

Abstract
Action myoclonus-renal failure syndrome (AMRF) is an autosomal-recessive disorder with the remarkable combination of focal glomerulosclerosis, frequently with glomerular collapse, and progressive myoclonus epilepsy associated with storage material in the brain. Here, we employed a novel combination of molecular strategies to find the responsible gene and show its effects in an animal model. Utilizing only three unrelated affected individuals and their relatives, we used homozygosity mapping with single-nucleotide polymorphism chips to localize AMRF. We then used microarray-expression analysis to prioritize candidates prior to sequencing. The disorder was mapped to 4q13-21, and microarray-expression analysis identified SCARB2/Limp2, which encodes a lysosomal-membrane protein, as the likely candidate. Mutations in SCARB2/Limp2 were found in all three families used for mapping and subsequently confirmed in two other unrelated AMRF families. The mutations were associated with lack of SCARB2 protein. Reanalysis of an existing Limp2 knockout mouse showed intracellular inclusions in cerebral and cerebellar cortex, and the kidneys showed subtle glomerular changes. This study highlights that recessive genes can be identified with a very small number of subjects. The ancestral lysosomal-membrane protein SCARB2/LIMP-2 is responsible for AMRF. The heterogeneous pathology in the kidney and brain suggests that SCARB2/Limp2 has pleiotropic effects that may be relevant to understanding the pathogenesis of other forms of glomerulosclerosis or collapse and myoclonic epilepsies.
Introduction
Action myoclonus-renal failure syndrome (AMRF [MIM 254900]) is a lethal inherited form of progressive myoclonus epilepsy associated with renal failure. It typically presents at 15–25 years with proteinuria evolving into renal failure or with neurological symptoms (tremor, action myoclonus, seizures, and later ataxia). The renal pathology is of focal glomerulosclerosis, sometimes with features of glomerular collapse. Brain pathology shows unusual and uncharacterized storage material. Initially identified in the French-Canadian isolate, the disorder has now been recognized in many countries.1–3
The autosomal-recessive gene defect underlying AMRF was unknown, and the lack of large pedigrees and lethality of the disorder precluded a conventional mapping strategy. Here, we identify a lysososmal-membrane gene responsible for the diverse pathologies in the kidney and brain in this condition. We used a novel strategy on just three unrelated affected subjects and compare the features of a knockout mouse with the human disease.
Material and Methods
Clinical Samples
Three unrelated Australian families with a single AMRF proband were used for identifying the gene. Case A was of Turkish-Cypriot origin; her parents were first cousins (Figure 1). Ancestors of families B and C came from different regions of Britain, and no inbreeding loops were known for either family.2,3 Peripheral blood lymphocytes were obtained by venepuncture for extraction of DNA, and lymphoblastoid cell lines were established for RNA and protein studies from cases A and B and selected relatives. Case C was deceased, but stored brain tissue in paraffin blocks was available for DNA extraction. Cases A and B underwent audiograms and electrophysiological investigations at ages 33 and 31, respectively, both 11 years from onset of the disease.
Figure 1.
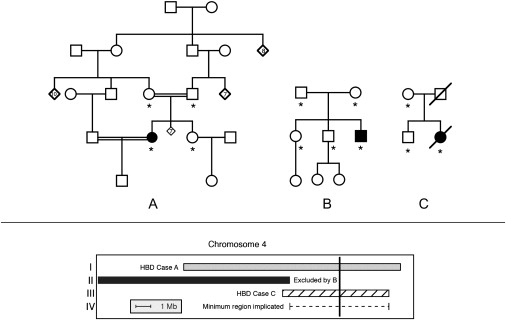
Linkage Analysis
Shown in the upper panel: pedigrees used to map AMRF. Families A and B were previously clinically described as families 2 and 1, respectively (3), and pedigree C here was family B in 2. Filled symbols indicate cases with AMRF. Asterisks indicate individuals used for genotyping.
Shown in the lower panel: localization of the critical region on chromosome 4 (see text). Region I (gray bar) was identified as homozygous by descent (HBD) in case A and not shared by the sibling of case A. In region II, case B shared both haplotypes identical by descent with unaffected siblings, thus excluding this region. Region III shows the homozygous region of case C. Region IV (dashed line) shows the deduced critical region of 6.6 Mb. The vertical black line indicates the position of SCARB2.
DNA samples from venous blood or postmortem tissue in previously reported Canadian AMRF cases2 and from families with a variety of other hereditary nephropathies were obtained. Clinical studies were approved by the Human Research Ethics Committee of Austin Health.
Genotyping
All 12 individuals labeled with asterisk in Figure 1 were genotyped with the Affymetrix XBA 50K (single-nucleotide polymorphism) SNP chip with standard protocols and software settings at the Australian Genome Research Facility (AGRF). We performed genotyping by using the Affymetrix GCOS software, with the Dynamic Model4 algorithm, with the default settings. DNA samples passed all quality-control checkpoints, and the call rate was high (>96%) for ten of the samples. Two samples had lower call rates of 86%. Data were assembled into LINKAGE5 style .dat and .pre files with in-house programs. Mendelian errors were detected and removed with PEDCHECK,6 and errors akin to unlikely double recombinants were found with MERLIN.7 Less than 0.5% of genotypes were deemed erroneous and recoded as missing data. Additionally, seven microsatellite markers were chosen with the UCSC genome browser (May 2004 assembly), and samples were genotyped at the AGRF.
Linkage Identical-by-Descent and Homozygous-by-Descent Analysis
All analysis was carried out with either ALLEGRO8 or MERLIN.7 Pairwise linkage identical by descent (IBD) sharing probabilities were calculated with MERLIN. Regions with homozygosity by descent (HBD) sharing were identified with the most likely inferred inheritance vector calculated by ALLEGRO.
Testing of Relatedness with GBIRP
Analysis with the software GBIRP9 was used to test for distant cousin-type relationships. The multipoint algorithm tests a likelihood ratio of an nth cousin-type relationship against a hypothesis of no relationship, assuming linkage equilibrium. dth degree relatives are expected to inherit a fraction 1/2d of their DNA IBD. To prevent potential bias due to linkage disequilibrium, we used a sparse subset of the total marker map, with each marker at a distance of at least 300 kb to its neighbors. This map included only 17% of available marker loci. Significance of the test statistic was calculated by simulation of genotype data for 10,000 pairs of unrelated individuals.
Expression Arrays
RNA was used for expression analysis from cases A and B and their gender-matched siblings. Each of the four samples was split and labeled and hybridized to two Affymerix U133 Plus2 arrays, giving eight arrays in total. Microarray analysis was performed with bioconductor packages10 for the R programming environment (see Web Resources). We performed quality assessment with diagnostic plots by using the affy package, and all arrays were found to be satisfactory. Background subtraction, normalization, and probe summarization were done with gcrma.11 We analyzed expression levels with genewise linear models with effects for the disease state (unaffected or affected) and the family (A or B) by using the limma package.12 The blocking introduced by the technical replication was handled by the method of Smyth et al.13 Tests of significance were conducted with empirical Bayes moderated t tests that ensure stable inference even with small sample size.14 Initially, this was done for the whole data set, and then we restricted the analysis to the 98 probe sets (which correspond to the 66 genes) in the mapped region of interest. Multiple testing adjustment was performed with the false-discovery-rate method.15
Sequencing of SCARB2 DNA and RNA
Genomic DNA was extracted from peripheral blood with the QIAamp DNA Blood Maxi Kit (QIAGEN). Genomic DNA was also extracted from formalin fixed paraffin-embedded tissue. The tissue was sequentially rehydrated, and per sample size of 0.5 cm2 was incubated in 1 mg/ml proteinase K, 1% SDS, and rotated at 37°C for at least 4 hr. After two extractions with phenol and chloroform and chloroform extraction, the DNA was ethanol precipitated. We used direct sequencing of PCR products to screen for mutations in the 12 exons and splice junctions of the SCARB2 gene. The NCBI accession number of the SCARB2 mRNA used for the analysis was NM_005506. The PCR primer sequences are available from L.D. on request.
