

Abstract
Childhood absence epilepsy (CAE) accounts for 10% to 12% of epilepsy in children under 16 years of age. We screened for mutations in the GABAA receptor (GABAR) β3 subunit gene (GABRB3) in 48 probands and families with remitting CAE. We found that four out of 48 families (8%) had mutations in GABRB3. One heterozygous missense mutation (P11S) in exon 1a segregated with four CAE-affected persons in one multiplex, two-generation Mexican family. P11S was also found in a singleton from Mexico. Another heterozygous missense mutation (S15F) was present in a singleton from Honduras. An exon 2 heterozygous missense mutation (G32R) was present in two CAE-affected persons and two persons affected with EEG-recorded spike and/or sharp wave in a two-generation Honduran family. All mutations were absent in 630 controls. We studied functions and possible pathogenicity by expressing mutations in HeLa cells with the use of Western blots and an in vitro translation and translocation system. Expression levels did not differ from those of controls, but all mutations showed hyperglycosylation in the in vitro translation and translocation system with canine microsomes. Functional analysis of human GABAA receptors (α1β3-v2γ2S, α1β3-v2[P11S]γ2S, α1β3-v2[S15F]γ2S, and α1β3-v2[G32R]γ2S) transiently expressed in HEK293T cells with the use of rapid agonist application showed that each amino acid transversion in the β3-v2 subunit (P11S, S15F, and G32R) reduced GABA-evoked current density from whole cells. Mutated β3 subunit protein could thus cause absence seizures through a gain in glycosylation of mutated exon 1a and exon 2, affecting maturation and trafficking of GABAR from endoplasmic reticulum to cell surface and resulting in reduced GABA-evoked currents.
Introduction
Childhood absence epilepsy (CAE [ECA1 (MIM 600131), ECA2 (MIM 607681), ECA3 (MIM 607682), ECA4 (MIM 611136)]),1–4 a common idiopathic generalized epilepsy (EIG [MIM 600669]), accounts for 10% to 12% of epilepsy in children under 16 years of age according to prospective community-based epidemiologic studies.5,6 Absence is characterized by frequent brief loss of consciousness lasting 3 to 10 s and occurring up to about 200 attacks per day. The electroencephalograph shows bilateral, symmetrical, synchronous, 3–4 Hz spike-and-wave bursts during absence seizures.7 CAE appears more frequently in girls,8,9 and when present as the sole phenotype, CAE has better prognosis10 and remits in 95% of cases in Loiseau's report.11 Historically, a strong genetic contribution to the spike-wave traits of CAE has been supported by 74% concordance for monozygotic twins and 27% concordance for dizygotic twins.12 Metrakos and Metrakos showed that siblings and offspring had (a) 50% risk of inheriting the 3Hz spike-wave trait, (b) 35% risk of expressing the EEG trait in their lifetime, and (c) 12% risk for tonic-clonic seizures and 8% risk for absences.13–15 These family and twin studies support the concept of a major gene interacting with additional genetic and environmental factors in CAE.
In 1999, Feucht et al.16 used a Monte Carlo version of the multiallele Transmission Disequilibrium Test to show possible association between GABRB3 (MIM 137192) and CAE in 50 Austrians. Urak et al.17 then replicated significant association between 45 CAE patients and 13 SNPs located between the exon 1a promoter and the beginning of intron 3 within GABRB3. Reporter-gene assays in NT2 cells (human neuronal-like cell lines) showed lower transcriptional activity of the disease-associated GABRB3 promoter haplotype. We screened for mutations in GABRB3 in families ascertained through a proband with remitting pyknoleptic CAE because of the findings in the above studies by Feucht et al.16 and Urak et al.,17 because typical and atypical absence attacks are present in Angelman syndrome patients whose chromosome 15q11–13 deletion includes GABRB3,18 and because heterozygous and homozygous null mutants for GABRB3 in mice show absence-like attacks with EEG characteristics and pharmacological responses similar to human absence seizures.19,20
Subjects and Methods
Family Material
We ascertained 48 families, through probands with remitting pyknoleptic childhood absence epilepsy, from Mexico City and Hermosillo, Mexico; Tegucigalpa, Honduras; and San Miguel, El Salvador (See Table 1 for inclusion and exclusion criteria). Initial diagnosis was made by neurologists at the study sites of the international consortium, GENESS (Genetic Epilepsy Studies) and then validated by M.T.M. and A.V.D.E. The diagnosis in probands and affected family members was based on the guidelines of the Commission on Classification of the International League Against Epilepsy.21 Each responsible person (parent or adult patient) in each family signed an informed-consent form that was approved by the Human Subject Protection Committee at the David Geffen School of Medicine at the University of California, Los Angeles; or by the National Institute of Neurology and Neurosurgery in Mexico City; or by the Secretary of Health of El Salvador; or by the Research Unit at the School of Medical Sciences at the National Autonomous University of Honduras. We obtained blood from 416 healthy Hispanic blood donors from Mexico. We also obtained blood samples from 190 healthy blood donors residing in Honduras and 24 individuals considered as “married-ins” belonging to Honduras families that had been recruited for genetic studies of childhood absence epilepsy and juvenile myoclonic epilepsy. Genomic DNA was extracted from EDTA-treated blood samples with the QIAamp DNA Blood Mini Kit (QIAGEN, Valencia, CA) and the Wizard Genomic DNA Purification System (Promega, Madison,WI).
Table 1.
Inclusion and Exclusion Criteria of Childhood Absence Epilepsy
| Inclusion Criteria for Remitting Pyknoleptic Absence Epilepsy (Modified from ILAE∗) |
|---|
| Age at onset between 2 and 12 years |
| Brief (3–20 seconds) and frequent (>10/day) absence seizures |
| EEG generalized high amplitude 2.5–3.5 Hz spike (maximum 3 spikes) and slow wave complexes lasting 3–20 seconds, spontaneously or on hyperventilation or on photic stimulation |
| Normal neurological state and development |
| Remission of absence seizure between 10 years and 18 years of age |
| Exclusion Criteria for Childhood Absence Epilepsy |
| Myoclonic jerks prior to or during active stage of absence |
| Symmetric synchronous or arrhythmic myoclonus of head, trunk or limb or a diagnosis of juvenile myoclonic epilepsy or a diagnosis of progressive myoclonus epilepsy |
| Progressive neurological deterioration |
∗International League Against Epilepsy Commission and Terminology 1989 Classification of Epilepsies and Epileptic Syndromes
When the proband is in late childhood or early adolescence still requiring treatment, we consider the family to have the remitting form of absence if an affected family member has remitting form of absence.
Linkage Analysis
We first used the computer-simulation method of Ott22,23 to assess the strength of genetic material for linkage in families M120, HMO10, H12, and H08, with the assumption that a dominant disease with 50% penetrance was present. We then genotyped eight microsatellite markers located in the vicinity of the UBE3A, GABRB3, α5 subunit gene (GABRA5), and γ3 subunit gene (GABRG3) cluster on chromosome 15q11.2-12. These markers were selected from the OMIM database and according to the report of Glatt et al.24,25 Two-point linkage analyses were performed with the MLINK and ILINK programs of the LINKAGE software package, version 5.21, under the assumption of autosomal-dominant inheritance with 50% penetrance, with the frequency of the disease allele at 0.001, phenocopy and gene-mutation rates of 1%. The allele frequencies were estimated with the use of the Centre d'Etude du Polymorphisme Humain (CEPH) database.
Mutation Analysis
We designed primers with the Primer 3 program to amplify the full region spanned from the 5′ UTR region to exon 3. This span starts from 1.5 kb upstream of exon 1a to 62 bp downstream from exon 3. We also amplified all coding regions from exon 4 to exon 9, including splice sites, and the selected regions from intron 3 and 3′ UTR. For PCR reactions, the AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA) or Fast Taq polymerase (Roche, Indianapolis, IN) was used according to manufacturer's instructions. Both patient and healthy-control genomic-DNA samples served as templates for PCR amplifications. Each PCR product of probands and controls was screened by heteroduplex analysis with the use of denaturing high-performance liquid chromatography (DHPLC WAVE, Transgenomic)26,27 or directly sequenced with the use of ABI 3700 capillary automated-sequencing system (Applied Biosystems, Foster City, CA). Suspected variants were subjected to PCR at least twice and confirmed by digestion or sequences of parents.
