

Abstract
Developmental ocular malformations, including anophthalmia-microphthalmia (AM), are heterogeneous disorders with frequent sporadic or non-Mendelian inheritance. Recurrent interstitial deletions of 14q22-q23 have been associated with AM, sometimes with poly/syndactyly and hypopituitarism. We identify two further cases of AM (one with associated pituitary anomalies) with a 14q22-q23 deletion. Using a positional candidate gene approach, we analyzed the BMP4 (Bone Morphogenetic Protein-4) gene and identified a frameshift mutation (c.226del2, p.S76fs104X) that segregated with AM, retinal dystrophy, myopia, brain anomalies, and polydactyly in a family and a nonconservative missense mutation (c.278A→G, p.E93G) in a highly conserved base in another family. MR imaging and tractography in the c.226del2 proband revealed a primary brain developmental disorder affecting thalamostriatal and callosal pathways, also present in the affected grandmother. Using in situ hybridization in human embryos, we demonstrate expression of BMP4 in optic vesicle, developing retina and lens, pituitary region, and digits strongly supporting BMP4 as a causative gene for AM, pituitary, and poly/syndactyly. Because BMP4 interacts with HH signaling genes in animals, we evaluated gene expression in human embryos and demonstrate cotemporal and cospatial expression of BMP4 and HH signaling genes. We also identified four cases, some of whom had retinal dystrophy, with “low-penetrant” mutations in both BMP4 and HH signaling genes: SHH (Sonic Hedgehog) or PTCH1 (Patched). We propose that BMP4 is a major gene for AM and/or retinal dystrophy and brain anomalies and may be a candidate gene for myopia and poly/syndactyly. Our finding of low-penetrant variants in BMP4 and HH signaling partners is suggestive of an interaction between the two pathways in humans.
Introduction
Developmental ocular malformations, including anophthalmia (MIM 206900), microphthalmia (MIM 206900), and coloboma (MIM 120200), affect ∼1 in 3–4000 individuals and are responsible for over a quarter of childhood blindness worldwide.1,2 Over half of anophthalmia microphthalmia (AM) cases are associated with systemic anomalies, although over 75% of these are not yet part of well-defined syndromes.2 Although recessive, dominant, and X-linked modes of inheritance of AM have all been described, sporadic or non-Mendelian inheritance patterns are highly prevalent, and this initially hampered the identification of genes responsible for AM. Recently, progress has been made through various methods including a positional candidate gene approach in which recurrent interstitial deletions in chromosome 14q22-q23 associated with AM led to the identification of OTX2 (MIM 600037) as a gene for microphthalmia and retinal dystrophy (MIM 120970).3 Although OTX2 has been identified as a causative gene for AM within this region,3 this may not account for all cases and does not explain a complex phenotype that includes hypopituitarism (MIM 313430) and digit anomalies4,5 because OTX2 is not expressed in the pituitary gland or digits during development.
BMP4 (MIM 112262), which is located in 14q22-q23, is an excellent candidate gene for both ocular malformation and digit anomalies including poly/syndactyly. It is a member of the BMP family and transforming growth factor-β1 (TGF-β1 [MIM 190180]) superfamily of secretory signaling molecules that play essential roles in embryonic development.6,7 The BMP4 gene is composed of four exons (although only exons 3 and 4 are translated); the protein is 408 amino acids long and consists of a TGF-β1 propeptide domain and a TGF-β domain that forms an active dimer.8 Bmp dimers initiate signaling by binding to both type I and type II serine or threonine kinase receptors. Upon ligand binding, the type II receptor phosphorylates a type I receptor, which then triggers an intracellular signaling cascade involving proteins of the SMAD family.
Bmp4 and Bmp7 (MIM 112267), another member of the Bmp family, are critical in dorsoventral patterning of the optic vesicle in mice.9,10 Both Bmp4 and Bmp7 cooperate with Pax6 (MIM 607108), a fundamental eye-development gene, in lens placode formation.11 Furthermore, Bmp4 can replace the activity of Sox2 (MIM 184429), another gene with a critical role in early ocular development,12–14 thereby allowing the progression of eye development in Sox2 null mice.9 In mice, although Bmp4−/− homozygotes die in early embryogenesis, Bmp4+/− heterozygotes exhibit AM (35%), failure of lens induction, anterior segment dysgenesis (MIM 107250), and retinal and optic nerve aplasia (MIM 179900 and 165550, respectively).9,15, 16 Extraocular features include cystic kidney, polydactyly (MIM 603596), and craniofacial malformations.15 Furthermore, excessive Bmp4 signaling can also lead to reduced ocular growth with small and misshapen eyes.17
We screened 215 individuals with ocular malformations, mainly AM, for cytogenetic defects by chromosome analysis, gene deletions by MLPA (multiplex-ligation-dependent probe amplification), and mutations in the BMP4 gene by direct sequencing. Here, we report on the identification of two individuals with a 14q22-q23 deletion, a frameshifting mutation in the coding region of the BMP4 gene in a familial case of anophthalmia, retinal dystrophy, brain malformation, and poly/syndactyly and a BMP4-coding-region missense mutation in another family. We performed structural magnetic resonance imaging (MRI) and connectivity studies of the brain of two individuals with the BMP4 frameshift mutation. We studied the expression patterns of BMP4, SHH (MIM 600725), PTCH1 (MIM 601309), GLI1 (MIM 165220), and GLI3 (MIM 165240) (downstream targets of SHH) in early human embryos and demonstrate cotemporal and cospatial expression of BMP4 and HH signaling partners, thereby highlighting their possible genetic interactions during eye, brain, and digit development. In addition, we describe four individuals with genetic variation in both BMP4 and SHH/PTCH1 genes.
Material and Methods
Patients and Controls
A total of 215 individuals with ocular malformation defects, principally anophthalmia, microphthalmia, and coloboma, were recruited as part of a national anophthalmia study based at Moorfields Eye Hospital, London and Birmingham Children's Hospital, Birmingham, UK. Where possible, ultrasound of the globes and visual electrophysiology were performed. Informed consent was obtained, and each patient was examined by an ophthalmic geneticist (NR) and pediatrician (AS). Ethics approval was obtained for the study from Huntingdon Ethics Committee 04/Q0104/129. DNA from 192 healthy adults (European collection of cell cultures [ECACC]) was screened as controls. Family members of positive variants were analyzed if DNA was available.
Visual Electrophysiology
International-standard full-field electroretinograms (ERGs) were recorded for assessment of generalized retinal function.18 The standard full-field ERG protocol involved the use of gold-foil corneal recording electrodes and incorporated the rod-specific and standard flash ERG, both recorded under conditions of dark adaptation, and the photopic 30 Hz flicker and single-flash ERGs, both recorded after a standard period of light adaptation. A stimulus 0.6 log units greater than the ISCEV-standard flash was also used, so that the bright flash ERG a-wave under conditions of dark adaptation could be better demonstrated. In some young individuals, and those unable to tolerate corneal electrodes and Ganzfeld stimulation, recordings were performed with periorbital skin electrodes, non-Ganzfeld (flash) stimulation, and a shortened pediatric ERG protocol,19 with the dark-adapted bright-flash ERG as an index of rod-system function and the light-adapted 30 Hz flicker ERG for assessment of the cone-system function. Pattern ERGs (PERGs) evoked by high-contrast checkerboard reversal were also recorded according to international standards.20 The PERG P50 component was used as an index of macular function. Pattern and flash visual evoked potentials (VEPs) were recorded in some cases to assess the integrity and function of the intracranial visual pathways.19
DNA Preparation, PCR, and MLPA
Genomic DNA was extracted from whole blood with a Nucleon DNA extraction Kit (Tepnel Life Sciences). Amplification of genomic DNA was performed with 100 ng DNA. Primers are outlined in Tables 1 and 2. The first A of the ATG initiating methionine of all genes described here is numbered nucleotide 1.
Table 1.
Primer Pairs Used to Amplify the BMP4 Gene
| Primer Sequence (5′–3′) |
|||
|---|---|---|---|
| Exon | Annealing Temp (°C) | Forward | Reverse |
| 1 | 63.5 | CTAGGCGAGGTCGGGCGGCTGGAG | CACCTTTTGCCTCGGTCACAGC |
| 2 | 55 | GCTCACGTGACTCCGAGGGGC | CAGGGAGCAGAGTGTGGATATTGTAAAGG |
| 3 | 50.5 | TGCCCCTCCATTTCTAGC | ACTGGGGCTTTGATGTAACC |
| 4a | 60 | TAGGTTTCCCCTGCATAAGC | GCTTAGGGCTACGCTTGG |
| 4b | 55 | ATTAGCCGATCGTTACC | GTCCAGCTATAAGGAAGC |
Table 2.
Primer Pairs Used to Amplify the SHH Gene
| Primer Sequence (5′–3′) |
|||
|---|---|---|---|
| Exon | Annealing Temp (°C) | Forward | Reverse |
| 1a | 60 | TGCCATTCCAGCCCCTGTCTGGGT | CCTAGGGTCTTCTCGGCCACATTGG |
| 1b | 60 | CTCCTCGCTGCTGGTATGCTCGGGAC | TTCCTATCTGCCCGAAGAGCAAACAGAG |
| 2a | 60 | GGCAGGCTGATGGAGGGGCCGGGA | GACGTGGTGATGTCCACTGCGCGGC |
| 2b | 60 | CCACTCAGAGGAGTCTCTGCACTA | AAACGCAGTCATCGCCCAGCGACCCTGC |
| 3a | 50 | TAGGAGCGCGGGCACCAAGCGT | AGTCGTTGTGCGGCGCCACAAAG |
| 3b | 54 | TCACTTTCCTGGACCGCGACGAC | AGTGCAGCCAGGAGCGCGTGCG |
| 3c | 56 | GGTCATCGAGGAGCACAG | GGTCCTTGTTTCCTTAGAGTCTA |
For BMP4 (Ensembl transcript ID ENST00000245451), PCR was carried out with the following: 100 μM dNTP's, 0.1 μM each primer, 5 μl of 10× PCR amplification buffer, and 1 U HotStar Taq DNA polymerase (QIAGEN). For exons 4A and 4B, an extra 3 mM and 1 mM MgCl2 were added, respectively. Thermocycling was performed as follows: step 1, 94°C for 15 min; step 2.1, 94°C for 30 s to 1 min; step 2.2, annealing temperatures as indicated in Table 1 for 30 s to 1 min; step 2.3, 72°C for 30 s to 1 min (35 cycles); and step 3, 72°C for 10 min.