Total RNA was isolated from lymphocytes and lymphoblastoid cells with the RNeasy kit (QIAGEN) according to the manufacturer's instructions or from fresh blood with the QIAamp RNA Blood Mini Kit (QIAGEN). We prepared cDNA from the RNA with Superscript reverse transcriptase (Invitrogen), using 10 ul of total RNA and oligo dT primer (Promega). Remaining single-stranded RNA was digested with RNaseH (Promega), and 2 μl of the cDNA reaction was used in a 50 μl PCR reaction. PCR was carried out with forward and reverse primers to the human SCARB2 sequence as follows:
F 5′-TTGCTAAAGCTTCATGGGCCGATGCTGCTTCTA-3′
R 5′-CGAACTGAATTCTTAGGTTCGAATGAGGGGTGCT-3′.
PCR was performed with Accusure Polymerase (Bioline). PCR products were resolved on 1% agarose gels, bands corresponding to the Limp2 sequence were excised, the DNA was extracted with QIAquick gel extraction kit (QIAGEN), and the PCR product was digested with HindIII and EcoRI. They were then cloned in frame into the pEGFP-C2 vector (Clontech).
For case A and case B, a discrete single band was not seen after the first round of PCR, so the bands were excised, the DNA was extracted, and this material was used as a template for a second round of PCR. In both cases, a band at approximately 1.4 kb (corresponding to the size of SCARB2 cDNA coding region) was seen. Ligations were transformed into competent E. coli, and plasmid DNA was extracted with the QIAprep spin mini prep kit (QIAGEN). Positive colonies were sequenced so that identity of the PCR product could be confirmed.
Analysis of SCARB2 Protein
Protein used for expression analysis was isolated from lymphoblastoid cell lines from cases A and B and their gender-matched siblings. An amount of 10–25 μg of total protein was then resolved on 10% SDS-PAGE gels, and the proteins were transferred to PVDF membranes. Membranes were probed with goat anti-Limp2 antibody (R&D Systems) and then peroxidase-labeled rabbit anti-goat Ig (Dako) and were developed with the SuperSignal West Pico Chemiluminescent substrate (Pierce). Membranes were stripped and reprobed with mouse mAb anti-β-tubulin (Sigma) as a control for protein loading.
Limp2-Deficient Mice
Limp2-deficient mice were described previously.16 All experiments using these mice were performed according to approved national animal-welfare regulations.
Histology
For conventional light and electron microscopic examination, six Limp2-deficient mice (ages 6–21.5 months) and four age-matched wild-type (WT) mice were used. Tissues were perfused with Bouin solution (diluted 20%–25% with PBS) or glutaraldehyde (6%, in 0.1 M phosphate buffer [pH 7.4]). Tissue blocks were processed for light- and electron-microscopic examination (embedding in paraffin or araldite) according to routine procedures.
Immunohistochemistry on Tissue Sections
Brains and kidneys of mice perfused with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB) were incubated in 30% sucrose in 0.1 M PB for at least 24 hr before preparing free-floating cryosections (50 μm). Sections were rinsed in 0.1 M PB and blocked in 0.1 M PB with 0.2% BSA, 4% NGS, and 0.5% Triton X-100 for 2 hr at RT. After incubation with the first antibody, sections were rinsed with 0.1 M PB/0.5% Triton X-100 and incubated with secondary fluorescent antibodies (Alexa fluor 488 and 594; Molecular Probes, 1:1000). The primary antibodies used were Limp2 (1:500) and DAPI (1:1000) for nuclei staining. Immunofluorescence was examined by the Zeiss Axiovert 200 M fluorescence microscope.
Behavioral Testing
A group of five Limp2−/− and five wild-type mice (both groups consisting of two females and three males) were examined in tests for neuromotor performance and locomotion, exploratory activity, and passive-avoidance learning. Cage activity was recorded with a laboratory-built activity logger connected to three infrared (IR) photo beams. Mice were put individually in 20 × 30 cm2 transparent cages, placed between the photo beams. Activity was expressed as beam crossings/30 min during a 24 hr interval.
Gait and walking abilities were evaluated for six 1 min trials over 2 days (intertrial interval, 1 hr) on a speed- and inclination-controlled treadmill connected to an electrified grid (PANLAB). Before testing, each mouse was adapted to the apparatus and explored it freely for 5 min. The treadmill inclination was set at 0, 5, and 10° slopes for each trial, with a constant speed of 10 cm/s. Next day, the same slopes were used, but the velocity was set to 15 cm/s. Mice were placed on the treadmill belt and had to move forward in order to avoid receiving foot shocks (0.4 mA). The number of shocks received was counted.
Grip strength was measured with a T shaped bar connected to a digital dynamometer (Ugo Basile). Mice were placed before the bar, which they grabbed spontaneously, and gently pulled backward until they released it. Ten measurements were averaged for each animal. Motor coordination and equilibrium were tested on an accelerating rotarod (MED Associates). Mice were first trained at constant speed (4 rpm, 2 min) before four test trials (intertrial interval, 10 min). During these test trials, the rotating rod accelerated in 5 min from 4 to 40 rpm. Time until they dropped from the rod was recorded (up to the 5 min cutoff).
Open-field exploration was examined with a 50 × 50 cm2 square arena. Each animal was dark adapted for 30 min and placed in the arena for 10 min. Movements of the mice in the arena were recorded with EthoVision video tracking equipment and software (Noldus). Total path length and corner crossings were included as measures of ambulatory activity. Entries into the center of the field were included as a measure of conflict resolution or anxiolysis. We also used the same arena to assess social exploration. In the social exploration condition, a round wire cage with two female mice was placed in the center of the arena, enabling visual, olfactory, and limited physical contact with the test mouse. Latency of the first approach and the number of approaches during the 10 min exploration trial were assessed.
Anxiety-related exploratory behavior was measured with the elevated plus maze. The maze was located 40 cm above the table surface. Mice were placed at the center of the maze and were allowed to explore freely for 10 min. Exploratory activity was recorded by five IR beams (four for arm entries and one for open arm dwell) connected to a computerized activity logger.
Learned passive avoidance and memory abilities were assessed in the mice with a step-through box, consisting of one small illuminated compartment and a dark chamber coupled with a grid floor. During training, dark-adapted mice (30 min) were put in the small illuminated compartment of the box. After 5 s, a sliding door to the larger dark chamber was opened, and entry latency was recorded. The door was closed as soon as all four feet were on the grid floor, and a slight foot shock (0.3 mA, 1 s) was delivered with a constant current shocker (Med Associates). Retention was tested 24 hr later with the same procedure, with the exception that the animals did not receive any shock. Entry was recorded up to a 300 s cutoff.
Results
Genomic Mapping of AMRF
Homozygosity mapping of the known consanguineous family A was carried out with multipoint analysis with MERLIN7 and identified 21 regions with evidence for homozygosity by descent (HBD) sharing in case A. The boundaries of these candidate regions were determined by flanking heterozygous SNPs. The regions ranged in size from 13 markers spanning 0.18 cM to 1103 markers spanning 62.4 cM. The average length of the regions was 12 cM, encompassing 213 markers.