Expression Constructs and Mutagenesis
Preparation of GABRB3 cDNAs for Expression
Full-length cDNA for human GABAA receptor β3 subunit in expression vector pCMV-SPORT 6 was obtained from American Type Culture Collection, Manassas, VA. We isolated the complete exon 1a and part of exon 2 by PCR, yielding one wild-type and two mutated versions of exon 1a from probands. We then cloned them into pCMV-SPORT after TOPO TA cloning (Invitrogen, Carlsbad, CA). The stop codon was removed from each transcript, which was inserted in the frame with the GFP coding sequence, in the vector pFP-N1 to produce three versions of C-terminal GFP-tagged GABRB3-exon 1a cDNA.
In Vitro Transcription, Translation, and Translocation
The isolated cDNA constructs from probands were cloned into pCMV-XL5 (Origene, Rockville, MD) after TOPO TA cloning (Invitrogen, Carlsbad, CA). A stop codon was introduced in exon 6 after amino acid Ile-206 by QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) in order to express only the extracellular domain of the GABRB3 protein. Each cRNA was transcribed with the use of the mMESSAGEmMACHINE T7 kit (Ambion, Austin, TX).
Immunoblotting Analysis
HeLa cells were transfected with expression constructs with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), and homogenized 11–48 hr after transfection in hypotonic buffer (0.25 M sucrose, 10 mM Tris–HCl, 10 mM NaCl, 1 mM EDTA, pH 7.5) supplemented with a mixture of protease inhibitors (Roche, Indianapolis, IN). The lysate was centrifuged at 3300 g for 5 min to remove nuclei, and the supernatant was used as total cytosolic-plus-membrane protein fraction according to Miyawaki28 and Ganesh.29 Protein samples were run on 10% Tris-HCl gels (BioRad, Hercules, CA) and transferred onto a nitrocellulose filter (BioRad, Hercules, CA) at 100 mA for one hour in transfer buffer (48 mM Tris base, 39 mM glycine, 0.037% [v/v] SDS [electrophoresis grade], 20% [v/v] methanol, pH 8.3). The filter was incubated in blocking solution (PBS + 0.05% Tween 20 pH 7.4) containing 5% nonfat dry milk powder for one hour at room temperature. The membrane was processed through sequential incubations with primary antibody (anti-GFP [Santa Cruz Biotechnology, Santa Cruz, CA], 1:200, dilution, monoclonal antibody against the GABAA receptor β3 subunit, bd17 [Chemicon, Temecula, CA], 1:500, and C20 [Santa Cruz Biotechnology, Santa Cruz, CA], 1:500) for one hour, then secondary antibodies were added at 1:3000 for one hour. Immunoreactive proteins on the filter were visualized by Typhoon software 9410. Immuno-quantitation was calculated by ImageQuant 5.2.
In Vitro Transcription, Translation, and Translocation
In vitro translation and translocation were performed with the use of the above plasmids in a coupled transcription and translocation rabbit reticulocyte lysate system or with the use of cRNAs in nuclease-treated rabbit reticulocyte lysate system (Promega, Madison, WI), with L-[35S]methionine (GE Healthcare Bio-Sciences, Piscataway, NJ) in accordance with the manufacturer's protocol. Canine pancreatic microsomal membranes (1.8 μl) (Promega, Madison, WI) were added directly to each reaction medium for translocation experiments. After incubation at 30°C for 60–90 min, aliquots of 3–10 μl were diluted into 100 μl of phosphate-buffered saline (PBS, pH 7.4) with 1 mM PMSF and kept on ice for 30 min. Microsomes were collected by centrifugation (20,000 × g for one hour, 4°C). The pellets were rinsed twice with 100 μl of PBS. Supernatant proteins or the reaction without microsomes were precipitated with 1000 μl of acetone with 10% trichloroacetic acid at −20°C overnight, centrifuged, then dissolved in sample buffer (S). For cleaving of whole N-linked carbohydrates, each microsomal pellet was resuspended in H2O and then treated with 100 units of PNGase F (NEB, Ipswich, MA) according to the manufacturer's instructions (37°C for 3 hr). Each pellet that was treated (D) and or not treated (P) with sample buffer was analyzed by SDS-PAGE on 12% or 6%–18% Tris-HCl gels (Biorad, Hercules, CA) after denaturing at 65°C for five min with supernatant protein. After electrophoresis, gels were soaked in 50% methanol, 7% glacial acetic acid for 30 min and then 7% methanol, 7% glacial acetic acid and 2% glycerol for 15 min. Gels were dried and exposed to a Phosphorimager cassette plate. Molecular weight was ascertained by the commercial protein standard (Invitrogen, Carlsbad, CA) and the size of the supplemental control of canine microsomes. Immuno-quantitation was calculated by ImageQuat 5.2. For mutations in exon 1a, the proportion of each band density was compared with the wild-type.
Cell Culture, Transfection, and Immunomagnetic Selection
HEK293T fibroblasts were maintained in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin (all cell-culture products from GIBCO, Carlsbad, CA) and incubated at 37°C with 5% CO2/95% air. GABAA receptor subunit cDNA was inserted into a pcDNA3.1 (α1 and γ2) or pCMV (β3) promoter, and point mutations were generated with the use of a Quikchange kit (Stratagene, La Jolla, CA). For transfection, 0.3 μg of each subunit cDNA (α1, β3, γ2S) was cotransfected with 1 μg of cDNA for the pHook antigen (Invitrogen, Carlsbad, CA) for selection. Twenty-four to thirty-six hours after transfection, positively transfected cells were selected with the use of ferromagnetic beads in a protocol described previously.30 After selection, cells were plated onto 35 mm dishes for electrophysiological recording the following day.
Electrophysiological Recording
Single cells were chosen for recording on the basis of the presence of two or more beads from the selection process. The external recording solution consisted of (in mM): NaCl 142, KCl 8, MgCl2 6, CaCl2 1, HEPES 10, glucose 10, pH 7.4 and 318–328 mOsm. The internal recording solution consisted of (in mM): KCl 153, MgCl2 1 MgATP 2, HEPES 10, EGTA 5, pH 7.3 and 305–312 mOsm (all reagents from Sigma-Aldrich, St. Louis, MO). Recording solutions were designed such that the Cl− reversal potential was 0 mV.
Whole-cell currents were low-pass filtered at 2kHz and recorded with the use of an Axopatch 200B Amplifier (Molecular Devices, Foster City, CA) and a Digidata 1332A (Molecular Devices) with pClamp9.1 software. Borosilicate recording pipettes (World Precision Instruments, Philadelphia, PA) were pulled with a Sutter P-2000 micropipette puller (Sutter Instruments, Novato, CA) and fire-polished with a Micro Forge (Narishige, Tokyo, Japan) to a resistance of 0.9 MΩ–1.5 MΩ. Once a seal was obtained on the cell and the membrane was perforated (whole-cell voltage clamp), the capacitance of the cell was recorded from the lab-bench tool bar in pClamp9.1 (Molecular Devices). The cells were lifted off the dish and placed in front of a glass multibarrel connected to a Piezo stepper used for rapid application of GABA. Perfusion from the barrel consisted of either external solution or external solution containing 1 mM GABA (Sigma). Open-tip exchange times between lumen of the barrel were determined by perfusion of low- and high-electrolyte solutions and were consistently less than 1 ms. For analysis of current density, the pipette capacitance (13 pF) was subtracted from the cell capacitance and the peak current amplitude was normalized to the capacitance of the cell. All data was compared with the use of a Student's paired t test, with Welch's correction when variances were significantly different, and plotted with the use of GraphPad Software (San Diego, CA).
In Silico Analysis
The genome information and homology searches were explored with the use of the UCSC genome browser (May 2006 assembly) and the NCBI website. GC percentage and GC-island prediction were performed by EMBL-EBI tools (EMBOSS CpGPlot/CpGReport/Isochore). The transmembrane helices were analyzed by SOSUI and TMHMM, version 2.0. The signal-peptide prediction was performed by the SignalP 3.0 server, and Signal CF. N-glycosylation and O-glycosylation sites were analyzed with NetNGlyc 1.0 and YinOYang, respectively. The secondary-structure analysis was performed by GOR IV secondary-structure-prediction method. The prediction of functional effect was analyzed by the Polyphen program.
Statistical Analysis
The Fisher exact χ2 test was used in comparison of the prevalence of mutations in patients with remitting CAE versus that in controls. The results of image densities were calculated and compared by the use of a chi-square goodness-of-fit test.
Results
Phenotypes
Figure 1 illustrates the pedigrees of four families (M120, HMO10, H12, and H08) that have mutations in GABRB3. Table 2 summarizes the clinical characteristics of absence seizures in probands. Table 3 identifies nonproband family members who carry GABRB3 mutations and are affected with absences. Table 3 also lists family members with GABRB3 mutations who are not affected by epilepsy or absences.
