

Abstract
The mutation 3243A→G is the most common heteroplasmic pathogenic mitochondrial DNA (mtDNA) mutation in humans, but it is not understood why the proportion of this mution decreases in blood during life. Changing levels of mtDNA heteroplasmy are fundamentally related to the pathophysiology of the mitochondrial disease and correlate with clinical progression. To understand this process, we simulated the segregation of mtDNA in hematopoietic stem cells and leukocyte precursors. Our observations show that the percentage of mutant mtDNA in blood decreases exponentially over time. This is consistent with the existence of a selective process acting at the stem cell level and explains why the level of mutant mtDNA in blood is almost invariably lower than in nondividing (postmitotic) tissues such as skeletal muscle. By using this approach, we derived a formula from human data to correct for the change in heteroplasmy over time. A comparison of age-corrected blood heteroplasmy levels with skeletal muscle, an embryologically distinct postmitotic tissue, provides independent confirmation of the model. These findings indicate that selection against pathogenic mtDNA mutations occurs in a stem cell population.
Introduction
Individuals with pathogenic mutations in their mitochondrial DNA (mtDNA) typically have a mixture of wild-type and mutant mtDNA in each cell, a condition called heteroplasmy.1 A heteroplasmic cell can compensate for the presence of mutant mtDNA until a certain threshold in the mutation level is reached, beyond which cell function is compromised.2 The level of mtDNA heteroplasmy can vary across the tissues in an individual3–6 and can even vary across the cells within a single tissue.7–9 For the most common heteroplasmic pathogenic mutation, 3243A→G (MIM #590050.0001),10,11 the general pattern to the tissue variation is that the level of this pathogenic mtDNA mutation is lower in dividing cell types, and cells recently derived from dividing cells, than it is in long-lived postmitotic cells in the same individual. Specifically, mtDNA 3243A→G mutation levels in blood samples are almost always lower than the mutation level determined from a muscle biopsy.3,4 The tissue variation and the variation with time of mtDNA heteroplasmy are dependent on the particular mutation. Most of the available clinical data is for the 3243A→G mutation heteroplasmy, and we focus primarily on this mutation in this analysis. Longitudinal clinical studies have shown that 3243A→G blood heteroplasmy levels tend to slowly decrease over time.12–14 This is generally thought to occur through selection at the cellular level, but it is not clear whether this occurs in the stem cell population or the proliferating and differentiating blood cell precursors. The pathogenesis of diseases caused by mtDNA mutations is driven by the loss of function of a cell type or the actual loss of the cells themselves. The specific phenotype depends on the specific cell type most strongly affected in that subject, due to intrinsic variability in the heteroplasmy levels across tissues, sensitivity of the cell (and the subject) to cofactors, or high energy requirements of the cell type. Understanding the long-time-scale dynamics of mtDNA heteroplasmy in different cell populations in the body is a basic step in understanding the pathology and the progression of these diseases.
In this paper, we analyze clinical data from two sets of patients: those with the 3243A→G mutation and those with the 8344A→G mutation (MIM #590060.0001). The 3243A→G mutation occurs in one of the mtDNA leucine transfer RNA (tRNA) genes (MTTL1). Although this mutation was first described15 in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS [MIM 540000]), the spectrum of phenotypes caused by this mutation is extremely broad, extending from diabetes and deafness to hypertrophic cardiomyopathy and retinitis pigmentosa. This high phenotypic variability might in part be due to tissue-specific differences within each individual in the heteroplasmy level of the mutation. The 8344A→G mutation occurs in the mtDNA lysine tRNA gene (MTTK). This mutation is associated with Myoclonic Epilepsy with Ragged-Red Fibers Syndrome (MERRF [MIM #545000]) and can also cause multiple lipomata, cardiomyopathy, optic atrophy, myopathy, ataxia, and dementia. The 8344A→G mutation is also one of many mutations associated with Leigh Syndrome (MIM #256000), a relapsing encephalopathy that usually presents in childhood and is characterized by lesions in the basal ganglia.
Although the presence of a mtDNA mutation in blood rarely causes a clinical problem, understanding why heteroplasmy changes in blood has important implications. First, this tissue type can be affected by mtDNA mutations because large-scale deletions of mtDNA can lead to pancytopenia due to bone marrow aplasia (Pearson syndrome [MIM #557000]16). Second, the decreasing level of mutant mtDNA in blood presents a diagnostic challenge because the mutation might not be detectable in blood samples from affected individuals with standard techniques.17 Third, changing heteroplasmy levels with time introduces a major confounding variable when one is studying the transmission of mtDNA heteroplasmy, compromising studies of the mtDNA genetic bottleneck in humans18 and limiting the development of genetic-counseling guidelines. Finally, understanding the mechanisms behind changing heteroplasmy levels in blood is likely to have broader implications for other stem cell populations and the role of age-related diseases.19
A few serial measurements of 3243A→G heteroplasmy have been made in blood samples from the same patient at different time points. These studies reported a decrease in the proportion of mutant mtDNA by approximately 0.5% to 2% per year,12 on the basis of pairs of measurements separated by several years. With this limited information, only a single value for the rate of decrease of mtDNA blood heteroplasmy can be calculated for each subject. It has not been known whether the variability in the measured rate of heteroplasmy decrease is due to variation across subjects or variation in the rate over time. However, on the basis of the simulation results reported here, we have reanalyzed published data to show that the rate of decrease in 3243A→G blood heteroplasmy is not constant, but is instead exponentially decreasing with time in a consistent manner across many subjects.
We have separated the problem of changes in blood heteroplasmy level into two questions. First, how does the heteroplasmy level in the hematopoietic stem cell population change over time? Second, how well does the heteroplasmy level in peripheral blood represent the heteroplasmy levels in the hematopoietic stem cell population? We have developed two separate simulations to address these two questions.
Material and Methods
Clinical Data
The clinical data were identified from a systematic search of the literature. Papers were identified that contained both the heteroplasmy level for an mtDNA point mutation measured in blood and the subject's age at sample collection. The focus was on identifying papers reporting data for multiple subjects. We concentrated on the 3243A→G and 8344A→G mutations because of the relatively large amount of data available for these two mutations. A total of 275 unique data pairs were identified for 3243A→G,3,4,12–14,20–37 and 48 unique data pairs were identified for 8344A→G.38–42 For the 3243A→G mutation, a subset of these studies was identified that also reported mtDNA heteroplasmy in muscle tissue.3,20–22,24,29,30,35,36 In cases where the data were only given in graphical form, the approximate numerical values were determined from these published graphs with the software Engauge Digitizer. For the analysis of changes in blood heteroplasmy levels over long time scales, one study was identified that had two measurements of 3243A→G in blood separated by 9 to 19 years, for six subjects.12 To extend this dataset, our collaborators gathered this data for a further 11 subjects.14
Simulations
We used two related simulations: a stem cell model (Figure 1A) and a model of the rapid expansion of blood progenitor cells (Figure 1B). The simulation parameters are summarized in Table 1. In the stem cell model, we simulated populations of 20,000 to 2,000,000 stem cells with up to approximately 1000 to 2000 mtDNA molecules per cell for a maximum duration of 100 years. The actual number of mtDNA per hematopoietic stem cell is unknown, so we used a range of values and used the simulation results to determine the behavior for an arbitrary mtDNA copy number per cell. For simplicity, the initial heteroplasmy level in all stem cells was set to a uniform value, P0. In the progenitor cell model, we began with a single cell and modeled a series of approximately 20 cell divisions43,44 to form the mature blood cells.