Pairwise identical by descent (IBD) sharing probabilities were calculated for all possible pairs of affected and unaffected siblings in each family. Candidate regions were eliminated or shortened if any of the unaffected siblings shared both haplotypes IBD with their affected sibling. This strategy assumes locus homogeneity and complete penetrance but makes no assumption about allelic homogeneity and allows for compound heterozygote affecteds. After application of this strategy, 12 regions remained with some of these regions now reduced in size (average length = 6.2 cM, spanning an average of 84 SNPs).
Given the apparent rarity of this disease, it was possible that only a single disease-causing mutation arose in a given population. Under the assumption of allelic homogeneity within families, cases B and C would also be homozygous for their private mutations caused by unknown inbreeding loops in the ancestry of the two families. Two methods were used to look for regions fitting this description. The first was to treat the parents of the two cases as second cousins and to conduct homozygosity analysis with these modified pedigrees. The second method is based on the observation that, if there were a single pathogenic mutation in a given population, the cases in the outbred families would be homozygous for all SNPs in the marker haplotype surrounding the mutation.
Regions fitting this description were identified as long runs of homozygous markers in the affected individuals from the outbred families. In family C, two regions were identified that overlapped with the set of regions identified by these methods and the set of 12 previously isolated candidate regions. A 5.3 cM critical region, which includes 175 SNPs, was found on chromosome 4. The second region on chromosome 20 produced a candidate intersection region covering 3.0 cM but only containing 13 SNPs.
We further investigated these two regions for true HBD by examining four polymorphic microsatellite markers in the chromosome 4 critical region and three in the chromosome 20 region. None of the three microsatellite markers in the region on chromosome 20 were homozygous in case C, indicating that this region was indeed only homozygous by state for the SNP markers and not HBD. The microsatellites in the chromosome 4 region were homozygous in cases A and C. This observation is consistent with the hypothesis that case C inherited this haplotype HBD through an unobserved (and possibly distant) inbreeding loop.
By using a multipoint maximum-likelihood method9 to test for the possibility of relatedness, we estimated that the parents of case C were fifth degree relatives (e.g., second cousins, LOD = 2.99, p value = 0.0001). Both parents of case C had ancestors from Tasmania, the island state of Australia. It is estimated that approximately two-thirds of Tasmania's current population of 500,000 are individuals descended from approximately 10,000 women of British origin who had migrated to the state by 1850.9
Thus, a 5.3 cM critical region (equivalent to 6.6 Mb) was defined by the overlap on chromosome 4q13-21 of regions in which case A and case C were HBD, narrowed slightly by a family B region in which the affected shared a segment IBD with an unaffected sibling (Figure 1).
Expression-Array Analysis
The critical region on 4q13-21 contains 66 known genes of which approximately half are expressed in both the brain and kidney. In order to prioritize candidate genes for sequencing, we hypothesized that the mRNA of the causative gene would be downregulated in affected subjects, possibly because of mutations causing RNA instability. We therefore analyzed RNA from lymphoblastoid cell lines derived from two living affected subjects and from a healthy gender-matched sibling of each (families A and B) with Affymetrix U133 Plus2 arrays.
The data were analyzed with a two-way empirical Bayes ANOVA approach for detecting significant differences between affected and unaffected individuals adjusted for differences between families (see Materials and Methods). Initial analysis of the data set on a genome-wide basis showed major differences in expression of a number of genes related to immune regulation. For example IGHG1, IGHV1-69, and IL16 were found to be significantly disregulated in affected individuals (data not shown). This was not surprising because the two affected individuals had undergone renal transplantation and were taking immunosuppressive drugs. Analysis was subsequently confined to the probe sets in the 6.6 Mb region defined by our mapping. SCARB2 (Scavenger Receptor B2) emerged as the leading candidate. It was downregulated ∼2-fold in the affected subjects compared to their healthy siblings (p < 0.001, Table 1). This was the probe set closest to the 3′ end, in which RNA degradation is least likely to have occurred and was therefore the most informative in indicating gene downregulation. Another probe set to the SCARB2 gene also showed differential downregulation in the affected individuals when compared to their unaffected siblings.
Table 1.
The Ten Most Differentially Expressed Probe Sets when Analysis Was Limited to the Critical Region on Chromosome 4
| ID | Symbol | logFC | AveExpr | t | FDR | B |
|---|---|---|---|---|---|---|
| 224983_at | SCARB2 | −1.109 | 4.26 | −10.07 | 0.001 | 4.32 |
| 235638_at | RASSF6 | 1.070 | 5.84 | 8.34 | 0.002 | 3.04 |
| 227119_at | CNOT6L | −0.960 | 3.68 | −6.99 | 0.005 | 1.81 |
| 204533_at | CXCL10 | −1.730 | 4.32 | −5.39 | 0.019 | 0.06 |
| 225548_at | SHRM | −0.674 | 2.76 | −5.38 | 0.019 | 0.05 |
| 226153_s_at | CNOT6L | −0.840 | 4.75 | −5.21 | 0.020 | −0.16 |
| 235653_s_at | THAP6 | 0.424 | 4.26 | 5.08 | 0.020 | −0.32 |
| 201646_at | SCARB2 | −0.741 | 2.22 | −4.98 | 0.020 | −0.45 |
| 208840_s_at | G3BP2 | 0.354 | 7.86 | 4.16 | 0.053 | −1.55 |
| 202770_s_at | CCNG2 | 0.550 | 8.17 | 4.02 | 0.058 | −1.76 |
“logFC” is the log2 fold change in expression between affected subjects and their unaffected siblings (a value of −1 indicates a 2-fold decrease in the affected). “FDR” is the upper bound for the expected false discovery rate after adjustment for multiple testing across the 98 probe sets in the critical region. “AveExpr” is the average log2 expression level across all subjects, “t” is the moderated t statistic testing differential expression, and “B” is the log-odds of differential expression. Note that some genes (including SCARB2) have two probe sets represented.
Mutational Analysis of SCARB2
We hypothesized from the clinical genetics (known consanguinity in family A) and the SNP-chip mapping that cases A and C would have mutations HBD, whereas case B would be a compound heterozygote.
We identified different homozygous mutations in cases A and C, consistent with our prediction. Case A had a homozygous splice-site mutation (c.1239+1G→T). RT-PCR analysis showed that this mutation leads to retention of intron 10 and the subsequent insertion of 20 amino acids and premature truncation of the protein at residue 433. The mutation in case C (c.435_436insAG W146SfsX16) results in a frameshift predicted to truncate the protein to 160 amino acids. From the sequence trace, this mutation appears homozygous, as predicted by the haplotype analysis, although the paternal DNA was not available for direct confirmation of carrier status. In case B, a heterozygous frameshift mutation (c.296 delA N99IfsX34) inherited from his unaffected mother was identified that predicts truncation of the protein to 131 amino acids, but no second mutation was found in the coding regions (Figure 2). RT-PCR analysis of RNA extracted from lymphocytes from case B showed the presence of wild-type cDNA as well as a number of additional RNA species compared to a single WT product from unaffected controls (data not shown). Sequencing of the additional multiple cDNA products suggested that a number of different exon-skipping events were occurring in case B, but we were unable to definitively characterize these. In our sequencing of the genomic DNA of this patient, we noted that he carried a number of intronic sequence variants not found in the other patients or reported in SNP databases. Together, this suggests that the patient's second SCARB2 allele may contain cryptic splice-site recognition sequences leading to aberrant RNA splicing. This suggests that the second mutation is outside the coding region, perhaps affecting translation or RNA stability, because no detectable SCARB2 protein is produced (see below) yet mRNA is present.
Figure 2.