Figure 1.
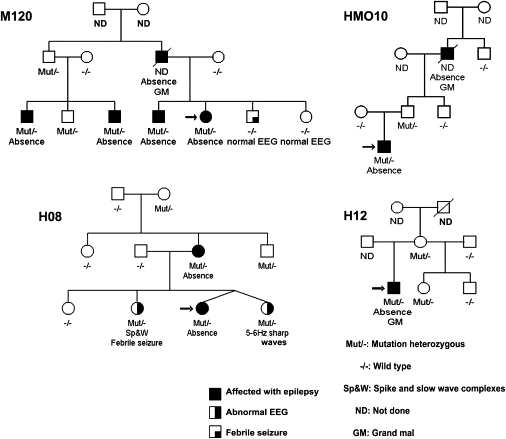
Four Families with GABRB3 Mutations
Each family number is placed beside each pedigree. Black circles or squares represent epilepsy affected females or males. Asymptomatic persons who have EEG 3 Hz diffuse bilateral spike wave complexes or 5 to 6 Hz sharp waves are represented by half black circles or squares.
Table 2.
Clinical Characteristics of Pyknoleptica Absences and Associated Seizure in Probands of Families with GABRB3 Mutations
| Family | Present Age (Yrs) and Years of Remission | Onset (Yrs) | Clinical Semiology |
|---|---|---|---|
| M120 | 30 (18 yrs w/o treatment, w/o seizures) | 5 | Staring with eyelid myocloniasb as eyeballs roll up. No grand mal seizure (GM). |
| HMO10 | 14 (2 yrs w/o treatment, w/o absence or atonic seizures) | 2 | Staring with 3 Hz eye blinks as eyeballs roll up, Rarely absences |
| Absences appears at 2 yrs of age, increased frequency (more than 20 attacks per day) between 4 and 6 yrs. | |||
| Rare episodes of atonic seizures with flaccid limbs and vomiting. No GM. | |||
| H12 | 18 (no seizures and GM seizures for 2 yrs but still on treatment) | 11 | Staring with eyelid myocloniab triggered by sunlight. GM at 12 yrs. |
| H08 | 15 (5 yrs w/o treatment, w/o absence seizures) | 7 | Staring as eyeballs roll up triggered by light, No GM. |
Pyknoleptic means more than one absence seizure per day, often 20 to 200 seizures per day.
Eyelid myoclonia consists of very rapid blinking and flickers of the eyelids as the eyes deviate upwards.
Table 3.
Family Members with Mutation and Absence
| Family Number | Family Members with Mutation | Absence-Affected Members with Mutation | Family Members Affected with Absence | Family Members with Mutation but No Epilepsy | Nucleotide Change | Effect on Protein |
|---|---|---|---|---|---|---|
| M120 | 6 | 4 | proband, brother of proband, two paternal male cousins | father of cousin; one brother of two affected paternal cousins | c.31C→T | Pro11Ser |
| HMO10 | 2 | 1 | proband | father of proband | c.31C→T | Pro11Ser |
| H12 | 3 | 1 | proband | mother of proband; half-sister of proband (with different father) | c.44C→T | Ser15Phe |
| H08 | 5 | 2 | proband, mother of proband | maternal grandmother of proband and two sistersa of proband | c.962G→A | Gly32Arg |
Two sisters with EEG abnormalities; one sister has 2–4 Hz diffuse spike- and slow-wave complexes and febrile convulsions, and the other sister has 5–6 Hz fronto-central sharp waves.
Absences with eyelid myoclonias that were sensitive to photic stimulation started at 5 and 11 years of age, respectively, in probands of families M120 and H12. The proband of family M120 only had absences and never developed grand mal seizures. On the other hand, five separate grand mal tonic-clonic seizures appeared at 12 years of age in the proband of family H12. Absences without eyelid myoclonias rarely appeared at 2 years of age in the proband of family HMO10. However, they increased to more than 30 absence attacks a day between 4 and 6 years of age. Rare sudden atonic falls appeared in early childhood, and grand mal or myoclonic seizures never developed. The proband of family H08 had absence without eyelid myoclonia as the sole phenotype, and absences started at 5 years of age. Absences and accompanying seizures disappeared after 12 years of age in all four probands.
Three probands of families M120, HMO10, and H08 remain without seizures and without treatment. Absences remained suppressed by valproate in the 18-year-old proband of family H12. When affected by seizures, family members most commonly had absences seizures. The father of the proband in family M120 and the grandfather of the proband in family HMO10 both had grand mal tonic-clonic seizures in addition to absences. A clinically asymptomatic sister of the proband in family H08 had epileptiform EEG 3–4 Hz polyspike wave complexes, whereas the asymptomatic twin sister of the proband had epileptiform EEG bifrontal 5–6 Hz sharp waves.
Genotypes
We observed three heterozygous missense mutations, namely, P11S and S15F in exon 1a and G32R in exon 2 (Figure 2). The P11S mutation in exon 1a segregated with four CAE-affected members of family M120. The deceased father of the proband of family M120 was not tested for mutations. The same P11S mutation was found in the proband and his clinically unaffected father in family HMO10. We considered family HM010 as a singleton because we were unable to screen his CAE-affected grandfather for mutations. Theoretically, family HM010 would be considered multigenerational if we had had access to the CAE-affected grandfather. Families M120 and HM010 are not related and reside in two separate cities of Mexico, namely, Mexico City and Hermosillo, and have different family names across three generations. Both have the heterozygous c.31C → T mutation in exon 1a at position 31 from the start codon.
Figure 2.
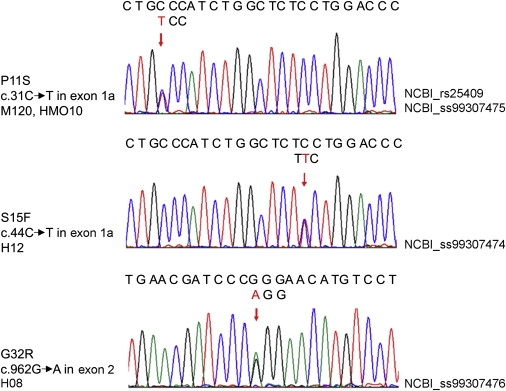
cDNA Sequencing in Each Proband
Each arrow shows the location of the mutation. The upper sequence represents wild-type. The lower triplet above the arrow represents the mutated code.
The S15F mutation in exon 1a is present in the proband, his asymptomatic mother, and his asymptomatic half brother in family H12 from Honduras. The G32R mutation in exon 2 segregated in four individuals who were clinically symptomatic or clinically asymptomatic with the EEG trait only; members of two generations in family H08, also from Honduras. P11S, S15F, and G32R mutations were not found in 630 healthy controls from Mexico and Honduras.
Linkage Analysis
Based on quadratic interpolation, the average simulated pooled LOD score was 1.9414 (SD 0.8848) and the maximum simulated pooled LOD score was 3.6339. The pooled maximum two point LOD score for all 4 families (M-120, H08, HMO10 and H12) was 1.022 for GABRB3, 2.019 for D15S1002 and 2.305 for 85CA at theta = 0 m = f. 85CA lies in 5′UTR of GABRB3 (see Figure 3 and Table 4). We obtained 0.351 for D15S122 and −3.918 for D15S1021.
Figure 3.
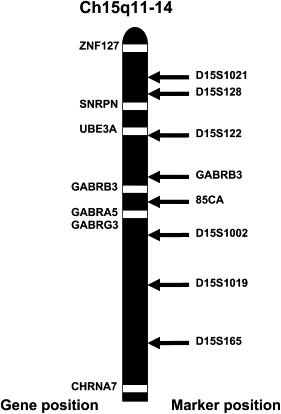
Gene and Marker Position on Chromosome 15q11-14
Figure 3 shows the relative position of markers in chr. 15q11-14. GABRB3 (marker) is about 60kb beyond the 3′ terminus of GABRB3, and 85CA is about 50kb from exon 1a of GABRB3.24,25
Table 4.
Summed Two-Point LOD Scores of Microsatellite Markers on Chromosome 15q11-14
| Microsatellite Marker |
θ |
||||
|---|---|---|---|---|---|
| 0 | 0.1 | 0.2 | 0.3 | 0.4 | |
| D15S1021 | −3.919 | −0.291 | −0.104 | −0.035 | −0.008 |
| D15S128 | −0.084 | −0.131 | −0.104 | −0.093 | −0.044 |
| D15S122 | 0.351 | 0.331 | 0.247 | 0.121 | 0.0054 |
| GABRB3 | 1.022 | 0.822 | 0.583 | 0.325 | 0.096 |
| 85CA | 2.306 | 1.78 | 1.233 | 0.688 | 0.219 |
| D15S1002 | 2.019 | 1.535 | 1.038 | 0.554 | 0.16 |
| D15S1019 | 0.712 | 0.554 | 0.384 | 0.211 | 0.064 |
| D15S165 | 2.117 | 1.64 | 1.138 | 0.632 | 0.193 |
θ: recombination fraction; m = f.