Figure 1.
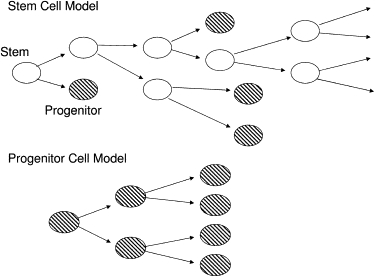
Schematic Diagram of the Two Cell-Division Models
Open ovals represent stem cells. Shaded ovals represent progenitor cells.
Table 1.
Simulation Parameter Values
| Parameter | Meaning | Values |
|---|---|---|
| D | Mean time between cell divisions | 1 year (stem cell model) or 1 day (progenitor model) |
| Thalf | mtDNA half-life | 10 to 20 days |
| mthresh | Cells with mutation level greater than mthresh were removed from the simulation. | 60% to 100% |
| Nt | Target number of mtDNA molecules in simulated cell (used to set replication rate). Also used to set the initial copy number in the cell. | 1000 to 2000 |
| P0 | Initial mutation level in cell | 0% to 100% |
| Δt | Simulation time step | 1 hr |
| No symbol | Total simulation duration | 100 years (stem cell model) or 20 to 25 days (progenitor model) |
| No symbol | Total number of cells | 20,000 to 2,000,000 (stem cell model) or 1 to 1,000,000 (progenitor model) |
Both simulation models contained many common features. Each mtDNA molecule in the simulation can be of either a wild-type mtDNA or a mutant-type mtDNA. We only considered point substitution mutations in this study because the dynamics of deletions might be different. Each simulated cell had integer values W and M representing the number of wild-type and mutant mtDNA in that cell. These numbers changed over time because of the processes of mtDNA replication (Figure 2A), degradation (Figure 2B), and cell division45,46 (Figure 2C). Note that although the total number of mtDNA molecules in the simulated cell is N = W + M, the total number N was not held constant over time and varied as W and M varied independently. The variables W and M were the basic variables of this model.
Figure 2.
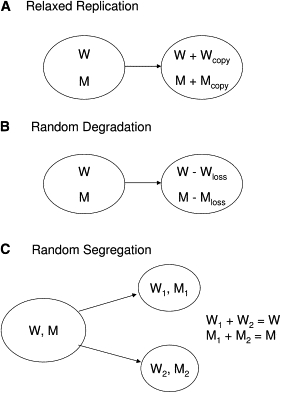
Schematic Diagrams of the mtDNA Dynamics in the Models
(A) In relaxed replication, the number of wild-type (W) and mutant (M) mtDNA molecules was increased over one time step by the amounts Wcopy and Mcopy, respectively.
(B) In the random-degradation model, the numbers W and M were decreased over one time step by Wloss and Mloss.
(C) At a cell-division event, the values for W and M were randomly distributed to the two daughter cells.
Cell Division
At each cell division, we assumed that the mtDNA molecules in the original cell were distributed randomly to each daughter cell with equal probability (Figure 2C). We modeled this with a Poisson distribution separately for the values W and M, with a mean value of W/2 and M/2 molecules distributed to each daughter cell. The sum of the mtDNA copy numbers in the two daughter cells equaled the copy number in the original cell. The timing of the cell division was stochastic, with mean value D for the time between cell divisions. For the hematopoietic stem cell model, we set D = 1 year,47 and for the blood progenitor model, we set D = 1 day.44 In the stem cell model (Figure 1A), the two daughter cells were randomly assigned identities as stem cells or progenitor cells, with equal probability48,49 (because we were modeling the consequences of stem cell division, we did not incorporate the complicated feedback mechanisms controlling the stem cell self-renewal50 because these detailed mechanisms were not considered relevant to this problem). The daughter cells labeled as “stem cells” were followed through subsequent cell divisions, and the daughter cells labeled “progenitor cells” were removed from this simulation, which was designed to only model the hematopoietic stem cell population. In the blood progenitor model (Figure 1B) all daughter cells were labeled as progenitor cells, and were retained in that simulation.
Degradation
Mitochondrial DNA molecules were removed from the simulation (Figure 2B) with a half-life, Thalf, which we typically set in the range 10 to 20 days.51 Both wild-type and mutant mtDNA were degraded with the same half-life. In one time step of length Δt, the number of wild-type mtDNA molecules lost to random degradation (Wloss) was calculated from a Poisson distribution with mean value of (ln(2) W Δt/Thalf), where W was the number of wild-type mtDNA in the cell. A similar calculation determined the number of mutant mtDNA molecules degraded.
Mitochondrial DNA Replication
The simulated cells copied their mtDNA (Figure 2A) at a rate R to compensate for the loss of mtDNA by degradation and to repopulate the cell with mtDNA after cell divisions. Let Nt be the target total number of mtDNA in the cell. Note that Nt was merely a parameter used for the calculation of the mtDNA replication rate and that it might be slightly different than the total number of mtDNA molecules actually present in the simulation at a given time, N = W + M. In steady state, the actual copy number N fluctuated about the target number Nt. The mtDNA replication rate R was modeled as a sum of two terms:
| (1) |
The first term was the replication needed to replace degraded mtDNA. The second term was the replication rate needed to compensate for the decrease in mtDNA copy number from cell divisions. This replication rate was distributed proportionally over the mutant and wild-type mtDNA in the cell. The total number of wild-type mtDNA copied (Wcopy) and mutant mtDNA copied (Mcopy) in one time step of length Δt was R Δt = Wcopy + Mcopy (Figure 2A). The separation into Wcopy and Mcopy was made through a binomial probability distribution.
Cell Loss
For the modeling of the pathogenic effect of the mutant mtDNA, any cell with a mutation level above a set threshold mthresh was removed from the simulation. Values in the range 60% < mthresh < 100% were used in the simulation. This range covers the range of reported values for the threshold of 3243A→G.52,53 This model of cell loss was used in both the hematopoietic stem cell simulation and in the blood precursor simulation.
Measurements of the Rate of Heteroplasmy Decrease
To remove the effect of an initial transient (the shoulder in the initial short period of simulation in Figure 3A) on the measurement of the heteroplasmy decay rate, we only used simulation data after the mutation level had dropped to below 90% of its initial value. Because the average mutation level in the simulation decreased over time, we only used the mutation-level values down to 10% mutant, to remove the effects of noise due to discreteness at low levels of mutant mtDNA copy number per cell.
Figure 3.
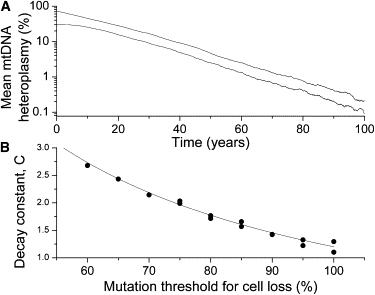
Simulation Results for the Blood Stem Cell Model
(A) An exponential decrease in mean mutation level in blood stem cell model. Results are shown for two simulations with different initial mutation levels, 70% and 30%. Other simulation parameters were Thalf = 10 days, N = 1000, and mthresh = 90%.
(B) Dependence of the decay constant C, defined in Equation 2, on the mutation threshold for loss of the stem cell, mthresh. The line is a fit to the data with Equation 3 (with R2 = 0.968 and p < 0.0001).