For SHH (Ensembl transcript ID ENST00000297261), PCR was carried out with the following: 200 μM dNTP's (100 μM for exon 3B), 0.1 μM each primer (0.05 μM for exons 2A and 3B and 0.2 μM for exon 3C), 5 μl of 10× PCR amplification buffer, and 1 U HotStar Taq DNA polymerase (QIAGEN). DMSO was added to 10% final concentration for exon 3B, and an extra 1 mM MgCl2 was added for exon 3A. Thermocycling (35–40 cycles) was performed as described above except for exon 3C when the denaturation steps were at 98°C (Table 2).
Regarding PTCH1 (Ensembl transcript ID ENSG00000185920): For case 7, all exons21 of the PTCH1 gene were screened by dHPLC analysis. Because exon 12b showed a variant band pattern (data not shown), it was sequenced.
PCR products were run on a 2% agarose gel for ensuring adequate yield and so that nonspecific products could be ruled out. Unincorporated primers and dNTPs were removed by incubation of 5 μl of PCR product with 1 μl of ExoSapIT (USB) for 1 hr at 37°C and then 15 min at 80°C. DNA sequencing was performed bidirectionally with Big Dye version 3.1 (Applied Biosystems) as recommended by the manufacturer.
Dosage analysis was carried out with MLPA probes designed specifically for exon 4 of both the BMP4 and OTX2 genes (sequences available on request). The probes were diluted to 1.33 fmol/μl, and 1.5 μl of this was used for MLPA in conjunction with a commercially available MLPA probe mix (Holoprosencephaly–P187; MRC-Holland) to provide controls. The standard protocol of MRC-Holland was used, and the initial DNA denaturation volume was reduced to 3.5 μl to allow for the extra 1.5 μl of BMP4/OTX2 probe mix.
Neuroimaging and Diffusion Tractography
Structural images of brain and eyes were obtained on a 3T Trio scanner (Siemens) including fat-suppressed T1-weighted imaging (2D-FSE, 0.6875 × 0.6875 × 3 mm3, TE 8 ms, and TR 750 ms) and proton-density-weighted imaging of the orbits (2D-TSE, 0.47 × 0.47 × 3.6 mm3, TE 13 ms, and 80 ms, TR 3110 ms) and high-resolution T1-weighted imaging of the brain (MP-RAGE, 1 × 1 × 1 mm3, TE 4.53 ms, TR 2200 ms, FA 8°, TI 900 ms). With noninvasive MRI, we assessed the macroscopic structure and connectivity between the thalamus and visual cortex as key structures connecting the visual system by employing probabilistic DT in the brains of case 3 and his affected grandmother and comparing them to imaging findings in nine healthy controls. All image processing was performed with software from FMRIB's Software Library (FSL). Probability distributions of fiber orientation were estimated at every voxel as described previously.22 Diffusion-weighted imaging (DWI) data acquisition was performed as described.23
For the visual masks, a threshold of 50% was applied to cytoarchitectonic probability data for visual areas V1 and V2 from FZ Jülich's SPM Anatomy toolbox.24 These data were then transformed into MNI152 space with affine registration. The two data sets were summed and binarized. Finally, the resulting mask was split up into a left- and right-hemispheric target mask. Standard space masks for thalami were obtained from a previous study, in which they had been defined manually.23 Laterality indices were calculated for assessment of symmetry of connectivity to visual cortex from thalamus. In a separate step, interhemispheric, transcallosal connectivity was assessed by establishment of connectivity probabilities between every voxel in each individual's manually defined callosal masks and predefined cortical targets in standard space.25 Connectivity was assessed as the percentage of connectivity to any of the predefined target masks, expressed as a fraction of the healthy control's overall callosal connectivity to that prespecified target.
In Situ Hybridization
The expression patterns of BMP4, SHH, PTCH1, GLI1, and GLI3 were investigated by nonradioactive RNA in situ hybridization in human embryos from Carnegie stages (CS) 13–23, which correspond approximately to mouse Theiler stages 16–23 (E10.25–E14.5). Human embryos were obtained from the Medical Research Council/Wellcome Trust Human Developmental Biology Resource, with full ethical approval. Preparation of embryo sections and nonradioactive RNA in situ hybridization was performed as described elsewhere.26 The templates for probes were as described previously as follows: BMP4,9 SHH,27 PTCH1,28 GLI1,29 and GLI3.30
Results
Patient Cohort
Of the 215 cases analyzed, 28 had bilateral anophthalmia, 40 had bilateral microphthalmia ± cataract; nine had unilateral anophthalmia with contralateral microphthalmia; 81 had unilateral AM ± coloboma with contralateral normal eye or very minor defect; 34 had unilateral AM with contralateral normal-sized eye but defect, e.g., retinal dystrophy and coloboma; 16 had isolated colobomas (nine bilateral and seven unilateral); and seven had other anomalies including cryptophthalmos (2) (MIM 123570), Peter's anomaly (2) (MIM 106210 and 180500), cone dystrophy (1), microcornea (1), and congenital aphakia (1) (MIM 610256).
MLPA Analysis
Out of a cohort of 215 cases with AM, we identified two cases with de novo cytogenetically visible 14q22-q23 deletions. Case 1 [46XX, del(14)(q22.3q23.2)] had bilateral anophthalmia, hypothyroidism, delayed motor development, partial callosal agenesis (MIM 217990), and cerebellar vermis hypoplasia and case 2 [46XY, del(14)(q22.2q23.1)] had bilateral anophthalmia, microcephaly, sensorineural deafness, cryptorchidism (MIM 219050), partial callosal agenesis, cerebellar and pituitary abnormalities, and developmental delay. The deletions were demonstrated by MLPA to include both BMP4 and OTX2 (Figure 1A and Table 3).
Figure 1.
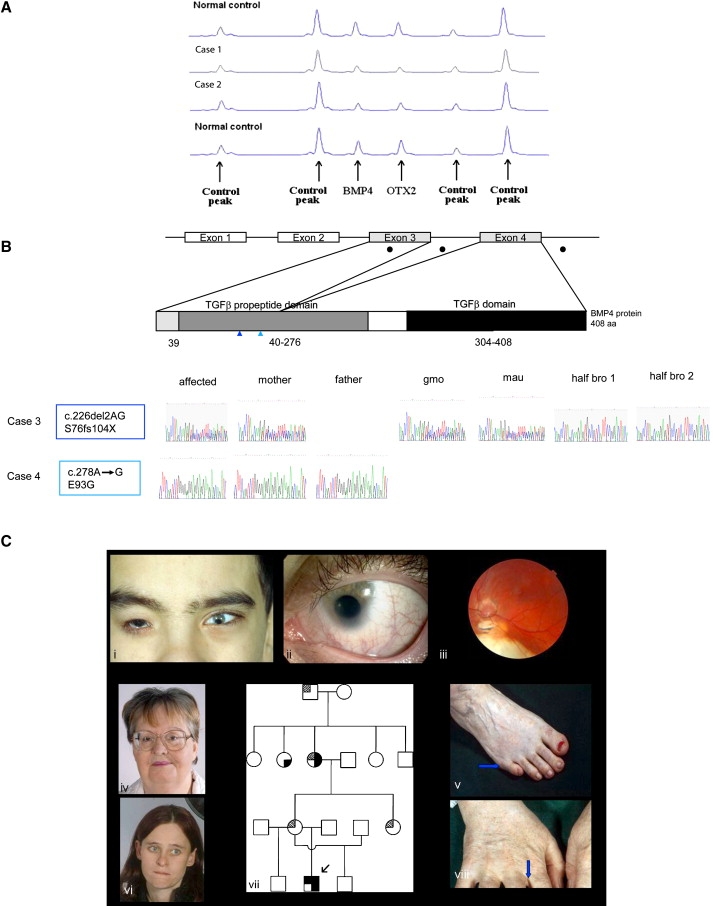
MLPA Analysis of Cases 1 and 2, BMP4 Mutational Analysis of Cases 3 and 4, and Clinical and Pedigree Data of Case 3
(A) MLPA of case 1 and 2. The figure shows a region of the Genotyper trace that includes the BMP4 and OTX2 peaks plus four control peaks. A deletion of the BMP4 and OTX2 MLPA probe binding site is demonstrated by a 50% reduction in peak height.
(B) BMP4 mutation analysis in two families with congenital eye abnormalities. The top row shows a schematic diagram of the human BMP4 cDNA with the positions of the exons and their corresponding translated protein domains. Filled black circles indicate locations of mutations; color-coded vertical arrowheads show the position of each mutation with the corresponding case number. The bottom row shows sequence traces of the affected individuals and their family members (gmo, grandmother; mau, maternal aunt; and bro, brother).
(C) Facial views of case 3 and his family members. (Ci) shows case 3 with right anophthalmia (wearing prosthesis), left microanterior segment with coloboma. (Cii) shows case 3 details of left eye with iris coloboma. (Ciii) shows case 3 left fundus with chorioretinal colobomas, hypoplastic tilted disc. Shown in (Civ), the facial view of case 3эs grandmother. Shown in (Cv), feet of case 3′s grandmother showing position of right postaxial polydactyly (arrow). In (Cvi) is the facial view of case 3′s mother. (Cvii) shows the pedigree of case 3, as follows:  or
or  = myopia;
= myopia;  or
or 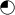 = anophthalmia, microphthalmia and retinal dystrophy;
= anophthalmia, microphthalmia and retinal dystrophy; 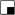 or
or 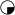 = polydactyly; and
= polydactyly; and 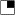 or
or 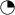 = brain anomalies. Shown in (Cviii), the right hand of case 3's grandmother with webbing.