In Silico Analysis
GABRB3, located on chromosome 15q11.2-q12, spans almost 230 kb (UCSC Genome Browser, March 2006). The mRNA of GABRB3 consists of nine exons. Two alternative first exons, exon 1a and exon 1, encode the signal peptides of GABRB3.31 Exon 1a to exon 3 spans a 1.4 kb genomic region (GenBank accession number L04311) and contains a GC-rich (55%–80%) region with high content of CpG islands. The P11S, S15F, and G32R missense mutations reside in evolutionarily conserved amino acid sequences of exon 1a and exon 2 (Figure 4A). All missense mutations are predicted to have the same cleavage site as the wild-type, cleaved between Gly22 and Ser23 amino acids as predicted by software programs Signal P 3.1 and Signal CF (Figure 4B). The G32R missense mutation is also predicted to have the same cleavage site as the wild-type pre-peptide, GABRB3 protein isoform 1 precursor that is translated from the exon 1 mRNA, namely GABRB3 transcript variant 1 (NM_000814). On the basis of the predicted cleavage site, we calculate the location of the G32R mutation in exon 2 to occur at position 10 from the N terminus of the mature polypeptide. This polypeptide is produced from the isoform 2 precursor translated by exon 1a mRNA, namely GABRB3 transcript variant 2 (NM_021912). The secondary structure of all mutations can be predicted to show significant changes in secondary structure, according to the software GOR4 IV (Figure 4C).
Figure 4.
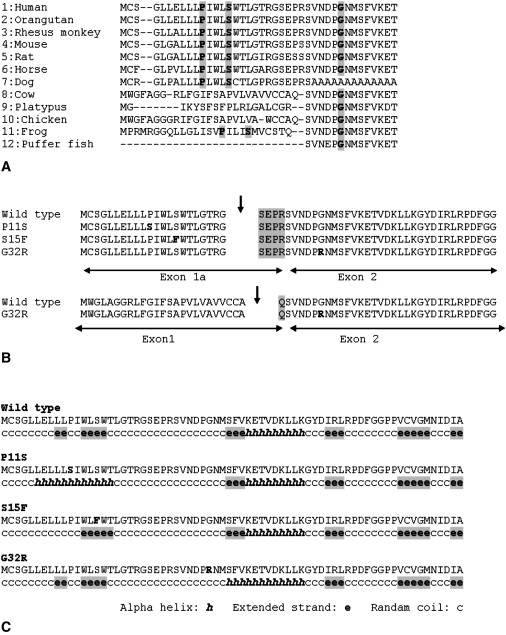
Conserved Amino Acid Sequence of Exon 1a and Exon 2 of GABRB3, Predicted Cleavage Site, and Predicted Secondary Structure of Each Mutation
(A) Conserved amino acid sequences of exon 1a and exon 2 of GABRB3. Each mutated amino acid position is indicated by gray shadow. 1. Homo sapiens: GABAA receptor, beta 3, (NP_068712), 2. Pongo pygmaeus: hypothetical protein, (CAH89717), 3. Macaca mulatta: PREDICTED GABAA receptor, beta 3, (XP_001109060), 4. Mus musculus: GABAA receptor, beta 3, (NP_0010337906), 5. Rattus norvegicus: GABAA receptor, beta 3, (EDL86448), 6. Equus caballus: PREDICTED: similar to GABAA receptor, beta 3, (XP_001493125), 7. Canis familiaris: PREDICTED: similar to GABAA receptor, beta 3, (XP_848482), 8. Bos Taurus: hypothetical protein, (NP_001092850), 9. Ornithorhynchus anatinus: PREDICTED: similar to GABAA receptor, beta 3, (XP_001505697), 10. Gallus gallus: GABAA receptor, beta 3, (NP_990677), 11. Xenopus tropicalis: Unknown protein, (AAI36050), 12. Tetraodon nigroviridis: unnamed protein product, (CAG06522).
(B) Predicted cleavage site. Arrows indicate each predicted cleavage site. Each mutation is predicted to have the same cleavage site as the wild-type exon 1a. G32R in exon 2 is predicted to have the same cleavage site even with exon 1 as the wild-type. The cleavage site of exon 1 is different from the exon 1a, therefore the N-termini differs (gray shadow).
(C) Predicted secondary structure of each mutation.
All mutations are predicted to change secondary structures.
The software programs NetNGlyc.1.0 and YinOYang predict with probability three N-glycosylation sites at asparagines and four O-glycosylation sites at serine and threonine to be located in the extracellular domain (Figure 5). Table 5 shows the potential score for the predicted N-glycosylation and O-glycosylation. There is no difference in potential for N-glycosylation between the wild-type and missense mutations in exon 1a. However, the G32R mutation has a slight but measurable lower potential at 33Asn for an N-glycosylation site and a higher potential for an O-glycosylation site at 23Ser (Table 5). In contrast, the S15F mutation has only a slightly lower potential than the wild-type and P11S for an O-glycosylation site at 23Ser.
Figure 5.

Predicted Glycosylation Site in Exon 1a–Exon 6 of GABRB3
Bold letters without shadow show the amino acids to be displaced by mutations.
Numbers show locations of amino acids.
Table 5.
Predicted Glycosylation Sites and Each Potential
| N-Glycosylation | |||
|---|---|---|---|
| Sample Position | Wild-Type, P11S, S15F | G32R | |
| 33 NMSF | 0.5857 | 0.5140 | |
| 105 NLTL | 0.7671 | 0.7671 | |
| 174 NCTL | 0.5487 | 0.5487 | |
| O-Glycosylation | |||
| Sample Position | Wild-Type, P11S | S15F | G32R |
| 23 S | 0.5166 | 0.4891 | 0.5336 |
| 157T | 0.4529 | 0.4529 | 0.4529 |
| 158T | 0.5198 | 0.5198 | 0.5198 |
| 201T | 0.5793 | 0.5793 | 0.5793 |
N-glycosylation is known to occur on Asparagines (N), which is located in the N-X-S/T stretch in which X is any amino acid except proline. The first column has the position number of the predicted glycosylation site in the amino acid sequences of exon 1a to exon 6 of GABRB3 (see fig. 5). Columns 2 and 3 represent their potential scores, derived from the averaged output of nine neural networks in NetNGlyc 1.0 Server. Intracellular O-glycosylation is characterized by the addition of N-acetylglucosamine, in a beta anomeric linkage (O-ß-GlcNAc), to Serine (S) and Threonine (T) residues in a protein. Each number shows each position number of the predicted O-glycosylation site. The YinOYang prediction server produces neural-network predictions for O-ß-GlcNAc attachment sites, incorporating predicted phosphorylated sites.
The prediction for functional effects by mutations with the use of the Polyphen program is applicable for the G32R exon 2 mutation in the mature polypeptide and the P11S exon 1a mutation but not for S15F. G32R is predicted to have a damaging effect on function, with a PSIC score difference of 1.973 by GABRB3 protein isoform 1 (NP_000805) and 1.78 by GABRB3 protein isoform 2 (NP_068712). P11S is predicted to be benign, with a PSIC score difference of 1.053.
Expression Study: Immunoblotting Analysis
For functional analysis, we studied first the expression level of full-length GABRB3-GFP fusion protein after transfection into HeLa cells by immunoblotting. We used GFP primary antibody and beta3 primary antibody to determine whether mutations in exon 1a and exon 2 had any consequences for expression. No differences in expression levels were observed between the mutated constructs and the wild-type when they were harvested at earlier times; 11–12 hr after transfection (Figure 6). At expression times of > 24 hr, the amount of each protein was variable, probably because they were digested.
Figure 6.
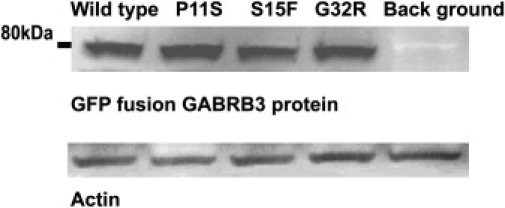
Western-Blot Analysis of Whole Cell Fractions Derived from HeLa Cell Expression 11 hr after Transfection
The GFP fusion GABRB3 protein was present at a slightly smaller molecular weight of 80kDa which was expected.