Results
Hematopoietic Stem Cell Simulation Results
The processes of relaxed replication of mtDNA,54 degradation of mtDNA, and random segregation between daughter cells all caused the heteroplasmy levels in this population of stem cells to spread out. As the distribution of heteroplasmy widened through random genetic drift,55,56 some cells developed a high mutation heteroplasmy level and were removed from the simulation, modeling the pathogenic effect of the mtDNA mutation. Over time, this loss of cells with high mtDNA mutation level gradually decreased the average mutation level of the simulated hematopoietic stem cell population. Decreasing heteroplasmy levels in blood have been reported for the 3243A→G mutation,12–14 based on two measurements spaced over a few years. These measurements are usually interpreted as a simple linear rate of decrease in the mutant mtDNA levels in blood samples. Our simulation results suggest that the rate of loss of heteroplasmy from blood stem cells is actually exponential (Figure 3A), not a constant linear rate.
In the model, the average mutation level as a function of time was described by a decaying exponential, after an initial transient period (Figure 3A). By varying the model parameters [mtDNA half-life Thalf and initial copy number per cell Nt = W(0) + M(0)], we found that the average mutation level m(t) as a function of time, t, had the functional form
| (2) |
where m0 was a constant approximately equal to the initial mutation level and C was a dimensionless constant, with the value of C = 1.45 ± 0.07 determined from the simulations (Table 2).
Table 2.
The Value of the Exponential-Decay Constant C in Equation 2 Computed for Various Simulation Parameters
| Number of Stem Cells | mtDNA Copy Number, Nt | mtDNA Half-Life, Thalf (Days) | Initial Mutation Level, m (%) | C |
|---|---|---|---|---|
| 20,000 | 1,000 | 10 | 30 | 1.4251 |
| 20,000 | 1,000 | 10 | 70 | 1.4212 |
| 20,000 | 1,000 | 20 | 30 | 1.5234 |
| 20,000 | 1,000 | 20 | 70 | 1.5459 |
| 20,000 | 2,000 | 10 | 30 | 1.3870 |
| 20,000 | 2,000 | 10 | 70 | 1.3662 |
| 20,000 | 2,000 | 20 | 30 | 1.5530 |
| 20,000 | 2,000 | 20 | 70 | 1.4423 |
| 2,000,000 | 1,000 | 10 | 30 | 1.4075 |
The parameters Nt, Thalf, and the initial heteroplasmy levels were varied. The mean decay constant in these simulations was C = 1.45, with standard deviation = 0.07.
Sets of simulations where the mutation-level threshold for cell removal, mthresh, was varied showed that the decay constant C is a function of the mutation threshold (Figure 3B), with lower thresholds causing faster loss of the mutation from the stem cell population, an intuitively reasonable behavior. In the range 60% ≤ mthresh ≤ 100%, this dependence was well described by function
| (3) |
where the constant values determined by a linear fit were C0 = −1.1 ± 0.1 and A = (230 ± 8)%. Even at a threshold of 100% (Figure 3B), requiring the simulated cell to fix on the mutation before the cell is removed, the simulation still showed an exponential decrease in heteroplasmy level in the hematopoietic stem cell population.
Blood Precursor Simulation Results
The hematopoietic stem cell simulation modeled the changes in the population of blood stem cells over time. To relate this to measurements taken from samples of peripheral blood, we needed to know how mtDNA heteroplasmy levels can change through the process of forming the mature blood cells from the stem cell population. We modeled this very simply as a series of rapid cell divisions with a mean cell division period of 1 day.44 For the purpose of this model, we ignored all of the complications of the different types of blood cells, and we focused just on the basic question of how a rapid expansion in the cell population could affect the mean mtDNA heteroplasmy level in these cells. Figure 4 shows a set of ten typical simulation results. Figure 4A shows the increase in the cell number as a function of time. The variation in this plot across repeated simulations shows the sensitivity of the total cell number to the stochastic timing of the first few cell divisions. This variation is not critical for the simulation results on mtDNA heteroplasmy levels. The primary result is in Figure 4B, where the mean mutant mtDNA level, m(t), is shown. Throughout this expansion from a single initial cell, the mean mutant mtDNA level remained relatively constant, varying only a few percent. In this model of an exponentially expanding population of cells, there was no consistent shift in heteroplasmy level, as there was in the stem cell model, even though both simulations included the threshold mechanism for removing cells with high mutation heteroplasmy. Our conclusion is that the mean mtDNA mutation level measured in peripheral blood is a good measure of the mean mutation level in the stem cell population from which those cells were derived. Moreover, the lack of any significant effect of selection in the differentiating (non-stem cell) blood precursors (Figure 4B) indicates that the selective mechanism must be acting at the stem cell level and not as the committed blood cell precursors mature. This is consistent with population genetic models and observations, which show that selection does not act significantly on an exponentially expanding population.57
Figure 4.
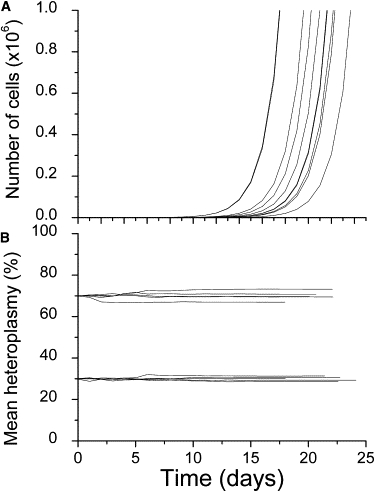
Simulation Results for the Progenitor Cell Model
Results are shown for five repetitions of the simulation starting at 70% mutant and five repetitions starting at 30% mutant.
(A) The exponential increase in the number of simulated cells, starting from a single progenitor cell and dividing on average once per day.
(B) The mean heteroplasmy of the cells over the course of the expansion in the number of cells.
Analysis of Experimental Data
The primary results of our modeling were qualitative ones: first, that the mean mtDNA mutation levels in hematopoietic stem cells should decrease exponentially over long time scales, and second, that measurements of mutation levels in peripheral blood samples should be a good proxy for the mutation level in the stem cell population. To apply these concepts to the published clinical data on mtDNA mutation levels in blood, we first needed a way to determine whether the observed mtDNA heteroplasmy decreases exponentially, as predicted by the simulations. The existing clinical data consists only of pairs of measurements spaced over a few years at most. With just pairs of measurements, not a long time series, we can only calculate the rate of change of heteroplasmy. For an exponential decay, like Equation 2, the rate of change of heteroplasmy is not constant, but is instead proportional to the heteroplasmy level. Therefore, a plot of the rate of mtDNA heteroplasmy change versus initial heteroplasmy level should be a linear plot with zero intercept and negative slope. If the decay is constant instead of exponential, then data on this plot will lie in a horizontal line with zero slope.
Because the decrease of blood mtDNA heteroplasmy in the simulation was very slow (Figure 3A), we need clinical data with measurements separated in time by approximately a decade. The best existing clinical data available was from a study by Rahman et al.12 where measurements of 3243A→G heteroplasmy in blood were separated by 9 to 19 years. However, that study only reported values for six individuals. To extend the dataset, we and our collaborators carried out repeat measurements of blood heteroplasmy on 11 individuals (all with the 3243A→G mutation) to compare to measurements that had been made 5 or more years previously. Those data have been published separately.14 The new data from the Pyle et al. study14 fit together well with the data from the Rahman et al. study12 (Figure 5, note that this is plotted so that an exponential decay in mtDNA heteroplasmy with time shows as a straight line). Taken together, these two clinical datasets give a consistent picture of an exponential decrease in blood mtDNA heteroplasmy levels over time in a consistent manner in different individuals from different studies.