= brain anomalies. Shown in (Cviii), the right hand of case 3's grandmother with webbing.
Table 3.
Clinical Characteristics and Genetic Findings of BMP4 Mutation Cases
| Mutation |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | BMP4 | Inheritance | SHH Pathway | Inheritance | Right Eye | Left Eye | Ultrasound | EDT | MRI | Refraction | Other | |
| 1 | 46XX,del(14)(q22.3q23.2) | De novo | An, Sc, ONH, and NLP | An, Co, ONH, and NLP | NT | BE: Flash VEP and flash ERG undetectable | Bilateral anophthalmia with no intracranial optic nerves or chiasm; partial agenesis of corpus callosum and hypoplasia of inferior cerebellar vermis; lateral ventricles mildly prominent | Delayed motor development and hypothyroidism | ||||
| 2 | 46XY,del(14)(q22.2q23.1) | De novo | An and NLP | An and NLP | R: ocular remnant, AL 6.9 mm; L: ocular remnant, AL 7.2 mm; no discernible structures within globe remnants | BE: Flash VEPs: undetectable | Marked plagiocephaly, partial cerebellar vermis aplasia, generalized lack of white matter, bilateral anophthalmia, absent optic nerves, chiasm, and tracts | Partial sensori-neural deafness, undescended testes, partial callosal agenesis, cerebellar and pituitary abnormalities, microcephaly, and developmental delay | ||||
| 3 | c.226del2AG (p.S76fs104X) | Maternal | SHH: c.1-125G→A | Unknown | An, NLP | Mc, Co, RD, tilted optic disc. 6/18 | L: AACD 2.1, AL 26.4, posterior staphyloma | LE Ganzfeld flash ERG: generalized rod and cone dysfunction. LE PERG: macular dysfunction. LE VEPs: preserved | Enlarged trigones, hypoplastic corpus callosum, and significant sulcal widening | L: −3.25/−1.75 × 65 | Mild learning difficulties and polydactyly | |
| 3 gmo | c.226del2AG (p.S76fs104X) | Unknown | 6/9 | 6/9 | NT | BE Ganzfeld flash ERG: borderline but no definite abnormality | Enlarged ventricles, hypoplastic corpus callosum, marked sulcal widening associated with diffuse brain atrophy | R: −9.00/−2.25 × 98 L: −8.50/−2.25 × 79 | Polydactyly | |||
| 4 | c.278A→G (p.E93G) | Paternal | Mi, Sc, and OC | Mi, Co, My, Mc, and Ny | R: AACD 0.8 mm, AL 9 mm; total funnel retinal detachment; L: AACD 1.1 mm, AL 20.9 mm | RE flash ERG: generalized rod and cone dysfunction. LE flash ERG; normal. | Delayed myelination and mild reduction in white matter | L: −6.50 | Developmental delay, seizures, undescended testes, simple prominent ears, broad hands, low placed thumbs, and dysplastic nails | |||
| 5∗ | c.370+28G→A | Paternal | MS. 6/36 | Mi and NLP | NT | RE Ganzfeld ERG: normal. RE PERG: subnormal due to macular scar. | NT | R: +0.50/−0.50 × 20 | Hearing loss, skin scars, and kidney and lung abnormalities | |||
| 6∗∗ | c.370+28G→A | Unknown | Ny, Mi, RD, anomalous optic disc, HM, and OC. 3/60 | Mi, Mc, OC, Cat, and NLP | R: AACD 2.2 mm, AL 19.6 mm; L: AACD 0.7 mm, AL 9.5 mm | RE Ganzfeld ERG: generalized retinal dysfunction affecting cones > rods. | Small chiasm, optic nerves on both sides and no cerebral abnormality | R: +4.00/−2.00 × 50 | Early motor and generalized delay, swallowing problems, and high arched palate | |||
| 7 | c.370+28G→A | Maternal | PTCH1: c.1835+1G→A | De novo | Mi and LP | Mi, OC, and NLP | R: long eye; L: short eye∗∗∗ | NT | NT | Mild developmental delay, periorbital BCCs | ||
| 8 | c.370+28G→A | Paternal | SHH: c.1-125G→A | Maternal | Co. 6/9 | Co and Mc. 1/60 | R: AACD 2.8 mm, AL 21.3 mm; L: AACD 2.0 mm, AL 21.2 mm, disc coloboma present | NT | NT | R: +0.75/+2.25 × 90 L: plano/+3.00 × 80 | Mild motor delay, learning difficulties, small septal defect, syndactyly of 2nd/3rd toes, reduced hearing | |
| 9 | c.1217+88C→T | Unknown | SHH: c.1-125G→A | Unknown | An | Mi, G, My, RD, Ny, and Emb. 6/18 | NT | LE Ganzfeld ERG: generalized rod and cone dysfunction; LE pattern and flash VEPs normal. | NT | L: −8.00/−4.00 × 87 | ||
| 10 | c.370+28G→A | Maternal | RPEM. 6/6 | Mi, Cor, and OC | L: AL 13.7 mm, posterior coloboma 9.2 mm | RE Ganzfeld ERG and flash VEP: normal; LE Ganzfeld ERG and flash VEP: undetectable. | Normal | R: +2.00/+0.75 × 90 | ||||
| 11 | c.370+28G→A | Paternal | Cat. 6/9 | Mi, Sc, and Aph | R: AACD 2.6 mm, AL 21.4 mm; L: AACD 0.5 mm, AL 22.0 mm. | NT | NT | R: +1.00/−0.50 × 70 | ||||
| 12 | c.1217+88C→T | Unknown | N. 6/6 | Mi, Cy, NPL | NT | NT | NT | Fifth finger in turning of distal phalanx. | ||||
| 13 | c.1217+88C→T | Maternal | Mi, Cat, PHPV, and NLP | 6/9 | R: AL 18 mm; L: AL 21.8 mm | NT | NT | L: +1.50/−0.75 × 180 | ||||
| 14 | c.1217+88C→T | Paternal | Mi and NLP | Mi, Co, Ny, and Aph. 0.8 at 1 m | R: AL 17.2 mm, L: AL 18.9 mm; R: disorganized globe/cyst extending to orbit, superior coloboma; L: aphakia, posterior coloboma | RE flash ERG: undetectable; LE flash ERG: mild generalized rod and cone system dysfunction | Normal | R: +5.00/−2.00 × 70 | Craniofacial dysmorphism, R dermoid cyst R eyebrow, hearing loss associated with middle ear effusion | |||
The following abbreviations are used: An, anophthalmia; Mi, microphthalmia; Sc, sclerocornea; Co, coloboma; RD, retinal dystrophy; OC, orbital cyst; Mc, microcornea; MS, macular scar; Ny, nystagmus; BCC, basal cell carcinoma; G, glaucoma; My, myopia; HM, hypermetropia; NLP, no light perception; and Cor, corectopia; Cat, cataract; PHPV, persistent hyperplastic primary vitreous; ONH, optic nerve Hypoplasia; NT, not tested; RPEM, retinal pigment epithelial mottling; Aph, aphakia; Emb, embryotoxon; AL, axial length (normal: 22.5–24.5 mm, normal average midrange: 23.5 mm); AACD, anatomical anterior chamber depth (posterior cornea to anterior lens, normal average: 2.5 mm); EDT, electrodiagnostic test; ERG, electroretinogram; PERG, pattern ERG; VEP, visual evoked potential; RE, right eye; LE, left eye; and BE, both eyes.
Varicella.
Carbamazepine exposure during pregnancy.
Ultrasound done as a baby in 1985.
BMP4 Mutation Analysis
Eighteen of the 215 probands showed sequence variation within the BMP4 gene. The clinical details of fourteen cases are shown in Table 3.
Case 3 (Figures 1Ci–1Ciii and Table 3) had right anophthalmia and left microanterior segment, with the following measurements: horizontal corneal diameter of 6 mm, anatomical anterior chamber depth of 2.1 mm (N = 2.5 mm), and overall axial length of 26.4 mm (normal range 22.5–24.5 mm, and normal average midrange 23.5 mm). He had iris and chorioretinal coloboma. There was no pigmentary retinopathy, but a retinal dystrophy was detected on electrodiagnostic testing. Full-field ERGs (Figure 2L) show moderate-to-severe amplitude reduction and peak-time delay, consistent with generalized retinal dysfunction affecting both rods and cones. Pattern ERG P50 reduction was consistent with severe macular involvement (Figure 2K). He also had learning difficulties, partial callosal agenesis, and postaxial polydactyly of the feet. He had a c.226del2, p.S76fs104X mutation within the N-terminal region of the TGF-β1 propeptide domain. This is predicted to truncate the protein at amino acid 104, excluding the functional C-terminal region of the TGF-β1 propeptide (amino acids 41–276) and TGF-β1 domains (amino acids 308–408). BMP4 haploinsufficiency is likely to occur through nonsense-mediated RNA decay. The proband's mother (Figure 1Cvi), grandmother (Figure 1Civ), and aunt, who all have high myopia but not AM, carry the same mutation; his normal two half-brothers do not. The grandmother's refraction is approximately −9.00 D, similar to that reported by the maternal aunt who had previously undergone refractive surgery; the mother has myopia by report but was unavailable for study. The myopia in the proband (−4.00 D) is lower than that expected for the large posterior segment, probably because of a small anterior segment. The grandmother's ERG demonstrated some decline in retinal function, but this was not definitely abnormal given her age and given that testing had to be performed under suboptimal conditions (surface electrodes). The grandmother also exhibited polydactyly and finger webbing (Figures 1Cv and 1Cviii); her sister (unavailable for study) has polydactyly.
Figure 2.