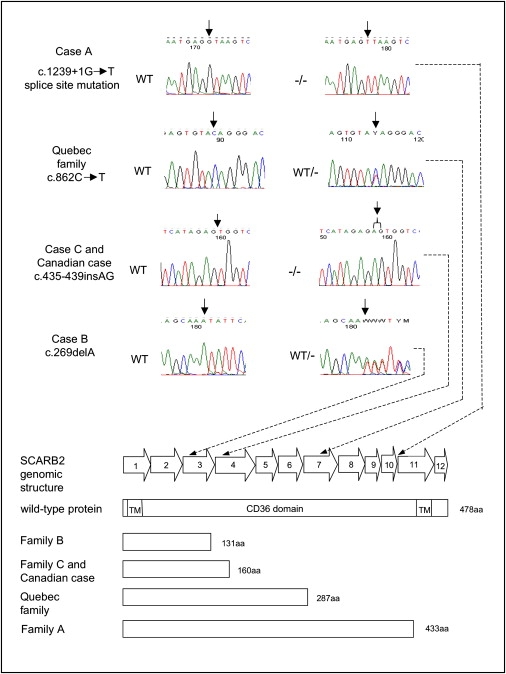
SCARB2 Mutations
Genomic structure of the SCARB2 gene and the position of the mutations found in the AMRF cases. The sequencing chromatograms indicate the mutations found in the affected individuals or in a carrier, and the predicted effects of the mutations on the SCARB2 protein are illustrated below. “WT” refers to an individual lacking a mutation, “WT/−” refers to a heterozygote, and “−/−” refers to an individual homozygous for a mutation.
We subsequently identified a SCARB2 mutation in one of the original AMRF families from Quebec (family F in 2). The three affected members are deceased, and no DNA is available for testing, but obligate carriers of this autosomal-recessive disorder have the nonsense mutation c.862C→T, Q288X, which is predicted to prematurely terminate the protein or lead to nonsense-mediated RNA decay of the transcript. Study of another previously reported Canadian case, without French-Canadian ancestry (case PO3 in 2) revealed a frameshift mutation (W146SfsX16), identical to case C. Parental DNA is not available for testing, so we are unable to distinguish whether the mutation is homozygous or hemizygous with deletion of a segment of the chromosome 4 homolog that would normally bear the second pathogenic allele.
In order to determine whether SCARB2 mutations were associated with other forms of inherited proteinuria, we studied eight families with nonsyndromic focal and segmental glomerulosclerosis (FSGS, “steroid-resistant nephrotic syndrome”) and five with minimal-change glomerulonephritis (“steroid-sensitive nephrotic syndrome”). Inheritance was consistent with autosomal-recessive disease in four families with FSGS and in two with minimal-change glomerulonephritis. NPHS2 mutations, which are the most common known cause of inherited FSGS, had been excluded previously. In addition, six patients with thin-basement-membrane nephropathy (defined on renal biopsy) and proteinuria >500 mg/day and three individuals from non-Alport families with haematuria and proteinuria (two of whom were consanguineous) were screened for SCARB2 mutations. No mutations were identified in the 22 index cases. Five variants were found (coding region variants c.445G→A, c.475A→G, and c.1265C→T; intronic variants c.1437+22G→A, c.995-49_53 delCAGCT) and were regarded as unlikely to be pathogenic because they did not segregate with disease, were found in controls, or were intronic.
Western Blots of SCARB2
Western blots of cell lysates prepared from EBV-transformed lymphoblastoid B cell lines from the two affected living subjects (A and B), and one unaffected sibling for each of the subjects were probed with antibodies to Limp2 (the mouse ortholog of human SCARB2) (Figure 3). The antibody was validated by transfection of GFP-tagged human SCARB2 cDNA into HEK293 cells; this was followed by western blotting (data not shown). SCARB2 was easily detectable in lymphoblastoid cell lines from healthy siblings as a single band at 75–80 kDa but was undetectable in both affected subjects.
Figure 3.
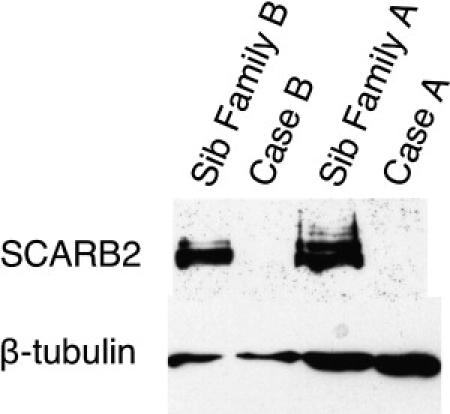
Western Blots with Lymphoblastoid Cell Lines from Cases A and B and Their Unaffected Siblings
SCARB2 was probed with a polyclonal goat antibody to Limp2 and is shown in the samples from the two siblings as a band at 75–80 kD; it was absent in the two cases. β-tubulin is shown in all four subjects at ∼50kD as a control.
Pleiotropic Effects of SCARB2/Limp2 Deficiency in Human and Mouse
A mouse deficient in Limp2 was developed previously,16 independent of clinical research on AMRF, and also has a phenotype predominantly involving the nervous system and kidney. The main phenotypic features were cochlear deafness, demyelinating peripheral neuropathy, and hydronephrosis due to ureteric-pelvic junction obstruction.16,17 We re-examined available AMRF cases in the light of observations in the mouse and reinvestigated the Limp2−/− mouse phenotype informed by observations in the patients (Table 2).
Table 2.
Pleiotropic Effects of SCARB2/Limp2 Deficiency in Man and Mice
| Action Myoclonus Renal Failure Syndrome | Limp2-Deficient Mouse | |
|---|---|---|
| Myoclonus and seizures | Central feature: progressive and severe | Absent |
| Ataxia | Moderate | Mild |
| Hearing | Normal hearing and audiograms | Deaf from 3 months |
| Peripheral nerves | Clinically normal; nerve-conduction studies in some subjects show evidence of a mild sensorimotor neuropathy | Demyelinating neuropathy |
| Brain pathology | Intracellular storage, apparently extra-neuronal | Storage in neurons and astrocytes |
| Pelvi-ureteric obstruction | Absent | Present in most mice |
| Glomerular pathology | Central feature: severe focal glomerulosclerosis with glomerular collapse | Minor mesangial proliferation, podocyte effacement; no glomerulosclerosis |
Deafness has not been reported in AMRF patients. Audiograms on cases A and B showed minor bilateral impairment at high frequencies, of doubtful significance. Patients have no clinical evidence of a sensory, motor, or autonomic peripheral neuropathy. Nerve-conduction studies previously reported in six patients were normal or showed mild abnormalities suggestive of a predominant axonal neuropathy.2,18 Nerve-conduction studies on case A were normal, and case B had evidence of a mild sensorimotor neuropathy. Whether this neuropathy is a core feature of AMRF or is due to renal failure and its treatment remains uncertain.
In the brains of AMRF patients, there is accumulation of autofluorescent pigment granules particularly in the laminae I and II of the cerebral cortex, basal ganglia, and in the Purkinje cell layer of the cerebellar cortex2 (Figure 4A). Examination of the brain in Limp2-deficient mice revealed abnormal autofluorescent cytoplasmic inclusions (not shown) in laminae II, III, and V of the cerebral cortex and in Purkinje cells, in some Bergmann astrocytes, and within the molecular layer of the cerebellum. Semithin sections and electron microscopy showed that the inclusions were intracellular (Figures 4B–4I). The human studies, performed on postmortem material, suggested that the granules were extraneuronal and probably in astrocytes.2 Immunohistological studies of cerebellum with Limp2 antibodies showed that the protein was concentrated in the Purkinje cell layer of normal mice and absent in Limp2−/− animals (Figure 4J).