Figure 5.
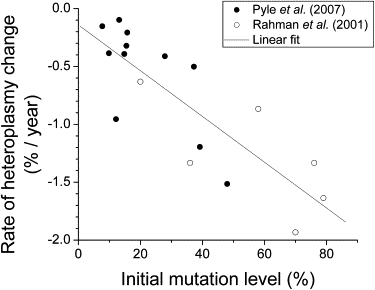
Analysis of Clinical Data to Test for an Exponential Decrease in Peripheral Blood Heteroplasmy
The data are taken from two independent experiments12,14 on the 3243A→G mutation. The line is a linear fit to the data, with slope −0.020 ± 0.003 (1/year) and intercept −0.14 ± 0.14 (%/year) (R2 = 0.68 and p < 10−4). A linear plot of this data with negative slope indicates an exponential decay of the blood heteroplasmy.
Serial measurements from the same patient are rare, particularly over a long time period. The vast majority of the available clinical data consist of heteroplasmy measurements at a single time or at relatively closely spaced times. Even those data can be used to test the predictions of this model. One intuitively obvious consequence of this exponential decay model is that we should not observe high 3243A→G blood heteroplasmy levels in older individuals. We can quantify this limitation by defining a maximum blood heteroplasmy level as
| (4) |
where S = 0.020 ± 0.003 (1/years) was the slope measured from the data in Figure 5. Blood heteroplasmy measurements for 3243A→G mtDNA mutations should fall under this maximum limit. We identified 23 published studies3,4,12–14,20–37 reporting 3243A→G blood mtDNA heteroplasmy levels together with the subjects' age, with a total of 275 unique data points. The data lie below the predicted maximum heteroplasmy (Figure 6A) except for five measurements that lie just above the line, and the data extend from zero mutation up to the predicted maximum. There is a region of the plot, at young age and low heteroplasmy, with very few data points. This is likely to be just an ascertainment effect, because most patients with 3243A→G present with symptoms in their teenage years or older.
Figure 6.
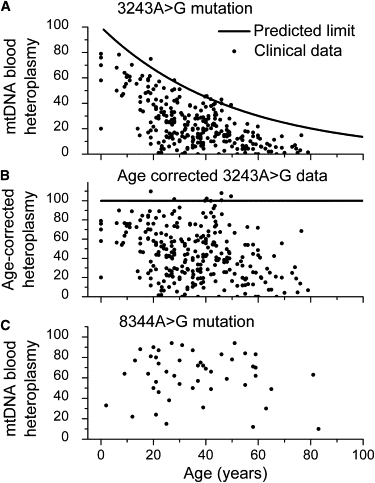
Comparison of Clinical Data to a Predicted Maximum Mutation Heteroplasmy in Peripheral Blood
(A) The theoretical maximum blood mutation level (Equation 4) and 275 measured values from 23 separate studies3,4,12–14,20–37 on subjects with the 3243A→G mutation. The theoretical upper limit is set with an initial heteroplasmy of 100%.
(B) The 3243A→G data modified by the age correction (Equation 5).
(C) Blood heteroplasmy data (48 measured values) for the 8344A→G mutation.38–42 The clinical results plotted here show no indication of a decrease in blood heteroplasmy with the subjects' age for this mutation.
The few data points in Figure 6A that lie just above the predicted maximum heteroplasmy are not a concern. These could easily be caused by very slight individual variations in the rate of decrease of the 3243A→G mutation. Only cases that lie far above the predicted maximum heteroplasmy limit would be of concern. Four such cases were reported in Hammans et al.,22 and they were recognized by those authors as being very unusual. All four cases came from a single small pedigree that also contained a homoplasmic T to C transition at position 3290, in the same tRNA gene as the pathogenic 3243A→G mutation. The 3290T→C sequence variant was not found in 140 controls or 50 patients. Hammans et al. noted that this family had a unique clinical phenotype and suggested that this 3290T→C sequence variant might have altered the pathology of the 3243A→G mutation. El Meziane et al.58 have also shown experimentally that a second mutation, at position 12300 in that case, can suppress the pathogenicity of the 3243A→G mutation.
On the basis of this analysis of the published data, we can suggest a simple method for correcting for the subject's age in blood heteroplasmy measurements. We defined an age-corrected mutation level as follows:
| (5) |
These age-corrected blood heteroplasmy measurements are plotted in Figure 6B.
For the 3243A→G mutation, the heteroplasmy levels clearly decrease with age, and we have shown that the loss of hematopoietic stem cells with high heteroplasmy levels can lead to this exponential decrease. However, it is possible that for some other pathogenic mtDNA mutations, these stem cells could remain viable even when homoplasmic for the mutation. In these cases, or in the case of neutral mutations, we would not expect to see a loss of the mtDNA mutation over time from the blood. These low-penetrance pathogenic mutations were excluded from the analysis of Figures 6A and 6B. One such mutation for which a significant amount of heteroplasmy data exists is the 8344A→G mutation. Indeed, when blood heteroplasmy measurements for subjects with this mutation38–42 were plotted as a function of age, an age-dependent upper limit to heteroplasmy did not appear (Figure 6C), indicating that this mutation is not significantly decreased in the blood over time.
In the 3243A→G mutation, measurements of blood heteroplasmy levels are almost always less than the heteroplasmy levels measured in muscle biopsies.3 For data sets where we have the measured heteroplasmy in blood and in muscle, along with the subjects' age, we can test to see whether the age-corrected blood heteroplasmy is consistent with the muscle heteroplasmy.3,20–22,24,29,30,35,36 In Figure 7A, we plot blood heteroplasmy versus muscle heteroplasmy for the 3243A→G mutation. The line marks equal heteroplasmy values. Although there is a significant correlation between the blood and muscle heteroplasmy values, all of the subjects had lower heteroplasmy in blood compared to muscle. Also, there is a pronounced age effect visible in this data, with older subjects generally having a greater difference between muscle and blood heteroplasmy. Assuming that muscle heteroplasmy is relatively constant over time, we applied the age correction of Equation 5 to the blood heteroplasmy levels (Figure 7B). Both the blood measurements and the age-corrected blood measurements are highly significantly correlated with the muscle heteroplasmy measurements (p < 0.0001 in both cases). The improvement is that the age-corrected data clustered around the line of equal heteroplasmy, though there was still some tendency for the age-corrected blood heteroplasmy to be lower than the muscle heteroplasmy, particularly for muscle heteroplasmy levels above 75%. However, the age stratification disappeared in the age-corrected data, with all three age groups overlapping.
Figure 7.
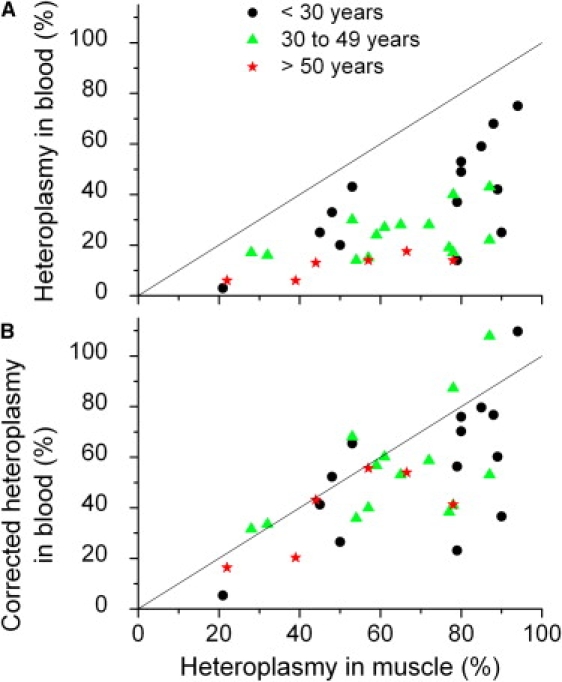
An Application of the Age Correction for Blood Heteroplasmy
(A) Comparison of 3243A→G heteroplasmy levels in muscle and in blood.