All expression levels of plasmids with mutated sequences were the same as the wild-type, even after normalization by Actin.
In Vitro Translation and Translocation
Next, we tested whether the observed mutations influenced the first steps of translation by using a cell-free in vitro translation and translocation system. In the absence of canine pancreatic microsomes, both wild-type and mutant cRNA (exon 1a to exon 6) were translated to GABRB3 proteins of the same size, namely 30 kDa (Figure 7, wild-type lane S, supernatant protein). Such proteins corresponded to the preproteins of the β3 subunit before cleavage and contain the signal peptides and the unglycosylated GABRB3 protein. Canine pancreatic microsomal membranes (1.8 μl) (Promega, Madison, WI) were then added directly to each reaction medium for translocation experiments. After one hour of centrifugation, pellets were separated from supernatant. The supernatant fluid of all samples did not contain GABRB3 protein (not shown). The pellets, on the other hand, contained the GABRB3 protein that had been translocated into the microsome (Figure 7—see translocated proteins represented as “P”). Translocation in the presence of 1.8 μl of canine pancreatic microsomes precipitated all beta 3 subunit protein into the pellet. This implied that all signal peptides were oriented toward the membrane and that all GABRB3 protein was translocated when incubating with 1.8 μl of canine pancreatic microsomes.
Figure 7.
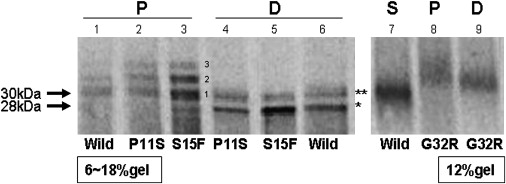
Increased Glycosylation of Mutated GABRB3 Protein
Products of in vitro translocation (P) and digestion (D) with N-glycosidase F, containing the exon 1a mutations P11S and S15F, were loaded on a 6%–18% gel (the left gel). Similar products containing the exon 2 mutation G32R were loaded on a 12% gel (the right gel). The supernatant protein (S) of only wild-type is shown (lane 7) in the 12% gel and is considered not to be translocated to microsomes and to present the 30kDa GABRB3 including the signal peptide. The 30 kDa supernatant protein therefore consists of untranslocated “exon 1a to exon 6.” Translocated proteins (P) are shown in lanes 1–3 and 8. The molecular weight of band 1 was 30 kDa. Bands 2 and 3 of both P11S and S15F mutations in in vitro translocation (P) revealed clearly higher density than wild-type, suggesting increased glycosylation. After digestion with N-glycosidase F, two smaller sized bands, 28 kDa (∗), and 30 kDa (∗∗) appeared. The 30 kDa bands represent incompletely digested protein and 28 kDa bands represent completely digested protein. The translocated protein of G32R (P, lane 8) had only bands larger than 30kDa, also suggesting increased glycosylation. The band of G32R has higher molecular weight than the supernatant protein from the wild-type even after digestion.
The pellet of the wild-type sample (Figure 7, lane 1) yielded a 30 kDa protein, which was the same size as the supernatant protein (lane 7). The pellet of the wild-type sample, similar to translocated proteins with P11S and S15F mutations, also yielded bands with molecular weights higher than 30 kDa (seen in lanes 2 and 3). However the image densities of bands 2 and 3 in wild-type samples were lower than the image densities of translocated proteins with P11S and S15F mutations. Figure 8 depicts results of three experiments in which we compared densities of bands 2 and 3 in GABRB3 containing P11S and S15F mutations versus wild-type GABRB3. Densities of bands 2 and 3 in mutated GABRB3 are clearly increased compared to bands 2 and 3 of wild-type GABRB3 (Figure 8). These larger band products represent glycosylated forms of the GABRB3 protein, and all GABRB3 proteins containing missense mutations were hyperglycosylated compared to wild-type GABRB3.
Figure 8.
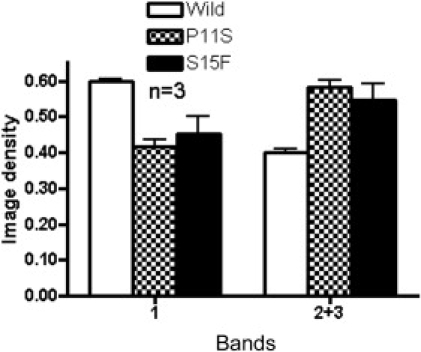
Quantitation of Density of Glycosylated Bands Comparing Wild-Type and Exon 1a Mutations Shown in Figure 7
Bands 2 and 3 are considered to be hyperglycosylated products. The image density of each band is the average of three experiments. Since bands 2 and 3 of one sample overlapped, the sum of bands 2 and 3 was compared. The proportion of the image density of band 1 and the sum of bands 2 and 3 were significantly different between the wild-type and mutations in exon 1a. (P11S, p = 0.0004, S15F, p = 0.005).
When samples are treated with N-glycosidase F, N-glycosylation chains are eliminated and all proteins are digested to the smaller molecular weight 28 kDa; “D” in Figure 7. “D” represents digested proteins. The 28 kDa size of the digested GABRB3 protein with exon 1a mutations P11S and S15F is the same molecular weight as the wild-type. It is a lower molecular weight than the supernatant proteins. However, the GABRB3 sample with exon 2 mutation G32R has a slightly higher molecular weight than do proteins in the supernatant (S), suggesting that the modification of this protein is different from that of exon 1a. This means that missense mutations in exon 1a have excess N-glycosylation and are able to be cleaved to the product of the same size as that of the wild-type. The missense mutation in exon 2 might have a different cleavage site, no cleavage, or normal cleavage with additional O-glycosylation. In silico analysis predicts normal cleavage and gain of O-glycosylation at 23Ser. This observation further suggests that the G32R mutation was subjected to more degradation than the exon 1a mutations, perhaps due to misfolding of GABRB3 protein containing the G32R mutation.
Larger Current Density from Cells Expressing Wild-Type β3-V2 Transcript than from Cells Expressing β3-v2(P11S), β3-v2(S15F), or β3-v2(G32R) Mutations
Cells were cotransfected with equivalent amounts of cDNA encoding for the α1, γ2S, and one of the β3-v2, β3-v2(P11S), β3-v2(S15F), or β3-v2(G32R) subunits. Saturating concentrations of GABA (1 mM) were applied to positively transfected cells for 4 s. Currents from α1β3-v2γ2S (wild-type β3 subunit with exon 1a) receptors and mutant α1β3-v2(P11S), α1β3-v2(S15F), and α1β3-v2(G32R) receptors all had a fast rate of rise, substantial multiphasic desensitization, and fast deactivation upon removal of GABA (Figure 9A). α1β3-v2γ2S receptors, however, had a mean current density (279.1 ± 34.0; n = 36) that was larger than the mean current densities of α1β3-v2(P11S)γ2S (118 ± 20.51; n = 18, p < 0.001), α1β3-v2(S15F)γ2S (134.1 ± 20.7; n = 25, p < 0.001), and α1β3-v2(G32R)γ2S receptors (174.7 ± 23.18; n = 41, p < 0.05) (Figure 9B). Peak-current amplitudes were reduced in each mutant condition as well (data not shown).
Figure 9.
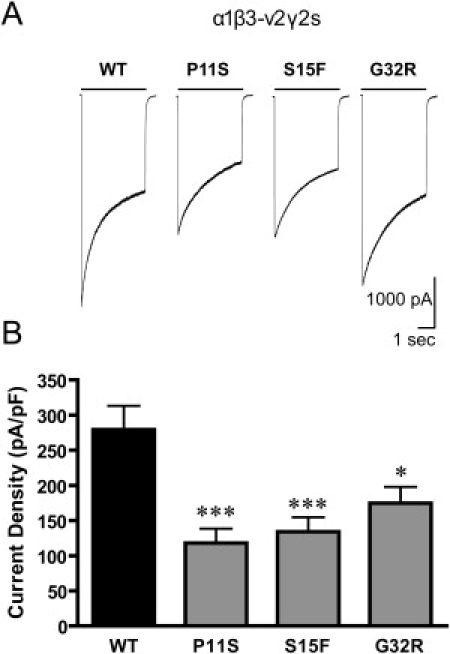
GABA-Evoked Currents of Mutations in Transfected HEK293 Cells
The current density from cells expressing the wild-type β3-v2 transcript was larger than in the cells expressing the β3-v2(PS), β3-v2(SF) or β3-v2(GR) mutations.