MR Imaging, Diffusion Tractography and Electrodiagnostic Results of Case 3 and Controls
MRI structural images (A–F) from case 3 (A and E) sagittal, (B) coronal, (C, D, and F) transaxial views demonstrating thinning of the corpus callosum in (A), and symmetric widening of the lateral ventricles, particularly the trigones in (B) and (C). The hippocampal (arrows, [D]) and sellar structures (E) appear intact. Orbital imaging (F) demonstrates right anophthalmia with a primary orbital implant behind the ocular remnant and slightly enlarged size of the left globe with hypoplasia of anterior structures including iris, lens, and ciliary apparatus. The right optic nerve and chiasm were atrophic, whereas the left optic nerve appeared intact. Thalamo-occipital connectivity scans (G and H) in a representative control subject (G) show symmetrically formed visual pathways (hot colors, for example, red or orange denotes strong evidence of connectivity) and normal structural imaging. In case 3 (H), asymmetry and disruption of the pathways are shown. Diffusion tractography of case 3 and controls (I and J), which assesses connectivity from a manually defined callosal mask to predefined cortical targets, namely prefrontal (PFC), premotor (PMC), primary motor (M1) and somatosensory (S1), occipital (OCC), temporal (TMP), and parietal cortex (PPC), shows reduced interhemispheric connectivity especially relating to M1 and S1 in case 3, whereas connectivity for OCC, TMP, and PPC appears relatively spared. Thalamocortical connectivity score in healthy controls = 1.7 ±0.48 (mean ± SD); case 3 = 4.34; Z = 5.47, p < 0.01). Visual evoked potentials and pattern ERGs (K) and full-field ERGs (L) from the left eye (LE) of case 3 are compared with representative normal examples. Full-field ERGs show moderate-to-severe amplitude reduction and peak-time delay, consistent with generalized retinal dysfunction affecting both rods and cones. Pattern ERG P50 component reduction is in keeping with severe macular involvement. Pattern and flash VEPs are relatively well preserved. LU indicates log units greater (+) or less (−) than the ISCEV standard flash.
Case 4 with bilateral microphthalmia, broad hands, low-placed thumbs, dysplastic nails, cryptorchidism, brain anomalies, seizures, and developmental delay had a c.278A→G, p.E93G transition. This mutation was also present in the father who does not have AM but demonstrates some mild inferior pigmentation of both retinas; this pigmentation could be a forme fruste coloboma. The substituted glutamic acid is highly conserved among multiple mammals and other vertebrates (Xenopus tropicalis and Tetraodon nigroviridis) and is within a stretch of five conserved glutamic acid residues. This suggests that this substitution is likely to be significant and to cause disruption to the structure of the BMP4 protein.
Six individuals (cases 5, 6, 7, 8, 10, and 11) had a c.370+28G→A substitution of a highly conserved base in intron 3, predicted to affect BMP4 splicing (also present in 3/186 control samples; data available upon request). Four individuals (cases 9, 12, 13, and 14) had a c.1217+88C→T change in the 3′ UTR of exon 4 (also present in 6/191 control samples; data available upon request). Other changes in BMP4 that are likely to be inconsequential include two cases with a c.371-24C→T change in intron 3, two cases with a c.76T→C, p.L26L change which was also present in 1/186 controls, two cases with a c.345C→T, p.N115N change, and 152 cases with a c.1-1025C→T change that was present in 60/89 controls.
Cerebral Imaging and Connectivity Studies of Case 3 and Grandmother
Case 3 demonstrated cerebral anomalies including enlarged trigones, widened cerebral sulci, and marked thinning of the corpus callosum on structural MRI (Figures 2A–2F). Furthermore, with DT, he showed pronounced asymmetry in connectivity between the thalamus and the occipital cortex when they were compared to imaging in the control population. Analysis of transcallosal connectivity showed reduced connectivity to cortical targets especially in primary sensorimotor and frontal areas (Figures 2G, 2H, 2I, and 2J).25 The thalamocortical asymmetry could not be the result of right anophthalmia or chiasmal hypoplasia because the changes would affect both pathways symmetrically. Structural MRI on the grandmother who carried the same BMP4 mutation revealed very similar findings to the proband, namely enlarged ventricles, marked widening of the cerebral sulci with generalized atrophy, and marked thinning of the corpus callosum. DT demonstrated asymmetry of the visual pathways. These findings show a remarkable consistency of brain phenotype.
Human Embryo In Situ Hybridization Studies
In light of evidence of an interaction between Bmp4 and Hh signaling partners in animal models,15,31 we investigated patterns of BMP4, SHH, PTCH1, GLI1, and GLI3 expression in the human developing eye, brain, and hand plate.
In the eye, human BMP4 expression is seen dorsally in the optic vesicle (CS13) and developing optic cup (CS14) in keeping with its role in lens induction in animals.9 Later at CS20, BMP4 is seen in the dorsal and ventral rim of the optic cup (Figure 3). PTCH1 expression is seen in the dorsal and ventral rim of the optic cup, whereas SHH is expressed strongly in the posterior optic cup and optic nerve. At CS17, SHH is expressed in the retinal ganglion cells, supporting a role for SHH in retinal organization.32 There is a reciprocal expression pattern of SHH and its antagonist receptor, PTCH1, in the neural retina. GLI1 and GLI3 are expressed posteriorly in the optic cup with reduced expression at the dorsal and ventral rim in a pattern similar to that of SHH. At later stages of development, expression of BMP4, SHH, and PTCH1 are confined to the developing lens (Figures 3L–3N).
Figure 3.
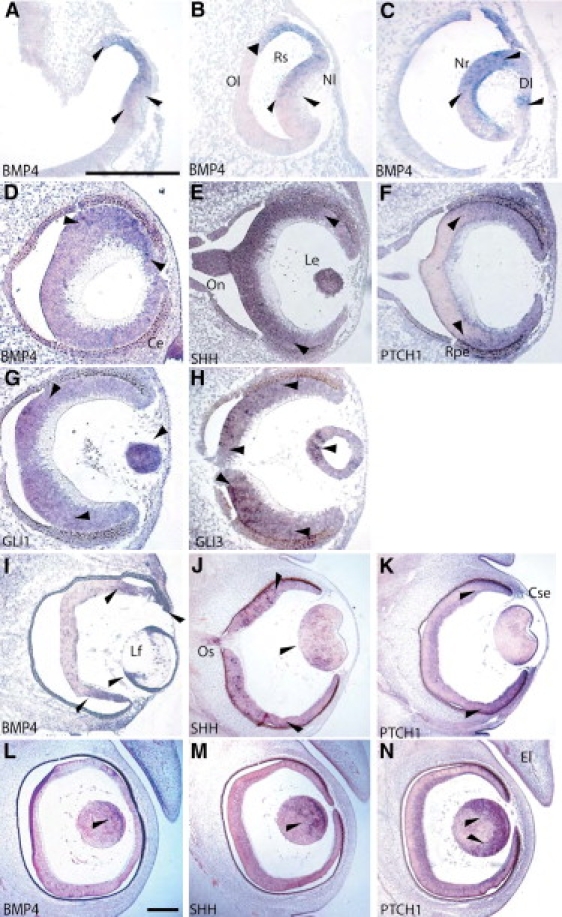
Expression Studies of BMP4, SHH, PTCH1, GLI1, and GLI3 in the Developing Human Eye
Eye formation in the human embryo at CS 13 (A and B), 14 (C), 17 (D–H), 20 (I–K), and 21 (L–N). Coronal sections through the eye were analyzed by in situ hybridization for the expression for BMP4 (A–D), SHH (E, J, and M), PTCH1 (F, K, and N), GLI1 (G), and GLI3 (H). The following abbreviations are used: El, eye lid; Lf, lens fiber; Rs, retina space; Nl, neural layer of optic cup; Ol, outer layer of optic cup; Nr, neural retina; Rpe, retinal pigment epithelium; Le, lens; Ce, corneal ectoderm; On, optic nerve; Os, optic stalk; Cse, corneal epithelium; and Dl, developing lens. The scale bar for (A)–(H) represents 1000 μm and that for (I)–(N) 1000 μm. Arrows indicate areas of strong expression.
In developing brain, BMP4 is strongly expressed in the diencephalic floor, consistent with a role in pituitary development (Figures 4A, 4F, and 4K), under the floor plate (Figures 4N and 4Q) and in the medial ganglionic eminence (MGE) (Figures 4A, 4F, and 4K). Strong expression of SHH is seen in the floor plate and the MGE of the diencephalon (Figures 4B, 4G, 4L, 4O, and 4R), and such a finding is consistent with its role in patterning of the central nervous system and oligodendrogenesis within the MGE.33 Expression of PTCH1 is seen in the neuroepithelium lateral to the floor plate, adjacent to the MGE, and very strongly in the roof plate (Figures 4C, 4H, 4M, 4P, and 4S). The MGE will develop into the amygdaloid nuclei in the adult brain. In the temporal lobes of the brain, these are small oval structures that are closely connected to the hypothalamus, the hippocampus, and the cingulate gyrus. They form part of the olfactory and limbic systems and play a role in the sense of smell, motivation, and emotional behavior. Expression of GLI1 is seen more strongly in the ventral diencephalon and within Rathke's pouch (Figures 4D and 4I), which develops into the adenohypophysis of the anterior pituitary. GLI3 expression is seen more strongly in the ventral diencephalon and within the infundibular recess (Figures 4E and 4J), which grows down to meet Rathke's pouch and will form the posterior lobe of the pituitary.
Figure 4.