Figure 4.
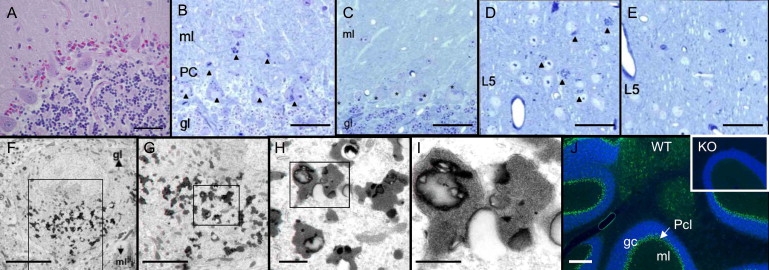
Neuropathology of AMRF and Limp2-Deficient Mice
(A) Human cerebellar vermis (autopsy sample; case C). Magenta-colored granules are seen between intact Purkinje cells (PAS stain; the scale bar represents 0.05 mm).
(B–E) Light microscopy of Limp2−/− brain in 16-month-old mice (semithin sections, toluidine blue). Cerebellar cortex of Limp2−/− mouse (B) shows all Purkinje cells (PC) as well as some neurons and glial cells in the molecular layer (ml) with numerous cytoplasmic inclusions (dense bodies) (arrowheads). In control wild-type mice (C), no inclusions are seen in the Purkinje cells (marked with asterisks), granular layer (gl), or ml. Sections of cerebral cortex from Limp2−/− show similar cytoplasmic inclusions that are in neuronal perikarya of laminae 2/3 and 5 (D) and that are absent in the cerebral cortex of wild-type mice (E). Layer 5 (L5) is shown. Scale bars represent 50 μm.
(F–I) Electron microscopy of Limp2−/− mouse brain. One Purkinje cell shown at increasing magnifications from left to right. The boxes indicate the region shown at higher magnification. The osmiophilic inclusions were most frequently observed within the perikarya and only very rarely within dendrites (not shown). The membrane-limited inclusions contain homogeneously granular material, some lipid droplets, and occasionally some lamellar structures. Scale bars represent 10 μm, 5 μm, 1 μm, and 0.5 μm, respectively.
(J) Immunohistological studies of mouse cerebellum. Wild-type cerebellar cortex (WT) is shown with nuclei stained with DAPI in blue. Limp2 is shown in green and is concentrated in the Purkinje cell layer (the scale bar represents 200 μm). The inset shows knockout (KO) with absence of Limp2 in the Purkinje cell layer.
In Limp2−/− mice, myoclonic jerks or epileptic seizures were never observed. Cage-activity recordings revealed slight hyperactivity in the Limp2−/− animals compared to controls. Both Limp2−/− and wild-type animals showed an initial explosion in exploratory activity upon their introduction into the cages, but the former were overall much more active between 21:00 p.m. and 5:00 a.m. (i.e., during most of their active phase). When placed on a tabletop, Limp2−/− mice clearly displayed ataxic gait, but treadmill gait performance was only decreased on the high-velocity trials (15 cm/s, not shown). Limp2−/− mice made more errors (8.2 ± 1.8 shocks) than wild-type mice (4.0 ± 0.5 shocks) at 5° and 10° treadmill inclination (one-tailed t test, p < 0.05). Grip-strength measurements were normal in Limp2−/− mice (660 ± 36 mN) compared to controls (688 ± 27 mN, not shown), but their performance on the rotarod test was severely impaired (Figure 5). On all trials, Limp2−/− animals fell from the rod much sooner than controls did (repeated-measures ANOVA, effect of genotype, F1,8 = 23.3, p = 0.001).
Figure 5.
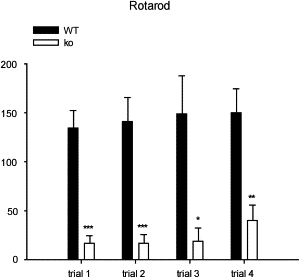
Rotarod Testing of Limp2−/− and Wild-Type Mice
Accelerating rotarod performance in wild-type (black bars) and Limp2−/− (white bars) mice. On each of the 5 min trials, Limp2−/− mice were unable to balance on the rod quite as long as wild-type (control) mice, and this demonstrates the ataxia and neuromotor impairment in the Limp2-deficient group. Data are means and SEM; asterisks indicate significance of difference with control values (post hoc Fisher LSD), ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p = 0.001.
In contrast, Limp2−/− mice performed normally on the remaining exploratory and learning tests, indicating that their ataxia might be an isolated impairment (Table 3). In accordance with the cage-activity measures, Limp2−/− mice were more active than controls in the open-field test (as indicated by their path length and corner-crossing measures), but differences did not reach significance. On the other hand, measures related to conflict resolution (i.e., latencies and number of approaches to the center) were very similar between genotypes; additionally, in the social-exploration task, Limp2−/− mice showed virtually the same exploratory responses as controls (Table 3). Limp2−/− mice also displayed normal elevated-plus-maze performance as we observed similar open-arm exploration to controls. Finally, passive-avoidance learning ability was not altered in the Limp2−/− group as demonstrated by closely matched step-through latencies between Limp2−/− and wild-type mice.
Table 3.
Cage and Exploratory Activity and Passive-Avoidance Learning in Limp2-Deficient Mice and Controls
| Test Measure | Limp2+/+ | Limp2−/− | p Value∗ |
|---|---|---|---|
| Cage activity (counts/24 hr) | 6135 ± 851 | 10342 ± 1225 | p = 0.022∗∗ |
| Open Field | |||
| Path length (cm) | 1197 ± 265 | 2731 ± 612 | p = 0.15 |
| Corner crossings | 37 ± 9 | 63.8 ± 14.6 | p = 0.17 |
| Latency of center entry (s) | 284 ± 130 | 166 ± 110.7 | p = 0.5 |
| Number of center entries | 4.8 ± 3.2 | 11.6 ± 5.2 | p = 0.3 |
| Social Exploration | |||
| Latency of center entry (s) | 172 ± 109 | 171 ± 113 | p = 0.99 |
| Number of center entries | 11 ± 5 | 14 ± 5 | p = 0.7 |
| Elevated Plus Maze | |||
| Percentage of time in open arms | 33 ± 8 | 32 ± 4 | p = 0.9 |
| Percentage of counts in open arms | 35 ± 5 | 26 ± 4 | p = 0.21 |
| Passive-Avoidance Learning | |||
| Step-through latency (s) | 156 ± 59 | 159 ± 57 | p = 0.97 |
All data are means ± SEM.
Values are determined by two-tailed t test except where indicated.
This value is determined by repeated measures ANOVA, effect of genotype.
One aspect of the renal pathology in Limp2-deficient mice is a hydronephrosis due to pelvi-ureteric obstruction that occurs unilaterally or bilaterally in most but not all mice. The obstruction was accompanied by hypertrophy of ureteric smooth muscle and epithelial metaplasia. The latter was associated with a disturbed apical expression of uroplakin suggesting an impairment of membrane transport processes.16 Kidneys in AMRF patients are small, with no evidence of obstruction, but pathological examination of the human ureter to find subtle mucosal thickening has not been possible to date. The renal lesion in AMRF is severe focal glomerulosclerosis sometimes with glomerular collapse presenting as proteinuria with acute or subacute renal failure (Figures 6A and 6B). Limp2-deficient mice have proteinuria, which was previously regarded as secondary to hydronephrosis.16 Examination of nonhydronephrotic kidneys showed definite glomerular lesions with mesangial hyper cellularity evident by light microscopy and severe effacement of foot processes at electron microcopy (Figures 6C–6G), but without frank changes of glomerulosclerosis. In contrast to other lysosomal-membrane proteins such as LAMP1 (data not shown), Limp2 is highly expressed in glomeruli of wild-type mice but is absent in Limp2-deficient mice (Figure 6H).