The current density recorded from cells expressing GABAA receptors with β3-v2 mutations associated with CAE was reduced. A. Representative traces of whole cell current elicited for 4 s with 1 mM GABA from cells expressing α1β3-v2γ2S (WT), α1β3-v2(P11S)γ2S, α1β3-v2(S15F)γ2S and α1β3-v2(G32R)γ2S receptors. B. Compared to cells expressing wild-type receptors (n = 36), the current densities of the cells expressing receptors containing the β3-v2 mutations P11S (n = 18), S15F (n = 25) and G32R (n = 41) were reduced (∗∗∗p < 0.001, ∗p < 0.05).
Discussion
We found three missense mutations of GABRB3 (Pro11Ser, Ser15Phe, Gly32Arg) in four out of 48 (8%) CAE-affected patients with American Indian and Spanish European ancestry. Two mutations (P11S and S15F) reside in the alternative signal peptide, exon 1a of the GABRB3 protein. A third mutation (G32R) is at amino acid 10 from the N terminus of the mature GABRB3 protein, which in turn is made from beta 3 isoform 2 precursor. These mutations segregated in clinically and EEG affected individuals and in asymptomatic persons belonging to two generations of these four families. Vertical transmission of the GABRB3 mutations in symptomatic and asymptomatic family members suggests a dominant trait with incomplete penetrance. We did not find the same mutations in 630 healthy ethnically and sex-matched controls.
P11S is listed as rs25409 in the SNP database of the National Center for Biotechnology Information (NCBI), where it is recognized as a minor allele in two out of 157 persons with autism. We do not know if these two persons with P11S have absence epilepsy. Thirty-five percent to sixty-five percent of patients with autism spectrum disorder have epileptiform EEG abnormalities, and 10% to 30% have seizures including absences.32–35 The same NCBI database contains the results of a HapMap study in which P11S is not found in 60 European, 44 Han Chinese, 43 Japanese, and 59 SubSaharan African persons. We did not find the P11S mutation in 630 controls, in sharp contrast to two patients with CAE (Fisher's exact test: p = 0.0049). The S15F and G32R missense mutations are both previously unreported in the NCBI databases. We did not find them in 416 controls from Mexico or the 214 controls from Honduras.
The GABAA receptor (GABAR) is a heteropentameric-membrane glycoprotein that is composed of five subunits.36,37 The first half of the polypeptide, which is translated from sequences of exon 2 to exon 7, forms a hydrophilic glycosylated extracellular domain. Several parts of the extracellular domain are important for receptor function, including GABA and allosteric modulator binding sites,36,37 as well as assembly signals.38 The last half of the polypeptide is translated from sequences of the remaining exon 7 to exon 9 and contains four hydrophobic sequences that form transmembrane domains. So far, the sites for binding GABA or allosteric modulators like benzodiazepines have been suggested to be somewhat removed from the N terminus in the extracellular domain, and binding sites for general anesthetic drugs are tentatively located in the transmembrane region.39–41 Thus, Pro11Ser and Ser15Phe mutations in the alternative signal peptide, exon 1a, and Gly32Arg mutation in exon 2 would not affect binding sites but instead influence protein maturation, topology, assembly, and subcellular localization of a GABAR.
In addition, two mutations in the signal peptide of the β3-v2 subunit, P11S and S15F, caused reductions in GABAA receptor current density when expressed as α1β3-v2(P11S)γ2S or α1β3-v2(S15F)γ2S receptors and compared to wild-type receptors (α1β3-v2γ2S). Similarly, cells expressing receptors containing a β3-v2 subunit mutation just beyond the signal peptide in exon 2, α1β3-v2(G32R)γ2S, also had smaller GABA-evoked current density than did cells expressing wild-type receptors. This is important because as a ligand-gated, chloride-selective ion channel, the function of the GABAA receptor is to provide the majority of synaptic inhibition in the central nervous system. GABAR expression and kinetic properties are determined by the subunit combination present in the receptor. Although there are numerous genes that encode for subunits and subunit subtypes of the GABAA receptor (α1-6, β1-3, γ1-3, δ, ɛ, θ, and π), the majority of the GABAA receptors in the central nervous system are composed of two α, two β, and a single γ or δ subunit.42,43 To date, several mutations have been identified in the α1, γ2, and δ subunits of the GABAA receptor associated with familial epilepsy syndromes. Although these families were classified under the generalized epilepsy with febrile seizures plus (GEFS+) (febrile and afebrile seizures) spectrum of epilepsy or juvenile myoclonic epilepsy, all had childhood absence seizures as a phenotype. These observations strongly suggest that GABAR might be the crucial pathogenic molecule for childhood absence epilepsy.44 The majority of these published GABAR mutations also cause altered subunit trafficking and, subsequently, expression of the mutated subunit, and others alter the function of the ion channel.45
Thus, several lines of evidence favor a pathogenic role for the P11S, S15F, and G32R mutations in absence seizures. First, all three mutations reside in evolutionarily conserved GC-rich regions and amino acid sequences of exon 1a and exon 2 (Figure 4A). Second, in silico analysis predicts the same cleavage sites for the wild-type as for P11S and S15F mutations in exon 1a. In silico analysis further predicts an alteration in secondary structure as a result of all mutations. More interestingly, both missense mutations in the signal peptide significantly increased N-glycosylation of the extracellular domain of the GABRB3 protein in actual experiments. G32R in the N terminus of the mature protein also increased in vitro glycosylation in actual experiments. N-glycosylation, conserved throughout evolution,46,47 is an essential modifier of protein folding and transport; maintenance of cell structure; and protein adhesion, recognition, and cell-surface trafficking.47,48 Increased glycosylation can thus affect processing and subsequent assembly of GABA receptors,49 possibly resulting in pathogenicity.50
Third, mutations in the β3-v2 subunit of the GABAA receptor in CAE show reduced α1β3-v2(P11S)γ2S, α1β3-v2(S15F)γ2S, and α1β3-v2(G32R)γ2S receptor currents.
Fourth, a deletion mutation in β3 subunit of the GABAA receptor is present in Angelman Syndrome, in which absence-like epilepsy is present.18
The fifth line of evidence supporting a pathogenic role for the P11S, S15F, and G32R mutations in absence seizures concerns GABRB3 homozygous null mice that have absence-like episodes. GABRB3 heterozygous null mice likewise show frequent absence-like arrests of movement with simultaneous theta bursts, suggesting an insufficient inhibition in the thalamocortical network.19,20,51 In GABRB3-deficient mice, ethosuximide stops seizures and CBZ aggravates seizures,52 the same pharmacological characteristics as those seen in human absence seizures. GABRB3, therefore, plays an important role in the thalamocortical network, which underlies absence seizures.51,53 Voltage-clamp recordings of reticular neurons and ventrobasal neurons of thalamic slices in GABRB3 homozygous null mice show nearly abolished GABA-mediated inhibition in the reticular nucleus. GABA-mediated inhibition was unaffected in ventrobasal relay neurons. Oscillatory synchrony dramatically increases, showing that the recurrent inhibitory connections in the reticular nucleus, which are lost in the GABRB3 null mice, actually result in desynchronization.54
Why do some childhood absences remit, such as those in families M120, H12, and HMO10? The two mutations (P11S and S15F) of these three families reside in exon 1a, located 543 base pairs upstream of exon 1. Exon 1a is richly expressed in whole fetal human brain, including the thalamus,31 whereas the adult brain contains a smaller amount of exon 1a. GABRB3 protein is highly expressed in almost all brain regions at birth and stays constant in all regions except the thalamus.55–58 After birth, the GABRB3 protein decreases rapidly in most thalamic nuclei but remains abundant in the reticular thalamic nucleus, where it is one of the main components of GABAR.57,59 However, the alternative signal peptide coded by exon 1a is eliminated developmentally in some areas like the thalamus. Perhaps as exon 1a is eliminated developmentally, so are the absence seizures that correlate with mutations in exon 1a.
We only investigated 7 kb (the sum of the upper region from exon 1a, the full region from exon 1a to exon 3, all coding region from exon 4 to exon 9 and the part of intron 3 and 3′ UTR) out of 230 kb for the full GABRB3 gene, and more mutations could be present in the other residues of GABRB3. Recently, the importance of epigenetic regulatory mechanisms for the expression of GABRB3 has been emphasized,60 in which a heritable change of GABRB3-gene expression could occur without a change in DNA sequence but with a change of DNA methylation on the CpG region. The mutations in GABRB3 described here are associated with a gain in glycosylation of the β3 subunit protein. Glycosylation of the β3 subunit protein is known to change its maturation and alter overall GABAR trafficking to the cell surface from the endoplasmic reticulum. We suggest that the resulting hyperglycosylation and reduced current densities of the mutated β3 subunit protein leads to absence seizures. Our results also allow us to hypothesize that mutated exon 1a leads to an abnormal isoform 2 precursor of GABRB3 polipeptide during development. This in turn might explain the decrease or disappearance of absence seizures in adolescents and adults.