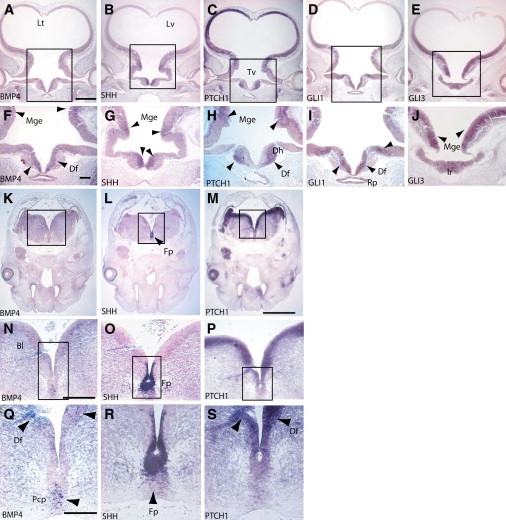
Expression Studies of BMP4, SHH, PTCH1, GLI1, and GLI3 in the Developing Human Brain
Coronal sections through the diencephalon of CS17 and 18 human embryos were analyzed by in situ hybridization for the expression of BMP4 (A, F, K, N, and Q), SHH (B, G, L, O, and R), PTCH1 (C, H, M, P, and S), GLI1 (D and I), and GLI3 (E and J). The following abbreviations are used: Rp, Rathke's pouch; Ir, Infundibular recess; Lt, lamina terminalis; Mge, medial ganglionic eminence; Df, diencephalic floor; Tv, third ventricle; Fp, floor plate; Dh, developing hypothalamus; Pcp, prechordal plate; Lv, lateral ventricle; and Bl, basal lamina. The scale bar for (A)–(E) represents 1000 μm, for (K)–(M), 1000 μm, for (F)–(J), 200 μm, for (N)–(P), 100 μm, and for (Q)–(R), 50 μm. Arrows indicate areas of strong expression.
In the hand, BMP4 is expressed in the interdigital mesenchyme and the joint primordium at CS20, the stage at which the limb buds have formed and are starting to differentiate into fingers (Figures 5A, 5D, and 5G). The extreme distal ends of the fingers are separate, but there is significant webbing more proximally between the digits of the hand. At CS23, which is the end of the embryonic period, the fingers are almost totally separate, and expression of BMP4 at this stage is restricted to the joint-forming region (Figures 5J, 5M, and 5P). There is stronger expression in the joint capsules and the dense connective tissue. PTCH1 and SHH are expressed in mutually exclusive areas in the hand plate (Figure 5). PTCH1 is expressed both proximally and dorsally to the developing digit, whereas SHH expression is seen within the perichondrium surrounding the distal tip of the digit.
Figure 5.
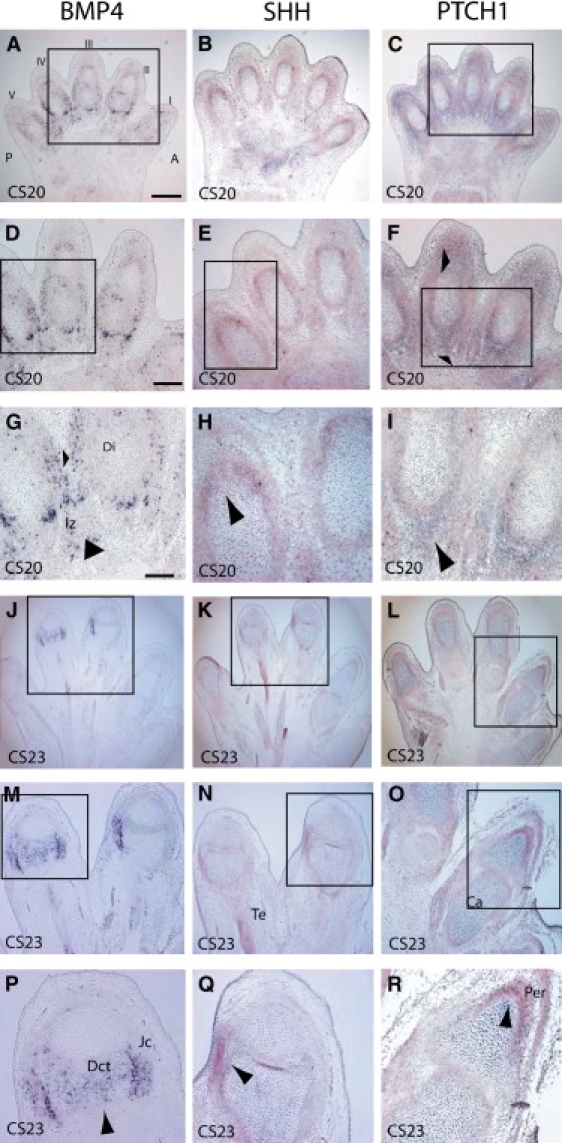
Digit Formation in the Human Embryo at CS20 and 23
All sections were cut across the dorsal ventral axis with respect to the hand plate. In situ analysis of mRNA expression for BMP4 (A, D, G, J, M, and P), SHH (B, E, H, K, N, and Q), and PTCH1 (C, F, I, L, O, and R). (A)–(I) show CS20 stages; (J)–(R) show CS23 stages. The following abbreviations are used: Iz, interdigital zone; Di, digit; Te, tendon; Ca, cartilage; Jc, joint capsule; and Per, pericardium; and Dct, dense connective tissue. The scale bar for (A)–(C) and that for (J)–(L) represent 1000 μm, for (D)–(F) and (M)–(O), 500 μm, and for (G)–(I) and (P)–(R), 50 μm. Arrows indicate areas of strong expression.
Our data show that patterns of expression in eye,9 brain,34 and hand plate35 in the human are similar to the mouse.
SHH and PTCH1 Mutation Analysis
Because cotemporal and cospatial expression of BMP4 and HH signaling genes were observed in the eye, brain, and hand plate, we next investigated whether individuals with mutations in BMP4 had coexisting mutations in SHH and PTCH1. We found three cases with mutations in BMP4 and SHH and one with mutations in BMP4 and PTCH1.
Cases 3 (c.226 del2), 8 (c.370+28G→A), and 9 (c.1217+88C→T) each had a different BMP4 mutation (Table 3), but all had a c.1-125G→A change in the 5′ UTR of SHH predicted to affect an exonic splicing enhancer.36 This SHH change was also present in 16/191 controls, compared to 32/215 individuals from our cohort. Interestingly, case 3′s ocular phenotype was much more severe than those of other family members with the same BMP4 mutation. Although, some family members of cases 3 and 8 had a mutation in BMP4 or SHH, only the affected probands had mutations in both genes; digenic mutations were not present in 191 controls tested. Case 9′s parents were unavailable for study. The clinical features of cases 3, 8, and 9 are shown in Table 3.
Case 7 had bilateral microphthalmia and developed two periorbital basal cell carcinomas in her teens. This phenotype is suggestive of Gorlin syndrome (MIM 109400), which is caused by a mutation in the PTCH1 gene, and we identified a de novo mutation in PTCH1 (c.1835+1G→A; data available upon request) predicted to destroy the intron 12b splice donor site. In addition to this, she had a c.370+28G→A BMP4 mutation. Her mother also had the BMP4 mutation, but only case 7 had mutations in BMP4 and PTCH1. Her eye phenotype is much more severe than that usually described for Gorlin syndrome.
Discussion
Interstitial deletions of chromosome 14q22-q23 are rare, and to date, only seven cases have been described, all with AM.4,5,37–40 We describe two additional cases of AM with extraocular anomalies associated with a de novo 14q22-q23 deletion. We undertook mutation analysis of the BMP4 gene in this region in our cohort of individuals with developmental eye anomalies and identified a previously unreported frameshift mutation in a family with AM, retinal dystrophy, myopia, poly/syndactyly, and brain anomalies that segregated with phenotype. We also identified a missense mutation in an individual with AM, brain, and hand anomalies. Furthermore, we found that BMP4 and HH signaling genes were expressed in a cospatial and cotemporal fashion in the eye, brain, and hand of developing human embryos. We also identified four cases with mutations in both BMP4 and SHH/PTCH1, suggesting the two pathways may act synergistically in eye development in humans.
Deletions in 14q22-q23 are known to be associated with AM, brain, pituitary, and ear anomalies (structural defects and hearing loss), hypothyroidism, poly/syndactyly, clinodactyly, high arched palate, cryptorchidism, and developmental delay.4,37 Similarly, our deletion cases had bilateral AM associated with hypothyroidism, cryptorchidism, partial callosal agenesis, sensorineural deafness, developmental delay, and cerebellar and pituitary abnormalities. The hypothyroidism and cryptorchidism may be related to hypothalamic-pituitary anomalies. Three eye development genes, SIX6 (MIM 606326), OTX2, and BMP4, reside within the 14q22-q23 region; Six6, which is expressed in the pituitary,41 could potentially explain the hypopituitarism in a contiguous gene syndrome; however, it does not appear to be important in human AM.42 Although OTX2 has been identified as one causative gene for AM,3 loss of function of OTX2 does not explain pituitary or digit anomalies.4,5 Bmp4 is important in eye and digit development in vertebrates, and therefore human BMP4 is an excellent candidate gene for ocular, pituitary, and digit anomalies.