Figure 6.
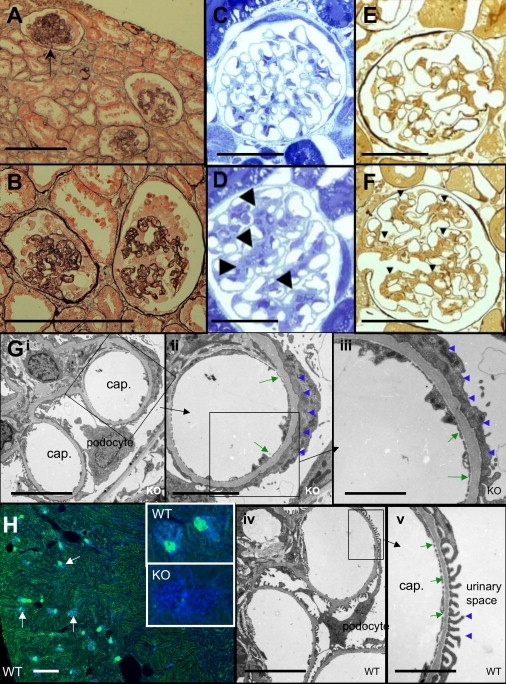
Glomerular Pathology in AMRF and Limp2-Deficient Mice
(A and B) Human glomerular pathology. Representative renal cortex (A) of human case C with AMRF showing an almost totally sclerosed glomerulus (arrow) because of global collapse and two glomeruli showing the early phases of collapse and sclerosis. Higher power (B) of the two glomeruli in the early phases of capillary collapse with partially obliterated lumina and prominent epithelial cells caused by hypertrophy and hyperplasia (silver methernamine-Marson's trichrome) is shown. Scale bars represent 50 μm.
(C–F) Light microscopy of Limp2−/− mice. Glomerulus of a wild-type (WT) mouse aged 16 months with the typical tissue structure (C) compared to Limp2−/− (KO) mouse (D), with arrowheads indicating an expanded messangium (semithin sections, toluidine blue). The same wild-type animal with silver stain for highlighting extracellular matrix and basal membranes (E) compared to the Limp2−/− mouse (F) with a mesangium that is expanded (arrowheads) is shown. Scale bars represent 50 μm.
(G) Electron microscopy of Limp2−/− (Gi–Giii) and wild-type (G iv–Gv) mice. Images show effacement of the foot processes of the podocytes (blue arrowheads) and thickening of the glomerular basal membrane (green arrows) in Limp2−/− (KO) mice compared to normal podocyte morphology and basement-membrane thickness in wild-type (WT) animals. Scale bars represent 10 μm (Gi and Giv), 5 μm (Gii), and 2.5 μm (Giii and Gv).
(H) Immunohistological studies of mouse renal cortex. The overview shows wild-type cortex with nuclei in blue (DAPI) and Limp2 in green; glomeruli are indicated by arrows (scale bar represents 200 μm). High-magnification insets of glomeruli show Limp2 in wild-type animals but absent in knockout animals.
Discussion
The discovery of mutations in SCARB2 responsible for AMRF highlights the power of recent advances in high-throughput genotype and expression platforms for the identification of genes responsible for rare autosomal-recessive diseases. A combination of SNP-chip linkage mapping, examination of haplotype sharing, and microarray-expression analysis on a sample of three unrelated individuals with AMRF led to the prioritization of SCARB2 as the leading candidate gene. This stepwise elimination of candidate regions represents a paradigm for other recessive Mendelian disorders with few affected individuals. This is a powerful approach for recessive-disease families with known or likely inbreeding loops in their ancestry.19
As demonstrated here, gene-expression studies can also aid in prioritizing genes for sequencing in which linkage mapping leads to an interval with a large number of candidate genes. This strategy is particularly applicable for recessive diseases, such as AMRF, for which a knockdown or knockout effect could produce a detectable difference in expression between cases and controls. This is one of the first studies that have successfully used gene-expression analysis to prioritize the best candidate gene for a recessive disease. Previous applications to genetic disorders have included a dominant disease with low penetrance20 and a complex disease.21 Success hinges on gene expression in the primary tissues of interest (in this case, brain and kidney) being mirrored in lymphocytes, when the primary tissues are not accessible. SCARB2 has low, but detectable, expression in lymphocytes. In AMRF, the analysis of lymphocyte expression was potentially compromised by the immunosuppressive therapies that patients were undergoing as part of their renal-transplant therapy. An essential point is that the differential-expression analysis does not need to be carried out genome wide. With the linkage-analysis data, only probe sets in 66 genes were assessed for expression changes. The success of the differential-expression study in identifying SCARB2 relied on microararray-analysis tools that maintain good precision even for lowly expressed genes and that can evaluate statistical significance reliably with small numbers of arrays. SCARB2 would not have been top ranked from the microarray study on the basis of more naive statistical criteria such as fold change or ordinary t tests.
These data establish mutations in SCARB2 as the major cause of AMRF. The same mutation responsible for AMRF in case C was also detected in a Canadian AMRF patient, highlighting that this mutation is likely to be an ancestral Anglo-Celtic mutation. The absence of mutations in other familial nephropathies suggests SCARB2 mutations may be specific for AMRF.
SCARB2 is an ancestral gene and has a ortholog in Caenorhabditis elegans. The human and mouse SCARB2/Limp2 share 84% nucleotide and 85% amino acid identity. SCARB2 encodes a 478 amino acid glycoprotein located in lysosomal membranes.22,23 The gene is expressed in a range of tissues including brain and kidney. The roles of SCARB2 are not well understood, but it is thought to play a role in the biogenesis and maintenance of endosomal and lysosomal compartments.24,25 The protein has an N- and C-terminal transmembrane domain and a large, well conserved CD36 domain.
Each of the SCARB2 mutations found in the AMRF patients fall within the CD36 domain (Figure 2). SCARB2/Limp2 is an abundant lysosomal-membrane protein. SCARB2/Limp2 may sometimes be expressed at the cell surface, but in the majority of cells, the steady-state distribution is intracellular.25 It may have pleiotropic functions as shown by the heterogeneous cellular pathology in mice16 and humans (this paper) deficient of Limp2/SCARB2, including a role in lysosomal biogenesis.24 Pleiotropy may be conferred by a gene product having multiple molecular functions or by multiple consequences of a single molecular function.26 Genes with pleiotropic effects are, in general, under strong stabilizing selection, but there may be compromises among adaptation of different traits because changes that are beneficial in one tissue may be deleterious in another.26,27 Dysfunction of SCARB2/Limp2 has maximum phenotypic effect on the nervous system and kidney, but the pathology is different, showing storage in the brain but not in the kidney. Moreover, across species, brain storage is predominantly intraneuronal in mice but extraneuronal in man. The renal pathology is glomerular in man but in mice predominantly affects the urothelium with subtle glomerular lesions.
The renal lesions seen in human and mouse kidneys with SCARB2/Limp2 mutations differed. Pelvi-ureteric obstruction, as observed in Limp2-deficient mice, was not seen in humans. Glomerular lesions were found in both, but of quite different severity. Whereas the human glomeruli showed collapsing glomerulopathy, the most severe variant of FSGS, this was not seen in the Limp2−/− mice. The mice did have proteinuria of presumed glomerular origin associated with foot-process effacement.
