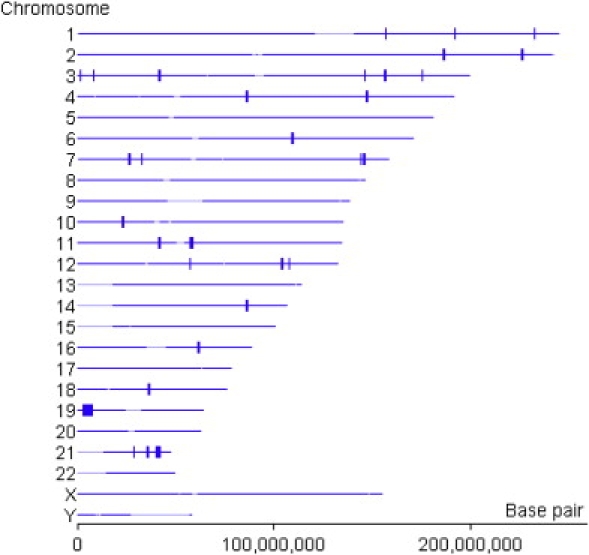
Autozygous Regions in NTE-MND Consanguineous Kindred Identified from 250 K SNP Chips
Output figure from a GeneSpring GT program.
The NTE gene was an obvious candidate because of its role in OPIDN and the similarity of symptoms seen in our patients to those reported for OPIDN. Analysis of NTE's coding sequence in the index family showed that each affected subject was homozygous for (and each obligate carrier heterozygous for) substitution of guanine for adenine at NTE cDNA 3034. This mutation was absent in 105 control subjects (data not shown) and caused substitution of valine for methionine at amino acid position 1012 (M1012V). This mutation disrupts an interspecies conserved residue within NTE's catalytic domain (Figure 4C).25 Mouse, Drosophila, and C. elegans species all contain M at residue 1012. Polyphen analysis of M1012V mutation gave a PSIC profile-difference score of 2.590 predicting that the mutation would be damaging. On the other hand, SIFT analysis predicted that M1012V substitution would be tolerated.
Figure 4.

Disease-Specific Mutations in NTE-MND Kindreds
(A) Homozygous NTE mutation in consanguineous NTE-MND kindred.
(B) Compound-heterozygous NTE mutations in nonconsanguineous NTE-MND kindred.
(C) Disease-specific mutations in NTE-MND kindreds disrupt interspecies conserved residues in the NTE catalytic domain (shown in part). The c.2946_2947insCAGC leads to a frameshift and premature truncation indicated by the sequence in red. Symbols below sequence indicate the following: “∗” indicates sequence identity; “:” indicates that one of strong groups is conserved; “.” indicates that one of weak groups is conserved.
We then analyzed the NTE coding sequence in the second, unrelated, nonconsanguineous family (Figure 1B). Analysis of NTE's coding sequence in affected subjects in this second family showed that they were compound heterozygotes for two NTE mutations. Like the mutation in the index family, these mutations also occurred within NTE's catalytic domain. One allele had a 2669G→A mutation corresponding to an R890H substitution in NTE's catalytic domain. Human and mouse contain R at residue 890, whereas Drosophila and C.elegans contain K at residue 890. Polyphen analysis (PSIC profile-difference score = 0.184) and SIFT analysis predicted that R890H would be a tolerated substitution. The other allele had a four base pair insertion (NTE mRNA position 2946) that caused a frameshift and protein truncation after residue 1019. The truncated protein is predicted to be missing the last 235 residues of NTE's catalytic domain (which extends from amino acid position 727 to position 1216). These mutations were present separately in each carrier parent and absent in 105 control subjects and the unaffected sibling.
In summary, we identified homozygous and compound heterozygous NTE mutations in subjects from two unrelated families with autosomal-recessive progressive spastic paraplegia associated with distal upper- and lower-extremity wasting. Because of its clinical similarity with SPG20 HSP (Troyer syndrome), we reserved for this disease the next-available HSP locus designation (SPG39) and refer to this disorder as NTE-related Motor Neuron Disorder (NTE-MND). These NTE mutations are disease-specific and considered pathogenic for several reasons. First, they were present in affected subjects in two unrelated kindreds and absent in control subjects. Second, they affect amino acid residues within NTE's catalytic domain. Third, NTE plays a central role in OPIDN, an upper- and lower-motor neuron disorder whose symptoms bear a striking resemblance to those exhibited by our NTE-MND patients.