(B) Comparison with the blood heteroplasmy corrected for age (Equation 5). The corrections were made with a value of S = 0.020 year−1, the value determined from the data in Figure 5. The solid lines are the lines of equal blood and muscle heteroplasmy.
Discussion
Our analysis of the clinical data relating mtDNA heteroplasmy levels of the 3243A→G mutation in blood to the subjects' age (Figures 5 and 6) indicates that the mutant heteroplasmy level does decrease exponentially with time, as predicted by the simulation. Our simulations of hematopoetic stem cells showed that the mechanism of loss of stem cells with high mutation level was sufficient to cause an exponential decrease in blood heteroplasmy. However, that does not mean that other mechanisms could not also lead to an exponential decrease. For example, Battersby et al.59 have reported a very fast exponential decrease in blood heteroplasmy in mice constructed to have a mixture of two naturally occurring nonpathogenic murine mtDNA strains, BALB/c and NZB. In these mice, the NZB mtDNA strain was lost from the blood with a decay constant Smouse = 1.387 year−1 (almost 70 times faster than the value Shuman = 0.02 year−1 from Figure 5). These authors test, and reject, the hypothesis that this selection against NZB mtDNA is occurring through an immune mechanism. They also did not find any functional difference in oxidative phosphorylation as a function of the cellular heteroplasmy level, indicating that a threshold effect and the cell-loss mechanism modeled in our simulation are likely not playing a role in those mice. The mechanism driving the rapid loss of NZB heteroplasmy in the blood of those mice is still unknown.
The data presented in Figures 5 and 6A, illustrating the exponential loss of pathogenic mtDNA from human blood, came exclusively from measurements on the 3243A→G mutation, the most commonly studied and the most commonly occurring10 heteroplasmic pathogenic mtDNA point mutation. Determination of whether this exponential loss occurs for other pathogenic mtDNA mutants will require either a number of pairs of longitudinal blood heteroplasmy measurements spaced by approximately 10 years (Figure 5) or a much larger number of single-time-point measurements (Figure 6). In the latter case, it is important that a wide range of subject ages are sampled, so that the upper limit on blood heteroplasmy as a function of age can be detected.
We have focused our analysis on the 3243A→G mtDNA point mutations. However, this does not mean that other mtDNA mutations causing low-penetrance diseases, such as Leber's Hereditary Optic Neuropathy (LHON [MIM #535000]), cannot also show a decrease in blood heteroplasmy in some individuals. For those individuals who have the currently unknown cofactors that lead to the disease state, it is possible that the loss of high-heteroplasmy blood stem cells will occur (if this stem cell population is affected by the cofactors). Indeed, sporadic cases of decreasing blood heteroplasmy of LHON mutations have been reported.60,61 We can speculate that the observance of a decrease in blood heteroplasmy over time, or of a lower heteroplasmy in blood compared to muscle, in carriers of LHON mtDNA mutations could indicate the presence of the pathogenic cofactors in those individuals, even in cases where the overall heteroplasmy level of the LHON mutation might not be high enough to cause the disease state (along with the presence of the cofactor). In LHON, an interacting nuclear genetic locus62 or transient exposure to environmental toxins (such as excess alcohol or tobacco, see 63 for a positive correlation and 64 for no correlation) might act as cofactors for the development of the disease state.
We have also focused our analysis on blood samples, for obvious practical reasons. The same behavior of exponentially decreasing mtDNA heteroplasmy would also occur in any other tissue that would experience the loss of cells with high heteroplasmy. Decreasing 3243A→G mutation levels with age have been reported in epithelial cells4,65 and in buccal mucosa.4 In contrast, skeletal muscle is a large multinucleate postmitotic cell, and there is minimal muscle cell loss in patients even with high levels of 3243A→G.66 This fundamental difference probably explains why mutant genomes tend to accumulate in nondividing tissues such as skeletal muscle,67,68 as we have discussed before.46 Despite the known variability of blood heteroplasmy with age, blood samples are still very commonly used for the determination of heteroplasmy levels in individuals, most commonly in asymptomatic individuals. The use of blood samples for the determination of heteroplasmy might lead to misleading artifacts, such as the reported increase in heteroplasmy of a pathogenic mtDNA mutation across generations,18 a measurement that naturally entails comparisons of individuals with radically different ages. The possibility of this artifact has been raised before;61 however, this is the first time that a viable method of correcting for this artifact has been suggested.
Do the observations in blood have a broader relevance? Recent observation in colonic19,69 and gastric70 tissue show that pathogenic point mutations of mtDNA accumulate with age within the stem cell niche of healthy subjects, causing mitochondrial dysfunction in the daughter cell population. The work described here is directly applicable to situations such as this if the mutation leads to cellular dysfunction or cell death in any self-renewing population. For hematological stem cells, at least, our observations do suggest that stem cell viability is partly dependent upon an intact mitochondrial respiratory chain. Our observations also provide some hope for developing autologous stem cell therapies for mtDNA diseases, where, paradoxically, disease progression is associated with the cleansing of the inherited mtDNA mutation from stem cells (an effect that is explained by the results of this study). These could be harnessed for the delivery of wild-type mtDNA to diseased cells, as has been demonstrated in human skeletal muscle.71–73
Web Resources
The URLs for data presented herein are as follows:
Engauge Digitizer, http://digitizer.sourceforge.net
Hematopoietic Stem Cell simulation codes, http://staff.vbi.vt.edu/dsamuels/2008_Blood_Stem_Cell_Paper/software/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
P.F.C. is a Wellcome Trust Senior Clinical Research Fellow. We are very grateful to Angela Pyle for allowing us advanced access to her data before publication.