Mutations of a lysosomal protein suggest a new mechanism for the genesis of proteinuria and FSGS. Inherited forms of FSGS that involve proteins expressed exclusively or strongly in the podocyte have previously been described. Many affect podocyte structure, and in particular, the maintenance of the slit diaphragm,28 and one affects a phospholipase (PLCE1) expressed in podocytes.29 The renal lesion in AMRF differs from the other inherited forms of FSGS in that it is a much more severe histological variant. SCARB2/Limp2 is expressed in podocytes (data not shown), although none of the proteins associated with congenital FSGS have a known role in lysosomal function, and it is not known whether SCARB2 interacts with any of them. A hypothesis to explain how abnormal lysosomal function due to lack of SCARB2 results in proteinuria is that altered recycling or degradation of one or more of these proteins contributes to glomerular damage.
Evidence that SCARB2 mutations contribute to the pathogenesis of collapsing glomerulopathy has implications for other diseases. Collapsing glomerulopathy is a FSGS variant that is associated with prominent podocyte hyperplasia and is commonly seen in HIV nephropathy. Nef is the protein critical for FSGS in HIV nephropathy.30 Interestingly, nef possesses a similar intracellular sorting motif to one present in SCARB2. It is possible that nef interacts with proteins that usually bind SCARB2, thereby giving rise to lysosomal abnormalities and changes in podocyte function.
Progressive myoclonus epilepsy (PME) can be caused by a variety of molecular lesions. These include mitochondrial tRNA mutations and autosomal-recessive mutations in a variety of genes including an intracellular protease inhibitor (Cystatin B) and molecules that lead to intracellular storage of glycoproteins (EPM2A and EPM2B) or lipofuscin-like material (CLN2-8).31 The addition of SCARB2 to this list further highlights the importance of lysosomal mechanisms in the genesis of PME. The final common pathway for the genesis of the PME syndrome by such diverse molecular causes is unknown but appears to involve the cerebellum and probably the rubral and oliviary connections. The prominent involvement of the cerebellum in the storage process in both human AMRF and the Limp2−/− mouse supports this. Interestingly, whereas both species have ataxia, myoclonus was not seen in the mouse. Similarly, in the prototypic PME, Unverricht-Lundborg disease, because of deficiency in cystatin B, the mouse model has prominent ataxia, but seizures and myoclonus are specific to certain genetic backgrounds.32,33 Moreover, cystatin B is known to be associated with lysosomes, at least at early stages of differentiation,34 so deficiencies in SCARB2 and cystatin B might act via a similar cellular pathway in causing PME.
The accumulation of storage material in brain is consistent with the known role of SCARB2/Limp2 as a lysosomal protein,22,23 perhaps affecting intracellular transport or endosome recycling. Lysosomes have a crucial role in autophagy, a process recently believed to be very important in neurodegenerative diseases.35 SCARB2/Limp2 might have a role in physiological autophagy, and its deficiency could thus lead to accumulation of normally recycled proteins or organelles manifesting as inclusions. Moreover, the neuropathology of SCARB2/Limp2 deficiency bears some resemblance to that of the family of neuronal ceroid lipofuscinoses in which a variety of lysosomal-related proteins are deficient36 and disturbed autophagy has been demonstrated;37–39 indeed, AMRF can be mistaken for the adult form (Kufs disease).40 Interestingly, Limp2 was also recently described as a receptor for β-glucocerebrosidase, the enzyme that is deficient in Gaucher disease.41 The lack of the Limp2 receptor in mice caused a missorting of the majority of glucocerebrosidase to the extracellular space, and the lysosomal level of this enzyme was found to be severely reduced. This lack of lysosomal β-glucocerebrosidase was apparently not sufficient to cause a robust Gaucher-like phenotype in Limp2-deficient mice. It needs to be elucidated whether the observed neuronal storage in Limp2-deficient mice is associated with a defect in glucosylceramide metabolism. In man, however, the type 3 form of Gaucher disease (MIM 231000) does have progressive myoclonus epilepsy as a key part of the phenotype.42 Finally, SCARB2/Limp2 has also been shown to have an extra-lysosomal role in the intercalated disc of the cardiac myocyte,43 so whether the renal lesions are the consequence of lysosomal or extra-lysosomal dysfunction needs to be determined and will be essential for designing therapeutic strategies.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for AMRF)
The R Project for Statistical Computing, http://www.r-project.org
Acknowledgments
We thank the patients and their families for participation. Sharon Bain, Mark Corbett, and Kathie Friend (Women's and Children's Hospital Adelaide) kindly assisted with the molecular genetic studies. We thank Dr. Michael Gonzales (Department of Anatomical Pathology, Royal Melbourne Hospital) for permission to restudy and photograph postmortem material and Dr. Stirling Carpenter (Portugal) for helpful discussions on the neuropathology. This paper was supported by the National Health and Medical Research Council (S.F.B., J.C.M., and M.B.), the Thyne Reid Charitable Trusts (S.F.B. and L.D.), and the Deutsche Forschungsgemeinschaft (P.S.; DFGSA683/5-1).
References
- 1.Andermann E., Andermann F., Carpenter S., Wolfe L.S., Nelson R., Patry G., Boileau J. Action myoclonus-renal failure syndrome: A previously unrecognized neurological disorder unmasked by advances in nephrology. Adv. Neurol. 1986;43:87–103. [PubMed] [Google Scholar]
- 2.Badhwar A., Berkovic S.F., Dowling J.P., Gonzales M., Narayanan S., Brodtmann A., Berzen L., Caviness J., Trenkwalder C., Winkelmann J. Action myoclonus-renal failure syndrome: Characterization of a unique cerebro-renal disorder. Brain. 2004;127:2173–2182. doi: 10.1093/brain/awh263. [DOI] [PubMed] [Google Scholar]
- 3.Vadlamudi L., Vears D.F., Hughes A., Pedagogus E., Berkovic S.F. Action myoclonus-renal failure syndrome: A cause for worsening tremor in young adults. Neurology. 2006;67:1310–1311. doi: 10.1212/01.wnl.0000238424.23177.5e. [DOI] [PubMed] [Google Scholar]
- 4.Di X., Matsuzaki H., Webster T.A., Hubbell E., Liu G., Dong S., Bartell D., Huang J., Chiles R., Yang G. Dynamic model based algorithms for screening and genotyping over 100 K SNPs on oligonucleotide microarrays. Bioinformatics. 2005;21:1958–1963. doi: 10.1093/bioinformatics/bti275. [DOI] [PubMed] [Google Scholar]
- 5.Terwilliger J.D., Ott J. Johns Hopkins University Press; Baltimore, London: 1994. Handbook of Human Genetic Linkage. [Google Scholar]
- 6.O'Connell J.R., Weeks D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 8.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 9.Stankovich J., Bahlo M., Rubio J.P., Wilkinson C.R., Thomson R., Banks A., Ring M., Foote S.J., Speed T.P. Identifying nineteenth century genealogical links from genotypes. Hum. Genet. 2005;117:188–199. doi: 10.1007/s00439-005-1279-y. [DOI] [PubMed] [Google Scholar]
- 10.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Z., Irizarry R., Gentleman R., Martinez Murillo F., Spencer F. A model based background adjustment for oligonucleotide expression arrays. Journal of the American Statistical Association. 2004;99:909–917. [Google Scholar]