Two mechanisms for NTE involvement in OPIDN have been proposed. The first mechanism proposes that neurotoxicity is the consequence of OP-induced inhibition of NTE. The second mechanism proposes that OP-NTE complexes are toxic. These proposed mechanisms are not mutually exclusive because OP interaction with NTE could both be toxic as well as interfere with NTE activity. The fact that identified NTE mutations in NTE-MND subjects disturb NTE's catalytic domain suggests that they could result in altered NTE activity in vivo. This suggests that modified NTE activity alone could be sufficient to cause corticospinal tract and peripheral motor axonopathy. Nonetheless, it is also possible that the NTE mutations identified result in neurotoxic or “aged” NTE.
Observations that NTE mutations are associated with progressive upper- and lower-motor neuron disease indicate the importance of NTE in maintaining the integrity of corticospinal tract and peripheral motor axons. The finding that NTE mutations underlie corticospinal tract and peripheral motor axon degeneration raises the possibility that other NTE polymorphisms (or genetic variation in factors that regulate or interact with NTE) could contribute to other motor neuron disorders including amyotrophic lateral sclerosis (ALS) and primary lateral sclerosis (PLS).
We have shown that two NTE mutations are the likely cause of autosomal-recessive motor neuron disease that closely resembles OPIDN. These NTE mutations were sufficient to cause the disorder even in the absence of apparent exposure to neurotoxic OP compounds. It will be important to determine whether other polymorphisms in NTE and/or proteins with which it interacts influence the susceptibility to OP-induced neurologic disease. Our findings, together with the recently identified association of PON1 polymorphisms with ALS,26 further support the possibility that neurotoxic OP compounds contribute to motor neuron disease.
Acknowledgments
This research is supported by grants from the University of Michigan Institute of Gerontology (to S.R.); and the Veterans Affairs Merit Review, the National Institutes of Health (National Institute of Neurological Disorders and Stroke [NINDS] R01-NS053917 and R01-NS045163), the Spastic Paraplegia Foundation, and the National Organization for Rare Disorders (to J.K.F.). Those funding this study did not play any role in design and conduct of the study, in the collection, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript. We are grateful for the Elderly Subjects Program of the University of Michigan Institute of Gerontology through which control subjects were ascertained, the technical assistance of Shalini Guduri, the expert secretarial assistance of Lynette Girbach, and the participation of research subjects and their families without whom this investigation would not be possible.
Supplemental Data
Web Resources
The URL for online data listed herein is as follows:
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/.
References
- 1.Abou-Donia M.B. Organophosphorus ester-induced delayed neurotoxicity. Annu. Rev. Pharmacol. Toxicol. 1981;21:511–548. doi: 10.1146/annurev.pa.21.040181.002455. [DOI] [PubMed] [Google Scholar]
- 2.Lotti M., Becker C.E., Aminoff M.J. Organophosphate polyneuropathy; pathogenesis and prevention. Neurology. 1984;34:658–662. doi: 10.1212/wnl.34.5.658. [DOI] [PubMed] [Google Scholar]
- 3.Woolf A.D. Ginger Jake and the blues: A tragic song of poisoning. Vet. Hum. Toxicol. 1995;37:252–254. [PubMed] [Google Scholar]
- 4.Morgan J.P., Penovich P. Jamaica ginger paralysis. Forty-seven-year follow-up. Arch. Neurol. 1978;35:530–532. doi: 10.1001/archneur.1978.00500320050011. [DOI] [PubMed] [Google Scholar]
- 5.Morgan J.P., Tulloss T.C. The Jake Walk Blues. A toxicologic tragedy mirrored in American popular music. Ann. Intern. Med. 1976;85:804–808. doi: 10.7326/0003-4819-85-6-804. [DOI] [PubMed] [Google Scholar]
- 6.Smith H.V., Spalding J.M. Outbreak of paralysis in Morocco due to ortho-cresyl phosphate poisoning. Lancet. 1959;2:1019–1021. doi: 10.1016/s0140-6736(59)91486-2. [DOI] [PubMed] [Google Scholar]