We identified a BMP4 frameshift mutation (c.226del2, p.S76fs104X), which would result in haploinsufficiency of the BMP4 protein in a family (case 3) with AM, retinal dystrophy, myopia, and poly/syndactyly (Figure 1). The affected family members have variable eye phenotypes ranging from high myopia in the mother, maternal aunt, and grandmother to unilateral anophthalmia and contralateral microanterior segment, iris and chorioretinal coloboma, and retinal dystrophy in the proband. The presence of retinal dystrophy is of great significance for two main reasons: First, it suggests a role for BMP4 in retinal development and maintenance and second, that individuals with BMP4 mutations may suffer a later, chronic deterioration of vision, sometimes in their only seeing eye, necessitating appropriate advice regarding, for instance, learning of Braille. Three other individuals (cases 6 [BMP4 (c.370+28G→A)], 9 and 14 [BMP4 (c.1217+88C→T)]) had ERGs in the better eye consistent with a retinal dystrophy. Case 6 had generalized retinal dysfunction affecting the cones more than the rods. Case 9 had moderate-to-severe generalized retinal dysfunction affecting rods and cones, similar to case 3, and case 14 had evidence of mild generalized rod- and cone-system dysfunction. Because the finding of AM and retinal dystrophy is not universal in our cohort of AM patients, it suggests a homogeneity of eye phenotype and suggests that these sequence variations in BMP4 may be significant. The proband, his maternal grandmother, and his grandmother's sister have poly/syndactyly, and interestingly, three 14q22-q23 deletion cases with AM also have hand and feet poly/syndactyly.4,5
Individuals with BMP4 mutations may have underlying brain anomalies and developmental delay. Case 3 and his grandmother (BMP4 c.226del2) had structural cerebral anomalies including enlarged trigones, widened sulci, and partial callosal agenesis on MR imaging, indicative of a primary-brain developmental disorder. The grandmother in addition showed marked diffuse brain atrophy, suggesting a progression of the disorder with age. Case 4 with a c.278A→G change showed mild reduction in white matter and delayed myelination. Furthermore, by using DT, which shows brain connections,22 we demonstrate significant asymmetry in thalamo-occipital connectivity in case 3 with diminished left thalamocortical visual pathways and reduced callosal connectivity, especially between primary sensorimotor cortices (Figures 2G–2J).25 The changes discovered were undetectable by conventional imaging, demonstrating the benefits DT offers in the study of complex brain disease. The cerebral findings point to a more complex underlying pathology, not only disturbing ocular development but also independently affecting brain development.
Our in situ hybridization studies show BMP4 is expressed in early developing human eye, brain, and digits, and its expression pattern correlates well with the range of phenotypes observed in individuals with BMP4 mutations. In the early human developing eye, we demonstrate expression of BMP4, SHH, PTCH1, GLI1, and GLI3 in the optic vesicle, optic cup, and retina. Bmp4 has been shown to be expressed in the optic vesicle and optic cup in a number of vertebrate species and is required to enable the optic vesicle to induce the lens by regulating Msx2 (MIM 123101).9 In Bmp4 homozygous null mutant embryos, lens induction is absent,9 and consistent with this, we demonstrate that BMP4 is expressed during early lens induction and later in the lens (Figure 3). Bmp4 and Shh induce expression of a number of transcription factors such as Tbx5 (MIM 601620), Pax2 (MIM 167409), and Vax2 (MIM 604295) within the eye17 and are believed to establish a dorsoventral gradient throughout the eye field.43 We demonstrate that BMP4 is expressed dorsally in the optic vesicle and developing optic cup and ventrally at CS20 when optic-fissure closure has completed, indicative of a requirement for Bmp4 signaling after formation of the optic cup. This is further supported by the observation of ocular coloboma in four cases (1, 3, 4, and 8) with deletion or mutations in BMP4. Interestingly, both a disruption of SHH and its antagonist PTCH1 can also lead to a failure of optic fissure closure and coloboma formation,44,45 suggesting that the delicate balance between agonist and antagonist is important.
In brain, we demonstrate that BMP4 expression is restricted to the diencephalic floor, and such a result would be consistent with a role in pituitary development (Figure 4) and would explain pituitary anomalies observed in 14q22-q23 deletion cases. Strong expression of SHH is seen in the floor plate and the MGE of the diencephalon, and such a finding is consistent with its role in patterning of the central nervous system and oligodendrogenesis within the MGE.33 The MGE, which gives rise to inhibitory neurons, has been shown to be absent in Shh−/− embryos.43 Ptch1 has been shown to act as a sponge for Shh, preventing it from reaching cells further away,46 and the expression of PTCH1 in the neuroepithelium lateral to the floor plate may therefore be limiting the expression of SHH. GLI3, which is a Shh antagonist47 and may directly regulate Bmp4 expression,48 is expressed in the ventral diencephalon. Gli3 is required for the specification of dorsal cell types and for the suppression of ventral cell types in the forebrain.49
Patterning of the limb is controlled by secreted signals (Bmp4, Shh, growth factors, and Wnt7a [MIM 601570]) that coordinate growth and morphogenesis along the anteroposterior, proximodistal, and dorsoventral axes. Digit number is related to the width of the limb bud, which in turn depends on the length of the apical ectodermal ridge,50 a specialized ectoderm that runs around the distal tip of the limb bud. In animals, both Bmp4 and Shh are expressed in the posterior part of the apical ectodermal ridge.51,52 Furthermore, both a reduction of Bmp4 expression in conditional knockouts or an altered Shh expression have been shown to result in polydactyly.53,54 BMP4 is expressed in the interdigital mesenchyme and the joint primordium (Figure 5). At later stages of limb development, mesoderm between the digits undergoes regression by apoptosis, which is controlled by Bmp4, and leads to freeing of the digits.55,56 The expression of BMP4 in the interdigital mesenchyme of humans is therefore consistent with a similar role. At CS23, when apoptosis in the interdigital zones has already occurred and the fingers are almost totally separate, BMP4 expression is restricted to the joint forming region (Figure 5). A decrease in Bmp4 levels leads to a lack of apoptosis in the mesoderm between the fingers and will result in syndactyly of the digits.55 Interestingly, we find that four of our cases (3, 4, 8, and 12) with BMP4 mutations had poly/syndactyly or other hand anomalies. From our human embryo data alone, it would be predicted that BMP4/SHH/PTCH1 are major genes in eye, pituitary, and hand development.
Non-Mendelian inheritance patterns and variable expressivity of AM and other ocular developmental disorders suggest complex mechanisms are at play, including multigenic inheritance and/or interaction with environmental factors. Case 3 (BMP4 c.226del2 mutation) is the only family member who has an additional c.1-125G→A change in the 5′ UTR of SHH (predicted to affect an exonic splicing enhancer)—a combination that may have contributed to a more severe eye phenotype of AM and retinal dystrophy compared with other family members who have myopia ± polydactyly. Our finding of cotemporal and cospatial expression of BMP4 and SHH in early developing human eye, including retina, supports this (Figure 3). Two of our cases (8 and 9) had a sequence variation in BMP4 with additional intronic or UTR changes in highly conserved residues in SHH, and one (case 7) had an additional mutation in PTCH1. The Hh signaling pathway is critical for specification of midline ventral structures, eyes, and digits.57–59 In humans, SHH mutations cause holoprosencephaly (MIM 236100), cyclopia (MIM 236100), and colobomatous microphthalmia,44,60,61 and mutations in the SHH antagonist receptor, PTCH1, can also cause colobomatous microphthalmia as part of Gorlin syndrome (naevoid basal cell carcinoma syndrome).45,62 Interestingly, Gorlin syndrome is only rarely associated with eye defects, and when they do occur, most appear to be mild, (e.g., isolated coloboma or epiretinal membranes) with normal ERGs;45,63 our subject (case 7) had severe bilateral eye anomalies, suggesting that the combination of a PTCH1 and BMP4 mutation may predispose to or worsen an ocular phenotype in Gorlin syndrome. Synergism between Bmp4 and Hh pathways has been observed in animal models whereby enhancement of preaxial polydactyly with additional syndactyly is seen when Bmp4+/− mice are backcrossed to Gli3+/− or Alx4+/− (MIM 605420) mice.15 More recently, Morcillo and colleagues have demonstrated that optic fissure closure in the mouse requires the sequential and independent activity of Bmp7 and Shh.31 Our finding of two cases who have mutations in BMP4 and SHH and who also have poly/syndactyly is highly significant and supports an interaction between these two pathways in humans.
There are common features between our cases and individuals with Bardet Biedl syndrome (BBS [MIM 209900]) including rod-cone dystrophy, polydactyly, hypogonadism, renal anomalies, reduced hearing, and developmental delay.64 However, our cases do not exhibit obesity, and BBS is not associated with AM. Nevertheless, there are common gene networks, e.g., the SHH signaling pathway, that could explain overlapping phenotypes.65 A further example of overlapping phenotypes and gene expression patterns is seen with PITX2 (the gene for Reiger syndrome) (MIM 601542) and BMP4.66,67 Mutations in the PITX2 homeobox gene cause ocular (anterior segment), brain (pituitary), and dental anomalies.68,69 These features are also observed in some of our cases; for instance, case 9 (BMP4 c.1217+88C→T) had glaucoma, and Bmp4+/− mice have anterior segment abnormalities16 and cases 1, 2 (whole gene deletions), and 3 (BMP4 c.226del2) had pituitary abnormalities. Both BMP4 (Figure 4) and Pitx268 are expressed in Rathke's pouch, consistent with a role in pituitary development.
The combination of a BMP4 sequence variation and environmental factors may also increase susceptibility to ocular malformation. Two cases (5 and 6) who have a c.370+28G→A mutation predicted to affect splicing in BMP4 were exposed to potential gestational insults (varicella exposure and maternal carbamazepine intake). Although, both mutations occurred in the noncoding region of BMP4, it is possible that these changes could still be relevant.70 Interestingly, case 5 and 14 have additional loss of hearing, and it has recently been reported that BMP4 heterozygous mice have structural and functional deficits in the inner ear71 and that hearing anomalies are observed in individuals with a 14q22-q23 deletion.5,40 Case 6 has a more severe eye phenotype, retinal dystrophy, developmental problems (motor and cognitive delay), and a high arched palate, which is also interesting because Bmp4 is implicated in animal palate development72 and one 14q22-q23 deletion case had a high arched palate.38 Recent exciting work has suggested that Hsp90AA1 (MIM 140571)/Hsp90AB1 (MIM 140572), one of a class of chaperone proteins, may play a protective role during development and oncogenesis and that its dysfunction may allow the manifestation of otherwise safe and probably highly prevalent low-penetrant mutations.73 Thus, gestational insults such as those described for cases 5 and 6 may be significant.
Our study is the first to combine the ex vivo examination of gene expression patterns with noninvasive in vivo imaging of white-matter pathways in the human brain, and most significantly, we demonstrate that BMP4 is an important gene for ocular malformations associated with poly/syndactyly, pituitary, and brain developmental anomalies. The identification of further low-penetrant gene variations may provide insights into human developmental eye disease and will be possible once large-scale screening of genes can be undertaken.