References
- 1.Lightowlers R.N., Chinnery P.F., Turnbull D.M., Howell N. Mammalian mitochondrial genetics: Heredity, heteroplasmy and disease. Trends Genet. 1997;13:450–455. doi: 10.1016/s0168-9525(97)01266-3. [DOI] [PubMed] [Google Scholar]
- 2.Rossignol R., Malgat M., Mazat J.P., Letellier T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J. Biol. Chem. 1999;274:33426–33432. doi: 10.1074/jbc.274.47.33426. [DOI] [PubMed] [Google Scholar]
- 3.Chinnery P.F., Zwijnenburg P.J.G., Walker M., Howell N., Taylor R.W., Lightowlers R.N., Bindoff L., Turnbull D.M. Nonrandom tissue distribution of mutant mtDNA. Am. J. Med. Genet. 1999;85:498–501. [PubMed] [Google Scholar]
- 4.Frederiksen A.L., Andersen P.H., Kyvik K.O., Jeppesen T.D., Vissing J., Schwartz M. Tissue specific distribution of the 3243A -> G mtDNA mutation. J. Med. Genet. 2006;43:671–677. doi: 10.1136/jmg.2005.039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirches E., Michael M., Warich-Kirches M., Schneider T., Weis S., Krause G., Mawrin C., Dietzmann K. Heterogeneous tissue distribution of a mitochondrial DNA polymorphism in heteroplasmic subjects without mitochondrial disorders. J. Med. Genet. 2001;38:312–317. doi: 10.1136/jmg.38.5.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossignol R., Letellier T., Malgat M., Rocher C., Mazat J.P. Tissue variation in the control of oxidative phosphorylation: implication for mitochondrial diseases. Biochem. J. 2000;347:45–53. [PMC free article] [PubMed] [Google Scholar]
- 7.Barron M.J., Chinnery P.F., Howel D., Blakely E.L., Schaefer A.M., Taylor R.W., Turnbull D.M. Cytochrome c oxidase deficient muscle fibres: Substantial variation in their proportions within skeletal muscles from patients with mitochondrial myopathy. Neuromuscul. Disord. 2005;15:768–774. doi: 10.1016/j.nmd.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Elson J.L., Samuels D.C., Johnson M.A., Turnbull D.M., Chinnery P.F. The length of cytochrome c oxidase-negative segments in muscle fibres in patients with mtDNA myopathy. Neuromuscul. Disord. 2002;12:858–864. doi: 10.1016/s0960-8966(02)00047-0. [DOI] [PubMed] [Google Scholar]
- 9.Shoubridge E.A. Mitochondrial-DNA diseases: Histological and cellular studies. J. Bioenerg. Biomembr. 1994;26:301–310. doi: 10.1007/BF00763101. [DOI] [PubMed] [Google Scholar]
- 10.Chinnery P.F., Johnson M.A., Wardell T.M., Singh-Kler R., Hayes C., Brown D.T., Taylor R.W., Bindoff L.A., Turnbull D.M. The epidemiology of pathogenic mitochondrial DNA mutations. Ann. Neurol. 2000;48:188–193. [PubMed] [Google Scholar]
- 11.Majamaa K., Moilanen J.S., Uimonen S., Remes A.M., Salmela P.I., Karppa M., Majamaa-Voltti K.A.M., Rusanen H., Sorri M., Peuhkurinen K.J., Hassinen I.E. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: Prevalence of the mutation in an adult population. Am. J. Hum. Genet. 1998;63:447–454. doi: 10.1086/301959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahman S., Poulton J., Marchington D., Suomalainen A. Decrease of 3243 A→G mtDNA mutation from blood in MELAS syndrome: A longitudinal study. Am. J. Hum. Genet. 2001;68:238–240. doi: 10.1086/316930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.‘t Hart L.M., Jansen J.J., Lemkes H.H.P.J., de Knijff P., Massen J.A. Heteroplasmy levels of a mitochondrial gene mutation associated with diabetes mellitus decrease in leucocyte DNA upon aging. Hum. Mutat. 1996;7:193–197. doi: 10.1002/(SICI)1098-1004(1996)7:3<193::AID-HUMU2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 14.Pyle A., Taylor R.W., Durham S.E., Deschauer M., Schaefer A.M., Samuels D.C., Chinnery P.F. Depletion of mitochondrial DNA in leucocytes harbouring the 3243A -> G mtDNA mutation. J. Med. Genet. 2007;44:69–74. doi: 10.1136/jmg.2006.043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obermaier-Kusser B., Paetzkebrunner I., Enter C., Mullerhocker J., Zierz S., Ruitenbeek W., Gerbitz K.D. Respiratory-chain activity in tissues from patients (Melas) with a point mutation of the mitochondrial genome. FEBS Lett. 1991;286:67–70. doi: 10.1016/0014-5793(91)80942-v. [DOI] [PubMed] [Google Scholar]
- 16.Rotig A., Cormier V., Blanche S., Bonnefont J.P., Ledeist F., Romero N., Schmitz J., Rustin P., Fischer A., Saudubray J.M., Munnich A. Pearson marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J. Clin. Invest. 1990;86:1601–1608. doi: 10.1172/JCI114881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chinnery P.F., Turnbull D.M., Walls T.J., Reading P.J. Driving and stroke - Reply. Lancet. 1997;350:1402–1403. doi: 10.1016/s0140-6736(97)05005-8. [DOI] [PubMed] [Google Scholar]
- 18.Chinnery P.F., Thorburn D.R., Samuels D.C., White S.L., Dahl H.H.M., Turnbull D.M., Lightowlers R.N., Howell N. The inheritance of mitochondrial DNA heteroplasmy: Random drift, selection or both? Trends Genet. 2000;16:500–505. doi: 10.1016/s0168-9525(00)02120-x. [DOI] [PubMed] [Google Scholar]
- 19.Taylor R.W., Barron M.J., Borthwick G.M., Gospel A., Chinnery P.F., Samuels D.C., Taylor G.A., Plusa S.M., Needham S.J., Greaves L.C. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 2003;112:1351–1360. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deschauer M., Wieser T., Neudecker S., Lindner A., Zierz S. Mitochondrial 3243 A -> G mutation (MELAS mutation) associated with painful muscle stiffness. Neuromuscul. Disord. 1999;9:305–307. doi: 10.1016/s0960-8966(99)00019-x. [DOI] [PubMed] [Google Scholar]
- 21.Devries D., Dewijs I., Ruitenbeek W., Begeer J., Smit P., Bentlage H., Vanoost B. Extreme variability of clinical symptoms among sibs in a MELAS family correlated with heteroplasmy for the mitochondrial A3243G mutation. J. Neurol. Sci. 1994;124:77–82. doi: 10.1016/0022-510x(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 22.Hammans S.R., Sweeney M.G., Hanna M.G., Brockington M., Morganhughes J.A., Harding A.E. The mitochondrial DNA transfer RNALeu(UUR) A-->G (3243) mutation. A clinical and genetic study. Brain. 1995;118:721–734. doi: 10.1093/brain/118.3.721. [DOI] [PubMed] [Google Scholar]
- 23.Hotta O., Inoue C., Miyabayashi S., Furuta T., Takeuchi A., Taguma Y. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 2001;59:1236–1243. doi: 10.1046/j.1523-1755.2001.0590041236.x. [DOI] [PubMed] [Google Scholar]
- 24.Huang C.C., Chen R.S., Chen C.M., Wang H.S., Lee C.C., Pang C.Y., Hsu H.S., Lee H.C., Wei Y.H. MELAS syndrome with mitochondrial tRNA(Leu(UUR)) gene mutation in a chinese family. J. Neurol. Neurosurg. Psychiatry. 1994;57:586–589. doi: 10.1136/jnnp.57.5.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato Y., Miura Y., Inagaki A., Itatsu T., Oiso Y. Age of onset possibly associated with the degree of heteroplasmy in two male siblings with diabetes mellitus having an A to G transition at 3243 of mitochondrial DNA. Diabet. Med. 2002;19:784–786. doi: 10.1046/j.1464-5491.2002.00777.x. [DOI] [PubMed] [Google Scholar]