Supplemental Data
One figure is available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
EMBL-EBI tools EMBOSS CpGPlot/CpGReport/Isochore, http://www.ebi.ac.uk/emboss/cpgplot/
GenBank, http://www.ncbi.nih.gov/Genbank/
GOR IV secondary structure prediction method, http://npsa-pbil.ibcp.fr/cgi/bin/npsa_automat.pl?page=/NPSA/npsa_seccons.html
NCBI website, http://www.ncbi.nlm.nih.gov/
NetNGlyc 1.0, www.cbs.dtu.dk/services/NetNGlyc/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Polyphen program, http://genetics.bwh.harvard.edu/pph/
Primer 3 program, http://frodo.wi.mit.edu/
SignalP 3.0, http://cbs.dtu.dk/services/SignalP
SOSUI, http://sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html
TMHMM, version 2.0, http://www.cbs.dtu.dk/services/TMHMM-2.0/
UCSC Genome Browser, http://genome.ucsc.edu/
YinOYang, www.cbs.dtu.dk/services/YinOYang
Accession Numbers
The S15F and G32R missense mutations reported in this paper have been deposited in the NCBI databases under accession numbers NCBI_ss99307474 and NCBI_ss99307476, respectively.
Acknowledgments
We thank all CAE patients and their families for their cooperation. We also thank R. Morita and H. Kim for their initial guidance, the Olsen laboratory members for their technical help, H. Shike for her advice, and Y. Ishikawa-Brush for her helpful information on bioinformatics to M.T. EEG technicians for their video-EEG monitoring studies, GENESS site neurologists and staff, UCLA sequencing core members for all their assistance for this study. This study were supported by (1) NIH grant NS35985 (to R.W.O.), (2) the Epilepsy Center of Excellence, Neurology and Research Services, VA Greater Los Angeles Healthcare System, West Los Angeles and a Veterans Administration Merit Review Grant (to A.V.D.E.), and (3) CONACYT grant 57919 in Mexico (to M.E.A.).
References
- 1.Fong G.C.Y., Shah P.U., Gee M.N., Serratosa J.M., Castroviejo I.P., Khan S., Ravat S.H., Mani J., Medina M.T., Delgado-Escueta A.V. Childhood absence epilepsy with tonic-clonic seizures and 3–4-Hz spike and multispike-slow wave complexes: linkage to chromosome 8q24. Am. J. Hum. Genet. 1998;63:1117–1129. doi: 10.1086/302066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace R.H., Marini C., Petrou S., Harkin L.A., Owser D.N., Panchal R.G., Williams D.A., Sutherland G.R., Mulley J.C., Berkovic S.F. Mutant GABA(A)receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- 3.Haug K., Warnstedt M., Alekov A.K., Sander T., Ramírez A., Poser B., Maljevic S., Hebeisen S., Kubisch C., Heils A. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat. Genet. 2003;33:527–532. doi: 10.1038/ng1121. [DOI] [PubMed] [Google Scholar]
- 4.Maljevic S., Krampfl K., Cobilanschi J., Tilgen N., Beyer S., Weber Y.G., Schlesinger F., Ursu D., Melzer W., Cossette P. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann. Neurol. 2006;59:983–987. doi: 10.1002/ana.20874. [DOI] [PubMed] [Google Scholar]
- 5.Callenbach P.M., Geerts A.T., Arts W.F., van Donselaar C.A., Peters A.C., Stroink H., Brouwer O.F. Familial occurrence of epilepsy in children with newly diagnosed multiple seizures: Dutch Study of Epilepsy in Childhood. Epilepsia. 1998;39:331–336. doi: 10.1111/j.1528-1157.1998.tb01382.x. [DOI] [PubMed] [Google Scholar]
- 6.Berg A.T., Shinnar S., Levy S.R., Testa F.M., Smith-Rapaport S., Beckerman B. How well can epilepsy syndromes be identified at diagnosis? A reassessment 2 years after initial diagnosis. Epilepsia. 2000;41:1269–1275. doi: 10.1111/j.1528-1157.2000.tb04604.x. [DOI] [PubMed] [Google Scholar]
- 7.Gibbs F.A., Davis H., Lennox W.G. The EEG in epilepsy and in conditions of impaired consciousness. Arch. Neurol. Psychiatry. 1935;34:1133–1148. [Google Scholar]
- 8.Currier R.D., Kooi K.A., Saidman L.J. Prognosis of “Pure” Petit Mal, a Follow-up Study. Neurology. 1963;13:959–967. doi: 10.1212/wnl.13.11.959. [DOI] [PubMed] [Google Scholar]
- 9.Hertoft P. The clinical, electroencephalographic and social prognosis in petit mal epilepsy. Epilepsia. 1963;74:298–314. doi: 10.1111/j.1528-1157.1963.tb05227.x. [DOI] [PubMed] [Google Scholar]
- 10.Gibberd F.B. The prognosis of petit mal. Brain. 1966;89:531–538. doi: 10.1093/brain/89.3.531. [DOI] [PubMed] [Google Scholar]
- 11.Loiseau P., Duche B., Pedespan J.M. Absence epilepsies. Epilepsia. 1995;36:1182–1186. doi: 10.1111/j.1528-1157.1995.tb01060.x. [DOI] [PubMed] [Google Scholar]
- 12.Lennox W.G., Lennox M.A., editors. The Genetics of Epilepsy. Epilepsy and Related Disorders. Volume 1. Little, Brown; Boston: 1960. [Google Scholar]
- 13.Metrakos J.D., Metrakos K. Childhood epilepsy of subcortical (“centrencephalic”) origin. Some questions and answers for the pediatrician. Clin. Pediatr. (Phila.) 1966;5:536–542. [PubMed] [Google Scholar]
- 14.Metrakos K., Metrakos J.D. Genetics of convulsive disorders. II. Genetic and electroencephalographic studies in centrencephalic epilepsy. Neurology. 1961;11:474–483. doi: 10.1212/wnl.11.6.474. [DOI] [PubMed] [Google Scholar]
- 15.Metrakos K., Metrakos J.D. Is the centrencephalic EEG inherited as a dominant? Electroencephalogr. Clin. Neurophysiol. 1961;13:289. doi: 10.1016/0013-4694(61)90146-8. [DOI] [PubMed] [Google Scholar]
- 16.Feucht M., Fuchs K., Pichlbauer E., Hornik K., Scharfetter J., Goessler R., Fureder T., Cvetkovic N., Sieghart W., Aschauer H. Possible association between childhood absence epilepsy and the gene encoding GABRB3. Biol. Psychiatry. 1999;46:997–1002. doi: 10.1016/s0006-3223(99)00039-6. [DOI] [PubMed] [Google Scholar]
- 17.Urak L., Feucht M., Fathi N., Hornik K., Fuchs K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum. Mol. Genet. 2006;15:2533–2541. doi: 10.1093/hmg/ddl174. [DOI] [PubMed] [Google Scholar]
- 18.Minassian B.A., DeLorey T.M., Olsen R.W., Philippart M., Bronstein Y., Zhang Q., Guerrini R., Van Ness P., Livet M.O., Delgado-Escueta A.V. Angelman syndrome: correlations between epilepsy phenotypes and genotypes. Ann. Neurol. 1998;43:485–493. doi: 10.1002/ana.410430412. [DOI] [PubMed] [Google Scholar]
- 19.DeLorey T.M., Handforth A., Anagnostaras S.G., Homanics G.E., Minassian B.A., Asatourian A., Fanselow M.S., Delgado-Escueta A., Ellison G.D., Olsen R.W. Mice lacking the beta3 subunit of the GABA(A) receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J. Neurosci. 1998;18:8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liljelund P., Handforth A., Homanics G.E., Olsen R.W. GABA(A) receptor beta3 subunit gene-deficient heterozygous mice show parent-of-origin and gender-related differences in beta3 subunit levels, EEG, and behavior. Brain Res. Dev. Brain Res. 2005;157:150–161. doi: 10.1016/j.devbrainres.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 21.ILAE Commission on Classification and Terminology of the International League Against Epilepsy, Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- 22.Ott J. Computer-simulation methods in human linkage analysis. Proc. Natl. Acad. Sci. USA. 1989;86:4175–4178. doi: 10.1073/pnas.86.11.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weeks D.E., Ott J., Lathrop G.M. SLINK: a general simulation program for linkage analysis. Am. J. Hum. Genet. 1990;47:A204. [Google Scholar]
- 24.Glatt K., Sinnett D., Lalande M. The human gamma-aminobutyric acid receptor subunit beta 3 and alpha 5 gene cluster in chromosome 15q11-q13 is rich in highly polymorphic (CA)n repeats. Genomics. 1994;19:157–160. doi: 10.1006/geno.1994.1027. [DOI] [PubMed] [Google Scholar]