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/
FMRIB's Software Library, http://www.fmrib.ox.ac.uk/fsl
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
MRC-Holland, http://www.mrc-holland.com/pages/indexpag.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We are grateful to all the referring clinicians, including Mrs. P. Bagga, North Lincolnshire, and Goole Hospital NHS Trust and Dr. O. Quarrell, Sheffield, and thank the families for their support and participation in this research. N.R. is a Senior Surgical Scientist supported by the Academy of Medical Sciences/The Health Foundation. We acknowledge with gratitude generous financial support from VICTA (P.B. and A.M.), the Polak Trust (A.W.), UK BBSRC (J.K.), UK MRC (H.J.B. and C.P.P). and Wellcome Trust (H.J.B.). We are grateful to Dr. Phil Anslow and Dr. Daniela Seixas for valuable comments on structural imaging. Human material was provided by the MRC/Wellcome-funded Human Developmental Biology Resource.
References
- 1.Clementi M., Turolla L., Mammi I., Tenconi R. Clinical anophthalmia: An epidemiological study in northeast Italy based on 368,256 consecutive births. Teratology. 1992;46:551–553. doi: 10.1002/tera.1420460604. [DOI] [PubMed] [Google Scholar]
- 2.Morrison D., FitzPatrick D., Hanson I., Williamson K., van Heyningen V., Fleck B., Jones I., Chalmers J., Campbell H. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: Investigation of genetic aetiology. J. Med. Genet. 2002;39:16–22. doi: 10.1136/jmg.39.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ragge N.K., Brown A.G., Poloschek C.M., Lorenz B., Henderson R.A., Clarke M.P., Russell-Eggitt I., Fielder A., Gerrelli D., Martinez-Barbera J.P. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005;76:1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad M.E., Dada R., Dada T., Kucheria K. 14q (22) deletion in a familial case of anophthalmia with polydactyly. Am. J. Med. Genet. 2003;120A:117–122. doi: 10.1002/ajmg.a.10146. [DOI] [PubMed] [Google Scholar]
- 5.Nolen L.D., Amor D., Haywood A., St Heaps L., Willcock C., Mihelec M., Tam P., Billson F., Grigg J., Peters G. Deletion at 14q22–23 indicates a contiguous gene syndrome comprising anophthalmia, pituitary hypoplasia, and ear anomalies. Am. J. Med. Genet. A. 2006;140:1711–1718. doi: 10.1002/ajmg.a.31335. [DOI] [PubMed] [Google Scholar]
- 6.Wozney J.M., Rosen V., Celeste A.J., Mitsock L.M., Whitters M.J., Kriz R.W., Hewick R.M., Wang E.A. Novel regulators of bone formation: Molecular clones and activities. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 7.Hogan B.L. Bone morphogenetic proteins: Multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- 8.Eimon P.M., Harland R.M. In Xenopus embryos, BMP heterodimers are not required for mesoderm induction, but BMP activity is necessary for dorsal/ventral patterning. Dev. Biol. 1999;216:29–40. doi: 10.1006/dbio.1999.9496. [DOI] [PubMed] [Google Scholar]
- 9.Furuta Y., Hogan B.L. BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 1998;12:3764–3775. doi: 10.1101/gad.12.23.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley A.T., Lyons K.M., Robertson E.J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995;9:2795–2807. doi: 10.1101/gad.9.22.2795. [DOI] [PubMed] [Google Scholar]
- 11.Wawersik S., Maas R.L. Vertebrate eye development as modeled in Drosophila. Hum. Mol. Genet. 2000;9:917–925. doi: 10.1093/hmg/9.6.917. [DOI] [PubMed] [Google Scholar]
- 12.Fantes J., Ragge N.K., Lynch S.A., McGill N.I., Collin J.R., Howard-Peebles P.N., Hayward C., Vivian A.J., Williamson K., van Heyningen V. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 13.Ragge N.K., Lorenz B., Schneider A., Bushby K., de Sanctis L., de Sanctis U., Salt A., Collin J.R., Vivian A.J., Free S.L. SOX2 anophthalmia syndrome. Am. J. Med. Genet. A. 2005;135:1–7. doi: 10.1002/ajmg.a.30642. [DOI] [PubMed] [Google Scholar]
- 14.Bakrania P., Robinson D.O., Bunyan D.J., Salt A., Martin A., Crolla J.A., Wyatt A., Fielder A., Ainsworth J., Moore A. SOX2 anophthalmia syndrome: Twelve new cases demonstrating broader phenotype and high frequency of large gene deletions. Br. J. Ophthalmol. 2007;91:1471–1476. doi: 10.1136/bjo.2007.117929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunn N.R., Winnier G.E., Hargett L.K., Schrick J.J., Fogo A.B., Hogan B.L. Haploinsufficient phenotypes in Bmp4 heterozygous null mice and modification by mutations in Gli3 and Alx4. Dev. Biol. 1997;188:235–247. doi: 10.1006/dbio.1997.8664. [DOI] [PubMed] [Google Scholar]
- 16.Chang B., Smith R.S., Peters M., Savinova O.V., Hawes N.L., Zabaleta A., Nusinowitz S., Martin J.E., Davisson M.L., Cepko C.L. Haploinsufficient Bmp4 ocular phenotypes include anterior segment dysgenesis with elevated intraocular pressure. BMC Genet. 2001;2:18. doi: 10.1186/1471-2156-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behesti H., Holt J.K., Sowden J.C. The level of BMP4 signaling is critical for the regulation of distinct T-box gene expression domains and growth along the dorso-ventral axis of the optic cup. BMC Dev. Biol. 2006;6:62. doi: 10.1186/1471-213X-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marmor M.F., Holder G.E., Seeliger M.W., Yamamoto S. Standard for clinical electroretinography (2004 update) Doc. Ophthalmol. 2004;108:107–114. doi: 10.1023/b:doop.0000036793.44912.45. [DOI] [PubMed] [Google Scholar]
- 19.Holder G.E., Robson A.G. Paediatric electrophysiology: A practical approach. In: Lorenz B., editor. Essentials in Ophthalmology. Springer-Berlag; Berlin: 2006. pp. 133–155. [Google Scholar]
- 20.Holder G.E., Brigell M.G., Hawlina M., Meigen T., Vaegan, Bach M. ISCEV standard for clinical pattern electroretinography–2007 update. Doc. Ophthalmol. 2007;114:111–116. doi: 10.1007/s10633-007-9053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahn H., Wicking C., Zaphiropoulous P.G., Gailani M.R., Shanley S., Chidambaram A., Vorechovsky I., Holmberg E., Unden A.B., Gillies S. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 22.Behrens T.E., Jenkinson M., Robson M.D., Smith S.M., Johansen-Berg H. A consistent relationship between local white matter architecture and functional specialisation in medial frontal cortex. Neuroimage. 2006;30:220–227. doi: 10.1016/j.neuroimage.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 23.Johansen-Berg H., Behrens T.E., Sillery E., Ciccarelli O., Thompson A.J., Smith S.M., Matthews P.M. Functional-anatomical validation and individual variation of diffusion tractography-based segmentation of the human thalamus. Cereb. Cortex. 2005;15:31–39. doi: 10.1093/cercor/bhh105. [DOI] [PubMed] [Google Scholar]
- 24.Eickhoff S.B., Stephan K.E., Mohlberg H., Grefkes C., Fink G.R., Amunts K., Zilles K. A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage. 2005;25:1325–1335. doi: 10.1016/j.neuroimage.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 25.Zarei M., Johansen-Berg H., Smith S., Ciccarelli O., Thompson A.J., Matthews P.M. Functional anatomy of interhemispheric cortical connections in the human brain. J. Anat. 2006;209:311–320. doi: 10.1111/j.1469-7580.2006.00615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai C.S., Gerrelli D., Monaco A.P., Fisher S.E., Copp A.J. FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003;126:2455–2462. doi: 10.1093/brain/awg247. [DOI] [PubMed] [Google Scholar]
- 27.Echelard Y., Epstein D.J., St-Jacques B., Shen L., Mohler J., McMahon J.A., McMahon A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 28.Goodrich L.V., Johnson R.L., Milenkovic L., McMahon J.A., Scott M.P. Conservation of the hedgehog/patched signaling pathway from flies to mice: Induction of a mouse patched gene by Hedgehog. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- 29.Hui C.C., Slusarski D., Platt K.A., Holmgren R., Joyner A.L. Expression of three mouse homologs of the Drosophila segment polarity gene cubitus interruptus, Gli, Gli-2, and Gli-3, in ectoderm- and mesoderm-derived tissues suggests multiple roles during postimplantation development. Dev. Biol. 1994;162:402–413. doi: 10.1006/dbio.1994.1097. [DOI] [PubMed] [Google Scholar]
- 30.Hui C.C., Joyner A.L. A mouse model of greig cephalopolysyndactyly syndrome: The extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 1993;3:241–246. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- 31.Morcillo J., Martinez-Morales J.R., Trousse F., Fermin Y., Sowden J.C., Bovolenta P. Proper patterning of the optic fissure requires the sequential activity of BMP7 and SHH. Development. 2006;133:3179–3190. doi: 10.1242/dev.02493. [DOI] [PubMed] [Google Scholar]
- 32.Dakubo G.D., Wang Y.P., Mazerolle C., Campsall K., McMahon A.P., Wallace V.A. Retinal ganglion cell-derived sonic hedgehog signaling is required for optic disc and stalk neuroepithelial cell development. Development. 2003;130:2967–2980. doi: 10.1242/dev.00515. [DOI] [PubMed] [Google Scholar]
- 33.Qi Y., Stapp D., Qiu M. Origin and molecular specification of oligodendrocytes in the telencephalon. Trends Neurosci. 2002;25:223–225. doi: 10.1016/s0166-2236(02)02145-8. [DOI] [PubMed] [Google Scholar]