- 26.Lien L.M., Lee H.C., Wang K.L., Chiu J.C., Chiu H.C., Wei Y.H. Involvement of nervous system in maternally inherited diabetes and deafness (MIDD) with the A3243G mutation of mitochondrial DNA. Acta Neurol. Scand. 2001;103:159–165. doi: 10.1034/j.1600-0404.2001.103003159.x. [DOI] [PubMed] [Google Scholar]
- 27.Lu J., Wang D.W., Li R.H., Li W.X., Ji J.Z., Zhao J., Ye W., Yang L., Qian Y.P., Zhu Y., Guan M.X. Maternally transmitted diabetes mellitus associated with the mitochondrial tRNA(Leu(UUR)) A3243G mutation in a four-generation Han Chinese family. Biochem. Biophys. Res. Commun. 2006;348:115–119. doi: 10.1016/j.bbrc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Majamaa-Voltti K., Peuhkurinen K., Kortelainen M.-L., Hassinen I.E., Majamaa K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc. Disord. 2002;2:12. doi: 10.1186/1471-2261-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinuzzi A., Bartolomei L., Carrozzo R., Mostacciuolo M., Carbonin C., Toso V., Ciafaloni E., Shanske S., Dimauro S., Angelini C. Correlation between clinical and molecular-features in 2 MELAS families. J. Neurol. Sci. 1992;113:222–229. doi: 10.1016/0022-510x(92)90250-o. [DOI] [PubMed] [Google Scholar]
- 30.Mosewich R.K., Donat J.R., Dimauro S., Ciafaloni E., Shanske S., Erasmus M., George D. The syndrome of mitochondrial encephalomyopathy, lactic-acidosis, and stroke-like episodes presenting without stroke. Arch. Neurol. 1993;50:275–278. doi: 10.1001/archneur.1993.00540030041012. [DOI] [PubMed] [Google Scholar]
- 31.Ng M.C.Y., Yeung V.T.F., Chow C.C., Li J.K.Y., Smith P.R., Mijovic C.H., Critchley J.A.J.H., Barnett A.H., Cockram C.S., Chan J.C.N. Mitochondrial DNA A3243G mutation in patients with early- or late-onset type 2 diabetes mellitus in Hong Kong Chinese. Clin. Endocrinol. (Oxf.) 2000;52:557–564. doi: 10.1046/j.1365-2265.2000.00989.x. [DOI] [PubMed] [Google Scholar]
- 32.Olsson C., Zethelius B., Lagerstrom-Fermer M., Asplund J., Berne C., Landegren U. Level of heteroplasmy for the mitochondrial mutation A3243G correlates with age at onset of diabetes and deafness. Hum. Mutat. 1998;12:52–58. doi: 10.1002/(SICI)1098-1004(1998)12:1<52::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 33.Saitoh S., Momoi M.Y., Yamagata T., Nakauchi H., Nihei K., Fujii M. Single-cell analysis of mitochondrial DNA in patients and a carrier of the tRNALeu(UUR) gene mutation. J. Inherit. Metab. Dis. 1999;22:608–614. doi: 10.1023/a:1005569711521. [DOI] [PubMed] [Google Scholar]
- 34.van Essen E.H.R., Roep B.O., ’t Hart L.M., Jansen J.J., Van den Ouweland J.M.W., Lemkes H., Maassen J.A. HLA-DQ polymorphism and degree of heteroplasmy of the A3243G mitochondrial DNA mutation in maternally inherited diabetes and deafness. Diabet. Med. 2000;17:841–847. doi: 10.1046/j.1464-5491.2000.00379.x. [DOI] [PubMed] [Google Scholar]
- 35.Vilarinho L., Santorelli F.M., Rosas M.J., Tavares C., Melo-Pires M., DiMauro S. The mitochondrial A3243G mutation presenting as severe cardiomyopathy. J. Med. Genet. 1997;34:607–609. doi: 10.1136/jmg.34.7.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilichowski E., Korenke G.C., Ruitenbeek W., De Meirleir L., Hagendorff A., Janssen A.J.M., Lissens W., Hanefeld F. Pyruvate dehydrogenase complex deficiency and altered respiratory chain function in a patient with Kearns-Sayre/MELAS overlap syndrome and A3243G mtDNA mutation. J. Neurol. Sci. 1998;157:206–213. doi: 10.1016/s0022-510x(98)00068-9. [DOI] [PubMed] [Google Scholar]
- 37.Zhong S., Ng M.C.Y., Lo Y.M.D., Chan J.C.N., Johnson P.J. Presence of mitochondrial tRNA Leu(UUR) A to G 3243 mutation in DNA extracted from serum and plasma of patients with type 2 diabetes mellitus. J. Clin. Pathol. 2000;53:466–469. doi: 10.1136/jcp.53.6.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canter J.A., Eshaghian A., Fessel J., Summar M.L., Roberts L.J., Morrow J.D., Sligh J.E., Hames J.L. Degree of heteroplasmy reflects oxidant damage in a large family with the mitochondrial DNA A8344G mutation. Free Radic. Biol. Med. 2005;38:678–683. doi: 10.1016/j.freeradbiomed.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 39.Hammans S.R., Sweeney M.G., Brockington M., Lennox G.G., Lawton N.F., Kennedy C.R., Morganhughes J.A., Harding A.E. The mitochondrial-DNA transfer RNA(Lys) A-]G(8344) mutation and the syndrome of myoclonic epilepsy with ragged-red fibers (MERRF). Relationship of clinical phenotype to proportion of mutant mitochondrial-DNA. Brain. 1993;116:617–632. doi: 10.1093/brain/116.3.617. [DOI] [PubMed] [Google Scholar]
- 40.Larsson N.G., Tulinius M.H., Holme E., Oldfors A., Andersen O., Wahlstrom J., Aasly J. Segregation and manifestations of the mtDNA trans RNA lys A->G(8344) mutation of myoclonus epilepsy and ragged-red fibers (MERRF) Syndrome. Am. J. Hum. Genet. 1992;51:1201–1212. [PMC free article] [PubMed] [Google Scholar]
- 41.Piccolo G., Focher F., Verri A., Spadari S., Banfi P., Gerosa E., Mazzarello P. Myoclonus epilepsy and ragged-red fibers: Blood mitochondrial-DNA heteroplasmy in affected and asymptomatic members of a family. Acta Neurol. Scand. 1993;88:406–409. doi: 10.1111/j.1600-0404.1993.tb05368.x. [DOI] [PubMed] [Google Scholar]
- 42.Traff J., Holme E., Ekbom K., Nilsson B.Y. Ekboms syndrome of photomyoclonus, cerebellar-ataxia and cervical lipoma is associated with the tRNA(Lys) A8344G mutation in mitochondrial-DNA. Acta Neurol. Scand. 1995;92:394–397. doi: 10.1111/j.1600-0404.1995.tb00153.x. [DOI] [PubMed] [Google Scholar]
- 43.Colijn C., Mackey M.C. A mathematical model of hematopoiesis - I. Periodic chronic myelogenous leukemia. J. Theor. Biol. 2005;237:117–132. doi: 10.1016/j.jtbi.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 44.Mackey M.C. Cell kinetic status of haematopoietic stem cells. Cell Prolif. 2001;34:71–83. doi: 10.1046/j.1365-2184.2001.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Capps G.J., Samuels D.C., Chinnery P.F. A model of the nuclear control of mitochondrial DNA replication. J. Theor. Biol. 2003;221:565–583. doi: 10.1006/jtbi.2003.3207. [DOI] [PubMed] [Google Scholar]