- 12.Smyth G.K. Limma: Linear models for microarray data. In: Gentleman R., Carey V., Dudoit S., Irizarry R., Huber W., editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 13.Smyth G.K., Michaud J., Scott H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 14.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 15.Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Statist. Soc. Ser. B. Methodological. 1995;57:289–300. [Google Scholar]
- 16.Gamp A.C., Tanaka Y., Lullmann-Rauch R., Wittke D., D'Hooge R., De Deyn P.P., Moser T., Maier H., Hartmann D., Reiss K. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum. Mol. Genet. 2003;12:631–646. [PubMed] [Google Scholar]
- 17.Knipper M., Claussen C., Ruttiger L., Zimmermann U., Lullmann-Rauch R., Eskelinen E.L., Schroder J., Schwake M., Saftig P. Deafness in LIMP2-deficient mice due to early loss of the potassium channel KCNQ1/KCNE1 in marginal cells of the stria vascularis. J. Physiol. 2006;576:73–86. doi: 10.1113/jphysiol.2006.116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothdach A.J., Dietl T., Kumpfel T., Gottschalk M., Schumann E.M., Trenkwalder C. Familiar myoclonus-renal failure syndrome. Nervenarzt. 2001;72:636–640. doi: 10.1007/s001150170065. [DOI] [PubMed] [Google Scholar]
- 19.Leutenegger A.L., Labalme A., Genin E., Toutain A., Steichen E., Clerget-Darpoux F., Edery P. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: Application to Taybi-Linder syndrome. Am. J. Hum. Genet. 2006;79:62–66. doi: 10.1086/504640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vierimaa O., Georgitsi M., Lehtonen R., Vahteristo P., Kokko A., Raitila A., Tuppurainen K., Ebeling T.M., Salmela P.I., Paschke R. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312:1228–1230. doi: 10.1126/science.1126100. [DOI] [PubMed] [Google Scholar]
- 21.Karp C.L., Grupe A., Schadt E., Ewart S.L., Keane-Moore M., Cuomo P.J., Kohl J., Wahl L., Kuperman D., Germer S. Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat. Immunol. 2000;1:221–226. doi: 10.1038/79759. [DOI] [PubMed] [Google Scholar]
- 22.Calvo D., Dopazo J., Vega M.A. The CD36, CLA-1 (CD36L1), and LIMPII (CD36L2) gene family: Cellular distribution, chromosomal location, and genetic evolution. Genomics. 1995;25:100–106. doi: 10.1016/0888-7543(95)80114-2. [DOI] [PubMed] [Google Scholar]
- 23.Fujita H., Takata Y., Kono A., Tanaka Y., Takahashi T., Himeno M., Kato K. Isolation and sequencing of a cDNA clone encoding the 85 kDa human lysosomal sialoglycoprotein (hLGP85) in human metastatic pancreas islet tumor cells. Biochem. Biophys. Res. Commun. 1992;184:604–611. doi: 10.1016/0006-291x(92)90632-u. [DOI] [PubMed] [Google Scholar]
- 24.Kuronita T., Eskelinen E.L., Fujita H., Saftig P., Himeno M., Tanaka Y. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J. Cell Sci. 2002;115:4117–4131. doi: 10.1242/jcs.00075. [DOI] [PubMed] [Google Scholar]
- 25.Eskelinen E.L., Tanaka Y., Saftig P. At the acidic edge: Emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13:137–145. doi: 10.1016/s0962-8924(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 26.He X., Zhang J. Toward a molecular understanding of pleiotropy. Genetics. 2006;173:1885–1891. doi: 10.1534/genetics.106.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodgkin J. Seven types of pleiotropy. Int. J. Dev. Biol. 1998;42:501–505. [PubMed] [Google Scholar]
- 28.Quaggin S.E. A new piece in the nephrotic puzzle. Nat. Genet. 2006;38:1360–1361. doi: 10.1038/ng1206-1360. [DOI] [PubMed] [Google Scholar]
- 29.Hinkes B., Wiggins R.C., Gbadegesin R., Vlangos C.N., Seelow D., Nurnberg G., Garg P., Verma R., Chaib H., Hoskins B.E. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat. Genet. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 30.Shah S.N., He C.J., Klotman P. Update on HIV-associated nephropathy. Curr. Opin. Nephrol. Hypertens. 2006;15:450–455. doi: 10.1097/01.mnh.0000232887.58271.67. [DOI] [PubMed] [Google Scholar]
- 31.Shahwan A., Farrell M., Delanty N. Progressive myoclonic epilepsies: A review of genetic and therapeutic aspects. Lancet Neurol. 2005;4:239–248. doi: 10.1016/S1474-4422(05)70043-0. [DOI] [PubMed] [Google Scholar]
- 32.Pennacchio L.A., Bouley D.M., Higgins K.M., Scott M.P., Noebels J.L., Myers R.M. Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice. Nat. Genet. 1998;20:251–258. doi: 10.1038/3059. [DOI] [PubMed] [Google Scholar]
- 33.Shannon P., Pennacchio L.A., Houseweart M.K., Minassian B.A., Myers R.M. Neuropathological changes in a mouse model of progressive myoclonus epilepsy: Cystatin B deficiency and Unverricht-Lundborg disease. J. Neuropathol. Exp. Neurol. 2002;61:1085–1091. doi: 10.1093/jnen/61.12.1085. [DOI] [PubMed] [Google Scholar]
- 34.Alakurtti K., Weber E., Rinne R., Theil G., de Haan G.J., Lindhout D., Salmikangas P., Saukko P., Lahtinen U., Lehesjoki A.E. Loss of lysosomal association of cystatin B proteins representing progressive myoclonus epilepsy, EPM1, mutations. Eur. J. Hum. Genet. 2005;13:208–215. doi: 10.1038/sj.ejhg.5201300. [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Vicente M., Cuervo A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 36.Mole S.E. Neuronal ceroid lipofuscinoses (NCL) Eur. J. Paediatr. Neurol. 2006;10:255–257. doi: 10.1016/j.ejpn.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Koike M., Shibata M., Waguri S., Yoshimura K., Tanida I., Kominami E., Gotow T., Peters C., von Figura K., Mizushima N. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am. J. Pathol. 2005;167:1713–1728. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao Y., Espinola J.A., Fossale E., Massey A.C., Cuervo A.M., MacDonald M.E., Cotman S.L. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2006;281:20483–20493. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- 39.Shacka J.J., Klocke B.J., Young C., Shibata M., Olney J.W., Uchiyama Y., Saftig P., Roth K.A. Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis. J. Neurosci. 2007;27:2081–2090. doi: 10.1523/JNEUROSCI.5577-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berkovic S.F., Carpenter S., Andermann F., Andermann E., Wolfe L.S. Kufs' disease: A critical reappraisal. Brain. 1988;111:27–62. doi: 10.1093/brain/111.1.27. [DOI] [PubMed] [Google Scholar]
- 41.Reczek D., Schwake M., Schroder J., Hughes H., Blanz J., Jin X., Brondyk W., Van Patten S., Edmunds T., Saftig P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131:770–783. doi: 10.1016/j.cell.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 42.Park J.K., Orvisky E., Tayebi N., Kaneski C., Lamarca M.E., Stubblefield B.K., Martin B.M., Schiffmann R., Sidransky E. Myoclonic epilepsy in Gaucher disease: Genotype-phenotype insights from a rare patient subgroup. Pediatr. Res. 2003;53:387–395. doi: 10.1203/01.PDR.0000049515.79882.94. [DOI] [PubMed] [Google Scholar]
- 43.Schroen B., Leenders J.J., van Erk A., Bertrand A.T., van Loon M., van Leeuwen R.E., Kubben N., Duisters R.F., Schellings M.W., Janssen B.J. Lysosomal integral membrane protein 2 is a novel component of the cardiac intercalated disc and vital for load-induced cardiac myocyte hypertrophy. J. Exp. Med. 2007;204:1227–1235. doi: 10.1084/jem.20070145. [DOI] [PMC free article] [PubMed] [Google Scholar]