- 34.Furuta Y., Piston D.W., Hogan B.L. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124:2203–2212. doi: 10.1242/dev.124.11.2203. [DOI] [PubMed] [Google Scholar]
- 35.Omi M., Fisher M., Maihle N.J., Dealy C.N. Studies on epidermal growth factor receptor signaling in vertebrate limb patterning. Dev. Dyn. 2005;233:288–300. doi: 10.1002/dvdy.20353. [DOI] [PubMed] [Google Scholar]
- 36.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett C.P., Betts D.R., Seller M.J. Deletion 14q (q22-q23) associated with anophthalmia, absent pituitary, and other abnormalities. J. Med. Genet. 1991;28:280–281. doi: 10.1136/jmg.28.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elliott J., Maltby E.L., Reynolds B. A case of deletion 14(q22.1→q22.3) associated with anophthalmia and pituitary abnormalities. J. Med. Genet. 1993;30:251–252. doi: 10.1136/jmg.30.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phadke S.R., Sharma A.K., Agarwal S.S. Anophthalmia with cleft palate and micrognathia: A new syndrome? J. Med. Genet. 1994;31:960–961. doi: 10.1136/jmg.31.12.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemyre E., Lemieux N., Decarie J.C., Lambert M. Del(14)(q22.1q23.2) in a patient with anophthalmia and pituitary hypoplasia. Am. J. Med. Genet. 1998;77:162–165. [PubMed] [Google Scholar]
- 41.Lopez-Rios J., Gallardo M.E., Rodriguez de Cordoba S., Bovolenta P. Six9 (Optx2), a new member of the six gene family of transcription factors, is expressed at early stages of vertebrate ocular and pituitary development. Mech. Dev. 1999;83:155–159. doi: 10.1016/s0925-4773(99)00017-9. [DOI] [PubMed] [Google Scholar]
- 42.Aijaz S., Clark B.J., Williamson K., van Heyningen V., Morrison D., Fitzpatrick D., Collin R., Ragge N., Christoforou A., Brown A. Absence of SIX6 mutations in microphthalmia, anophthalmia, and coloboma. Invest. Ophthalmol. Vis. Sci. 2004;45:3871–3876. doi: 10.1167/iovs.04-0641. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X.M., Yang X.J. Temporal and spatial effects of Sonic hedgehog signaling in chick eye morphogenesis. Dev. Biol. 2001;233:271–290. doi: 10.1006/dbio.2000.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schimmenti L.A., de la Cruz J., Lewis R.A., Karkera J.D., Manligas G.S., Roessler E., Muenke M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am. J. Med. Genet. 2003;116A:215–221. doi: 10.1002/ajmg.a.10884. [DOI] [PubMed] [Google Scholar]
- 45.Ragge N.K., Salt A., Collin J.R., Michalski A., Farndon P.A. Gorlin syndrome: The PTCH gene links ocular developmental defects and tumour formation. Br. J. Ophthalmol. 2005;89:988–991. doi: 10.1136/bjo.2004.061390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y., Struhl G. Dual roles for patched in sequestering and transducing Hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- 47.Ruiz i Altaba A. Gli proteins encode context-dependent positive and negative functions: Implications for development and disease. Development. 1999;126:3205–3216. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- 48.Kawai S., Sugiura T. Characterization of human bone morphogenetic protein (BMP)-4 and −7 gene promoters: Activation of BMP promoters by Gli, a sonic hedgehog mediator. Bone. 2001;29:54–61. doi: 10.1016/s8756-3282(01)00470-7. [DOI] [PubMed] [Google Scholar]
- 49.Motoyama J. Essential roles of Gli3 and sonic hedgehog in pattern formation and developmental anomalies caused by their dysfunction. Congenit. Anom. (Kyoto) 2006;46:123–128. doi: 10.1111/j.1741-4520.2006.00114.x. [DOI] [PubMed] [Google Scholar]
- 50.Brickell P.M., Tickle C. Morphogens in chick limb development. Bioessays. 1989;11:145–149. doi: 10.1002/bies.950110508. [DOI] [PubMed] [Google Scholar]
- 51.Pizette S., Niswander L. BMPs negatively regulate structure and function of the limb apical ectodermal ridge. Development. 1999;126:883–894. doi: 10.1242/dev.126.5.883. [DOI] [PubMed] [Google Scholar]
- 52.Drossopoulou G., Lewis K.E., Sanz-Ezquerro J.J., Nikbakht N., McMahon A.P., Hofmann C., Tickle C. A model for anteroposterior patterning of the vertebrate limb based on sequential long- and short-range Shh signalling and Bmp signalling. Development. 2000;127:1337–1348. doi: 10.1242/dev.127.7.1337. [DOI] [PubMed] [Google Scholar]
- 53.Selever J., Liu W., Lu M.F., Behringer R.R., Martin J.F. Bmp4 in limb bud mesoderm regulates digit pattern by controlling AER development. Dev. Biol. 2004;276:268–279. doi: 10.1016/j.ydbio.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 54.Panman L., Zeller R. Patterning the limb before and after SHH signalling. J. Anat. 2003;202:3–12. doi: 10.1046/j.1469-7580.2003.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guha U., Gomes W.A., Kobayashi T., Pestell R.G., Kessler J.A. In vivo evidence that BMP signaling is necessary for apoptosis in the mouse limb. Dev. Biol. 2002;249:108–120. doi: 10.1006/dbio.2002.0752. [DOI] [PubMed] [Google Scholar]
- 56.Tang M.K., Leung A.K., Kwong W.H., Chow P.H., Chan J.Y., Ngo-Muller V., Li M., Lee K.K. Bmp-4 requires the presence of the digits to initiate programmed cell death in limb interdigital tissues. Dev. Biol. 2000;218:89–98. doi: 10.1006/dbio.1999.9578. [DOI] [PubMed] [Google Scholar]
- 57.Cohen M.M. The hedgehog signaling network. Am. J. Med. Genet. A. 2003;123:5–28. doi: 10.1002/ajmg.a.20495. [DOI] [PubMed] [Google Scholar]
- 58.Litingtung Y., Dahn R.D., Li Y., Fallon J.F., Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979–983. doi: 10.1038/nature01033. [DOI] [PubMed] [Google Scholar]
- 59.Harfe B.D., Scherz P.J., Nissim S., Tian H., McMahon A.P., Tabin C.J. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517–528. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 60.Belloni E., Muenke M., Roessler E., Traverso G., Siegel-Bartelt J., Frumkin A., Mitchell H.F., Donis-Keller H., Helms C., Hing A.V. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- 61.Dubourg C., Lazaro L., Pasquier L., Bendavid C., Blayau M., Le Duff F., Durou M.R., Odent S., David V. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum. Mutat. 2004;24:43–51. doi: 10.1002/humu.20056. [DOI] [PubMed] [Google Scholar]
- 62.Wicking C., Gillies S., Smyth I., Shanley S., Fowles L., Ratcliffe J., Wainwright B., Chenevix-Trench G. De novo mutations of the Patched gene in nevoid basal cell carcinoma syndrome help to define the clinical phenotype. Am. J. Med. Genet. 1997;73:304–307. [PubMed] [Google Scholar]
- 63.Scott A., Strouthidis N.G., Robson A.G., Forsyth J., Maher E.R., Schlottmann P.G., Michaelides M. Bilateral epiretinal membranes in Gorlin syndrome associated with a novel PTCH mutation. Am. J. Ophthalmol. 2007;143:346–348. doi: 10.1016/j.ajo.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 64.Tobin J.L., Beales P.L. Bardet-Biedl syndrome: Beyond the cilium. Pediatr. Nephrol. 2007;22:926–936. doi: 10.1007/s00467-007-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haraguchi R., Mo R., Hui C., Motoyama J., Makino S., Shiroishi T., Gaffield W., Yamada G. Unique functions of Sonic hedgehog signaling during external genitalia development. Development. 2001;128:4241–4250. doi: 10.1242/dev.128.21.4241. [DOI] [PubMed] [Google Scholar]
- 66.Lin D., Huang Y., He F., Gu S., Zhang G., Chen Y., Zhang Y. Expression survey of genes critical for tooth development in the human embryonic tooth germ. Dev. Dyn. 2007;236:1307–1312. doi: 10.1002/dvdy.21127. [DOI] [PubMed] [Google Scholar]
- 67.Gould D.B., Smith R.S., John S.W. Anterior segment development relevant to glaucoma. Int. J. Dev. Biol. 2004;48:1015–1029. doi: 10.1387/ijdb.041865dg. [DOI] [PubMed] [Google Scholar]
- 68.Hjalt T.A., Semina E.V., Amendt B.A., Murray J.C. The Pitx2 protein in mouse development. Dev. Dyn. 2000;218:195–200. doi: 10.1002/(SICI)1097-0177(200005)218:1<195::AID-DVDY17>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 69.Idrees F., Bloch-Zupan A., Free S.L., Vaideanu D., Thompson P.J., Ashley P., Brice G., Rutland P., Bitner-Glindzicz M., Khaw P.T. A novel homeobox mutation in the PITX2 gene in a family with Axenfeld-Rieger syndrome associated with brain, ocular, and dental phenotypes. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2006;141:184–191. doi: 10.1002/ajmg.b.30237. [DOI] [PubMed] [Google Scholar]
- 70.Cartegni L., Chew S.L., Krainer A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 71.Blauwkamp M.N., Beyer L.A., Kabara L., Takemura K., Buck T., King W.M., Dolan D.F., Barald K.F., Raphael Y., Koenig R.J. The role of bone morphogenetic protein 4 in inner ear development and function. Hear. Res. 2007;225:71–79. doi: 10.1016/j.heares.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Z., Song Y., Zhao X., Zhang X., Fermin C., Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- 73.Yeyati P.L., Bancewicz R.M., Maule J., van Heyningen V. Hsp90 selectively modulates phenotype in vertebrate development. PLoS Genet. 2007;3:e43. doi: 10.1371/journal.pgen.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]