- 46.Chinnery P.F., Samuels D.C. Relaxed replication of mtDNA: A model with implications for the expression of disease. Am. J. Hum. Genet. 1999;64:1158–1165. doi: 10.1086/302311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shepherd B.E., Guttorp P., Lansdorp P.M., Abkowitz J.L. Estimating human hematopoietic stem cell kinetics using granulocyte telomere lengths. Exp. Hematol. 2004;32:1040–1050. doi: 10.1016/j.exphem.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 48.Ho A.D. Kinetics and symmetry of divisions of hematopoietic stem cells. Exp. Hematol. 2005;33:1–8. doi: 10.1016/j.exphem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Ogawa M. Differentiation and proliferation of hematopoietic stem-cells. Blood. 1993;81:2844–2853. [PubMed] [Google Scholar]
- 50.Akala O.O., Clarke M.F. Hematopoietic stem cell self-renewal. Curr. Opin. Genet. Dev. 2006;16:496–501. doi: 10.1016/j.gde.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 51.Gross N., Getz G., Rabinowitz M. Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J. Biol. Chem. 1969;244:1552–1562. [PubMed] [Google Scholar]
- 52.Matthews P.M., Brown R.M., Morten K., Marchington D., Poulton J., Brown G. Intracellular heteroplasmy for disease-associated point mutations in mtDNA: Implications for disease expression and evidence for mitotic segregation of heteroplasmic units of mtDNA. Hum. Genet. 1995;96:261–268. doi: 10.1007/BF00210404. [DOI] [PubMed] [Google Scholar]
- 53.Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J.P., Letellier T. Mitochondrial threshold effects. Biochem. J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Birky C.W. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu. Rev. Genet. 2001;35:125–148. doi: 10.1146/annurev.genet.35.102401.090231. [DOI] [PubMed] [Google Scholar]
- 55.Brown D.T., Samuels D.C., Michael E.M., Turnbull D.M., Chinnery P.F. Random genetic drift determines the level of mutant mtDNA in human primary oocytes. Am. J. Hum. Genet. 2001;68:533–536. doi: 10.1086/318190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jenuth J.P., Peterson A.C., Fu K., Shoubridge E.A. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat. Genet. 1996;14:146–151. doi: 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- 57.Avise J.C., Neigel J.E., Arnold J. Demographic influences on mitochondrial DNA lineage survivorship in animal populations. J. Mol. Evol. 1984;20:99–105. doi: 10.1007/BF02257369. [DOI] [PubMed] [Google Scholar]
- 58.El Meziane A., Lehtinen S.K., Hance N., Nijtmans L.G.J., Dunbar D., Holt I.J., Jacobs H.T. A tRNA suppressor mutation in human mitochondria. Nat. Genet. 1998;18:350–353. doi: 10.1038/ng0498-350. [DOI] [PubMed] [Google Scholar]
- 59.Battersby B.J., Redpath M.E., Shoubridge E.A. Mitochondrial DNA segregation in hematopoietic lineages does not depend on MHC presentation of mitochondrially encoded peptides. Hum. Mol. Genet. 2005;14:2587–2594. doi: 10.1093/hmg/ddi293. [DOI] [PubMed] [Google Scholar]
- 60.Howell N., Ghosh S.S., Fahy E., Bindoff L.A. Longitudinal analysis of the segregation of mtDNA mutations in heteroplasmic individuals. J. Neurol. Sci. 2000;172:1–6. doi: 10.1016/s0022-510x(99)00207-5. [DOI] [PubMed] [Google Scholar]
- 61.Jacobi F.K., Leo-Kottler B., Mittelviefhaus K., Zrenner E., Meyer J., Pusch C.M., Wissinger B. Segregation patterns and heteroplasmy prevalence in Leber's hereditary optic neuropathy. Invest. Ophthalmol. Vis. Sci. 2001;42:1208–1214. [PubMed] [Google Scholar]
- 62.Hudson G., Keers S., Man P.Y.W., Griffiths P., Huoponen K., Savontaus M.L., Nikoskelainen E., Zeviani M., Carrara F., Horvath R. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am. J. Hum. Genet. 2005;77:1086–1091. doi: 10.1086/498176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sadun F., De Negri A.M., Carelli V., Salomao S.R., Berezovsky A., Andrade R., Moraes M., Passos A., Belfort R., Da Rosa A.B. Ophthalmologic findings in a large pedigree of 11778/Haplogroup J Leber hereditary optic neuropathy. Am. J. Ophthalmol. 2004;137:271–277. doi: 10.1016/j.ajo.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 64.Kerrison J.B., Miller N.R., Hsu F.C., Beaty T.H., Maumenee I.H., Smith K.H., Savino P.J., Stone E.M., Newman N.J. A case-control study of tobacco and alcohol consumption in Leber hereditary optic neuropathy. Am. J. Ophthalmol. 2000;130:803–812. doi: 10.1016/s0002-9394(00)00603-6. [DOI] [PubMed] [Google Scholar]
- 65.Olsson C., Johnsen E., Nilsson M., Wilander E., Syvanen A.C., Lagerstrom-Fermer M. The level of the mitochondrial mutation A3243G decreases upon ageing in epithelial cells from individuals with diabetes and deafness. Eur. J. Hum. Genet. 2001;9:917–921. doi: 10.1038/sj.ejhg.5200742. [DOI] [PubMed] [Google Scholar]
- 66.Sciacco M., Fagiolari G., Lamperti C., Messina S., Bazzi P., Napoli L., Chiveri L., Prelle A., Comi G.P., Bresolin N. Lack of apoptosis in mitochondrial encephalomyopathies. Neurology. 2001;56:1070–1074. doi: 10.1212/wnl.56.8.1070. [DOI] [PubMed] [Google Scholar]
- 67.Durham S.E., Samuels D.C., Chinnery P.F. Is selection required for the accumulation of somatic mitochondrial DNA mutations in post-mitotic cells? Neuromuscul. Disord. 2006;16:381–386. doi: 10.1016/j.nmd.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 68.Bidooki S.K., Johnson M.A., Chrzanowska-Lightowlers Z., Bindoff L.A., Lightowlers R.N. Intracellular mitochondrial triplasmy in a patient with two heteroplasmic base changes. Am. J. Hum. Genet. 1997;60:1430–1438. doi: 10.1086/515460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Greaves L.C., Preston S.L., Tadrous P.J., Taylor R.W., Barron M.J., Oukrif D., Leedham S.J., Deheragoda M., Sasieni P., Novelli M.R. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc. Natl. Acad. Sci. USA. 2006;103:714–719. doi: 10.1073/pnas.0505903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McDonald S.A.C., Preston S.L., Greaves L.C., Leedham S.J., Lovell M.A., Jankowski J.A.Z., Turnbull D.M., Wright N.A. Clonal expansion in the human gut: Mitochondrial DNA mutations show us the way. Cell Cycle. 2006;5:808–811. doi: 10.4161/cc.5.8.2641. [DOI] [PubMed] [Google Scholar]
- 71.Taivassalo T., Fu K., Johns T., Arnold D., Karpati G., Shoubridge E.A. Gene shifting: A novel therapy for mitochondrial myopathy. Hum. Mol. Genet. 1999;8:1047–1052. doi: 10.1093/hmg/8.6.1047. [DOI] [PubMed] [Google Scholar]
- 72.Clark K.M., Bindoff L.A., Lightowlers R.N., Andrew R.M., Griffiths P.G., Johnson M.A., Brierley E.J., Turnbull D.M. Reversal of a mitochondrial DNA defect in human skeletal muscle. Nat. Genet. 1997;16:222–224. doi: 10.1038/ng0797-222. [DOI] [PubMed] [Google Scholar]
- 73.Fu K., Hartlen R., Johns T., Genge A., Karpati G., Shoubridge E.A. A novel heteroplasmic tRNA(leu(CUN)) mtDNA point mutation in a sporadic patient with mitochondrial encephalomyopathy segregates rapidly in skeletal muscle and suggests an approach to therapy. Hum. Mol. Genet. 1996;5:1835–1840. doi: 10.1093/hmg/5.11.1835. [DOI] [PubMed] [Google Scholar]