- 7.Taylor P. Ninth Edition. McGraw Hill; New York: 1996. Anticholinesterase agents. Goodman and Gilman's Pharmacologic Basis of Therapeutics. [Google Scholar]
- 8.Sorokin M. Orthocresyl phosphate neuropathy: Report of an outbreak in Fiji. Med. J. Aust. 1969;1:506–508. doi: 10.5694/j.1326-5377.1969.tb92252.x. [DOI] [PubMed] [Google Scholar]
- 9.Srivastava A.K., Das M., Khanna S.K. An outbreak of tricresyl phosphate poisoning in Calcutta, India. Food Chem. Toxicol. 1990;28:303–304. doi: 10.1016/0278-6915(90)90047-q. [DOI] [PubMed] [Google Scholar]
- 10.Sarkar J.K. Outbreaks of paralystic disease in West Bengal due to tricresyl phosphate poisoning. J. Indian Med. Assoc. 1974;63:359–361. [PubMed] [Google Scholar]
- 11.Mehta R.S., Dixit I.P., Khakharia S.J. Toxic neuropathy in Raipur due to triorthocresylphosphate (TOCP) J. Assoc. Physicians India. 1975;23:133–138. [PubMed] [Google Scholar]
- 12.Inoue N., Fujishiro K., Mori K., Matsuoka M. Triorthocresyl phosphate poisoning - A review of human cases. J. UOEH. 1988;10:433–442. doi: 10.7888/juoeh.10.433. [DOI] [PubMed] [Google Scholar]
- 13.Fink J.K. Hereditary spastic paraplegia. In: Rimoin D., Connor J.M., Pyeritz R.E., Korf B.R., editors. Emery and Rimoin's Principles and Practice of Medical Genetics. 5 ed. Churchill Livingstone Elsevier; Philadelphia: 2007. pp. 2771–2801. [Google Scholar]
- 14.Patel H., Cross H., Proukakis C., Hershberger R., Bork P., Ciccarelli F.D., Patton M.A., McKusick V.A., Crosby A.H. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- 15.van Tienhoven M., Atkins J., Li Y., Glynn P. Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J. Biol. Chem. 2002;277:20942–20948. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- 16.Forshaw P.J., Atkins J., Ray D.E., Glynn P. The catalytic domain of human neuropathy target esterase mediates an organophosphate-sensitive ionic conductance across liposome membranes. J. Neurochem. 2001;79:400–406. doi: 10.1046/j.1471-4159.2001.00562.x. [DOI] [PubMed] [Google Scholar]
- 17.Glynn P. Neuropathy target esterase. Biochem. J. 1999;344:625–631. [PMC free article] [PubMed] [Google Scholar]
- 18.Glynn P. Neural development and neurodegeneration: Two faces of neuropathy target esterase. Prog. Neurobiol. 2000;61:61–74. doi: 10.1016/s0301-0082(99)00043-x. [DOI] [PubMed] [Google Scholar]
- 19.Cross H.E., McKusick V.A. The Troyer syndrome. A recessive form of spastic paraplegia with distal muscle wasting. Arch. Neurol. 1967;16:473–485. doi: 10.1001/archneur.1967.00470230025003. [DOI] [PubMed] [Google Scholar]
- 20.Lathrop G.M., Lalouel J., Julient C., Ott J. Multipoint linkage analysis in humans: Detection of linkage and estimation of recombination. Am. J. Hum. Genet. 1985;37:482–498. [PMC free article] [PubMed] [Google Scholar]
- 21.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: A unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 22.Sobel E., Lange K. Descent graphs in pedigree analysis: Applications to haplotyping, location scores, and marker sharing statistics. Am. J. Hum. Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 23.Sobel E., Sengul H., Weeks D.E. Multipoint estimation of identity-by-descent probablilities at arbitrary positions among marker loci on general pedigrees. Hum. Hered. 2001;52:121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 24.Sobel E., Papp J.C., Jange K. Detection and integration of genotyping errors in statistical genetics. Am. J. Hum. Genet. 2002;70:496–508. doi: 10.1086/338920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lush M.J., Li Y., Read D.J., Willis A.C., Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem. J. 1998;332:1–4. doi: 10.1042/bj3320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slowik A., Tomik B., Wolkow P.P., Partyka D., Turaj W., Malecki M.T., Pera J., Dziedzic T., Szczudlik A., Figlewicz D.A. Paraoxonase gene polymorphisms and sporadic ALS. Neurology. 2006;67:766–770. doi: 10.1212/01.wnl.0000219565.32247.11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.