- 25.Glatt K., Glatt H., Lalande M. Structure and organization of GABRB3 and GABRA5. Genomics. 1997;41:63–69. doi: 10.1006/geno.1997.4639. [DOI] [PubMed] [Google Scholar]
- 26.Liu W., Smith D.I., Rechtzigel K.J., Thibodeau S.N., James C.D. Denaturing high performance liquid chromatography (DHPLC) used in the detection of germline and somatic mutations. Nucleic Acids Res. 1998;26:1396–1400. doi: 10.1093/nar/26.6.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morita R., Miyazaki E., Shah P.U., Castroviejo I.P., Delgado-Escueta A.V., Yamakawa K. Exclusion of the JRK/JH8 gene as a candidate for human childhood absence epilepsy mapped on 8q24. Epilepsy Res. 1999;37:151–158. doi: 10.1016/s0920-1211(99)00061-3. [DOI] [PubMed] [Google Scholar]
- 28.Miyawaki A., Furuichi T., Maeda N., Mikoshiba K. Expressed cerebellar- type inositol 1,4,5-trisphosphate receptor, P400, has calcium release activity in a fibroblast L cell line. Neuron. 1990;5:11–18. doi: 10.1016/0896-6273(90)90029-f. [DOI] [PubMed] [Google Scholar]
- 29.Ganesh S., Agarwala K.L., Ueda K., Akagi T., Shoda K., Usui T., Hashikawa T., Osada H., Delgado-Escueta A.V., Yamakawa K. Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 2000;9:2251–2261. doi: 10.1093/oxfordjournals.hmg.a018916. [DOI] [PubMed] [Google Scholar]
- 30.Greenfield L.J., Sun F., Neelands T.R., Burgard E.C., Donnelly J.L., Macdonald R.L. Expression of functional GABAA receptors in transfected L 929 cells isolated by immunomagnetic bead separation. Neuropharmacology. 1997;36:63–73. doi: 10.1016/s0028-3908(96)00150-5. [DOI] [PubMed] [Google Scholar]
- 31.Kirkness E.F., Fraser C.M. A strong promoter element is located between alternative exons of a gene encoding the human gamma-aminobutyric acid-type A receptor beta 3 subunit (GABRB3) J. Biol. Chem. 1993;268:4420–4428. [PubMed] [Google Scholar]
- 32.Minshew N.J. Indices of neural function in autism: clinical and biologic implications. Pediatrics. 1991;87:774–780. [PubMed] [Google Scholar]
- 33.Rapin I. Autistic children: diagnosis and clinical features. Pediatrics. 1991;87:751–760. [PubMed] [Google Scholar]
- 34.Gabis L., Pomeroy J., Andriola M.R. Autism and epilepsy: cause, consequence, comorbidity, or coincidence? Epilepsy Behav. 2005;7:652–656. doi: 10.1016/j.yebeh.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Childs J.A., Blair J.L. Valproic acid treatment of epilepsy in autistic twins. J. Neurosci. Nurs. 1997;29:244–248. doi: 10.1097/01376517-199708000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Olsen R.W., Tobin A.J. Molecular biology of GABA(A) receptors. FASEB J. 1990;4:1469–1480. doi: 10.1096/fasebj.4.5.2155149. [DOI] [PubMed] [Google Scholar]
- 37.DeLorey T.M., Olsen R.W. Gamma-aminobutyric acidA receptor structure and function. J. Biol. Chem. 1992;267:16747–16750. [PubMed] [Google Scholar]
- 38.Taylor P.M., Thomas P., Gorrie G.H., Connolly C.N., Smart T.G., Moss S.J. Identification of amino acid residues within GABA(A) receptor beta subunits that mediate both homomeric and heteromeric receptor expression. J. Neurosci. 1999;19:6360–6371. doi: 10.1523/JNEUROSCI.19-15-06360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith G.B., Olsen R.W. Functional domains of GABA(A) receptors. Trends Pharmacol. Sci. 1995;16:162–168. doi: 10.1016/s0165-6147(00)89009-4. [DOI] [PubMed] [Google Scholar]
- 40.Mihic S.J., Ye Q., Wick M.J., Koltchine V.V., Krasowski M.D., Finn S.E., Mascia M.P., Valenzuela C.F., Hanson K.K., Harrison N.L. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- 41.Li G.D., Chiara D.C., Sawyer G.W., Husain S.S., Olsen R.W., Cohen J.B. Identification of a GABA(A) receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korpi E.R., Grunder G., Luddens H. Drug interactions at GABA(A) receptors. Prog. Neurobiol. 2002;67:113–159. doi: 10.1016/s0301-0082(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 43.McKernan R.M., Whiting P.J. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 44.Audenaert D., Van Broeckhoven C., De Jonghe P. Genes and loci involved in febrile seizures and related epilepsy syndromes. Hum. Mutat. 2006;27:391–401. doi: 10.1002/humu.20279. [DOI] [PubMed] [Google Scholar]
- 45.Macdonald R.L., Gallagher M.J., Feng H.J., Kang J. GABA(A) receptor epilepsy mutations. Biochem. Pharmacol. 2004;68:1497–1506. doi: 10.1016/j.bcp.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 46.Burda P., Aebi M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta. 1999;1426:239–257. doi: 10.1016/s0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- 47.Conde R., Cueva R., Pablo G., Polaina J., Larriba G. A search for hyperglycosylation signals in yeast glycoproteins. J. Biol. Chem. 2004;279:43789–43798. doi: 10.1074/jbc.M406678200. [DOI] [PubMed] [Google Scholar]
- 48.Scheiffele P., Peränen J., Simons K. N-glycans as apical sorting signals in epithelial cells. Nature. 1995;378:96–98. doi: 10.1038/378096a0. [DOI] [PubMed] [Google Scholar]
- 49.Buller A.L., Hastings G.A., Kirkness E.F., Fraser C.M. Site-directed mutagenesis of N-linked glycosylation sites on the gamma-aminobutyric acid type A receptor alpha 1 subunit. Mol. Pharmacol. 1994;46:858–865. [PubMed] [Google Scholar]
- 50.Vogt G., Chapgier A., Yang K., Chuzhanova N., Feinberg J., Fieschi C., Boisson-Dupuis S. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat. Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 51.Crunelli V., Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat. Rev. Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- 52.Handforth A., Deloreym T.M., Homanics G.E., Olsen R.W. Pharmacologic evidence for abnormal thalamocortical functioning in GABA(A) receptor beta3 subunit- deficient mice, a model of Angelman syndrome. Epilepsia. 2005;46:1860–1870. doi: 10.1111/j.1528-1167.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 53.Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–324. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 54.Huntsman M.M., Porcello D.M., Homanics G.E., DeLorey T.M., Huguenard J.R. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- 55.Laurie D.J., Seeburg P.H., Wisden W. The distribution of 13 GABA(A) receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J. Neurosci. 1992;12:1063–1076. doi: 10.1523/JNEUROSCI.12-03-01063.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laurie D.J., Wisden W., Seeburg P.H. The distribution of thirteen GABA(A) receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J.H., Sato M., Tohyama M. Different postnatal ontogenic profiles of neurons containing beta (beta 1, beta 2 and beta 3) subunit mRNAs of GABA(A) receptor in the rat thalamus. Brain Res. Dev. Brain Res. 1991;58:289–292. doi: 10.1016/0165-3806(91)90017-d. [DOI] [PubMed] [Google Scholar]
- 58.Zhang J.H., Sato M., Tohyama M. Different postnatal development profiles of neurons containing distinct GABA(A) receptor beta subunit mRNAs (beta 1, beta 2, and beta 3) in the rat forebrain. J. Comp. Neurol. 1991;308:586–613. doi: 10.1002/cne.903080407. [DOI] [PubMed] [Google Scholar]
- 59.Pirker S., Schwarzer C., Wieselthaler A., Sieghart W., Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- 60.Hogart A., Nagarajan R.P., Patzel K.A., Yasui D.H., Lasalle J.M. 15q11–13 GABA(A) receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 2007;16:691–703. doi: 10.1093/hmg/ddm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.