

Abstract
Autosomal-recessive inheritance is believed to be relatively common in mental retardation (MR), although only four genes for nonsyndromic autosomal-recessive mental retardation (ARMR) have been reported. In this study, we ascertained a consanguineous Pakistani family with ARMR in four living individuals from three branches of the family, plus an additional affected individual later identified as a phenocopy. Retinitis pigmentosa was present in affected individuals, but no other features suggestive of a syndromic form of MR were found. We used Affymetrix 500K microarrays to perform homozygosity mapping and identified a homozygous and haploidentical region of 11.2 Mb on chromosome 4p15.33-p15.2. Linkage analysis across this region produced a maximum two-point LOD score of 3.59. We sequenced genes within the critical region and identified a homozygous splice-site mutation segregating in the family, within a coiled-coil and C2 domain-containing gene, CC2D2A. This mutation leads to the skipping of exon 19, resulting in a frameshift and a truncated protein lacking the C2 domain. Conservation analysis for CC2D2A suggests a functional domain near the C terminus as well as the C2 domain. Preliminary functional studies of CC2D2A suggest a possible role in Ca2+-dependent signal transduction. Identifying the function of CC2D2A, and a possible common pathway with CC2D1A, in correct neuronal development and functioning may help identify possible therapeutic targets for MR.
Main Text
Mental retardation (MR) or developmental delay is a heterogeneous group of disorders that are defined by deficits in cognitive and adaptive development. The prevalence of MR is commonly given as 1%–3% of the population,1 with a higher proportion of males to females affected (1.4:1).2 MR is subclassified according to intelligence quotient (IQ) as mild MR in the IQ range 50–55 to 70, moderate MR as 35–40 to 50–55, severe MR as 20–25 to 35–40, and profound MR as below 20–25.3 Understanding the etiological basis of MR in affected individuals is important because it may provide diagnostic clues for possible comorbid features (e.g., scoliosis in Rett syndrome), as well as providing information for genetic counseling, or may help in identifying a treatable condition such as phenylketonuria.4 In addition, understanding the cause of MR may help families to cope with an affected child and may help them gain access to support infrastructure.
Genetic factors are involved in the etiology of approximately two-thirds of MR cases.5 In inherited forms of MR, X-linkage or autosomal-recessive inheritance patterns are the most plausible because procreation from affected individuals is not common. To date, more than 60 loci have been reported for X-linked MR;6 however, the molecular genetic basis for ARMR is still poorly understood. Although autosomal-recessive inheritance is estimated to be involved in nearly a quarter of all individuals with nonsyndromic MR (reviewed in Basel-Vanagaite et al.7), only four autosomal genes, PRSS12 on 4q26 (neurotrypsin [MIM #606709]), CC2D1A on 19p13.12 [MIM #610055], CRBN on 3p26 (cereblon [MIM #609262]), and very recently GRIK2 on 6q16.3 (ionotropic glutamate receptor 6 [MIM #138244]) have been reported so far to cause nonsyndromic ARMR (NS-ARMR).8–11 However, only a very few families or unrelated individuals with ARMR have been confirmed for each of these genes (PRSS12, n = 2; CRBN, n = 1; CC2D1A, n = 9; GRIK2, n = 1). The most recent of these genes, GRIK2, was discovered at one of eight loci for NS-ARMR recently mapped by homozygosity mapping in 78 consanguineous Iranian families. However, no disease gene or mutation has yet been reported for the other seven loci.12 Another recently published study has mapped a new locus for NS-ARMR to 1p21.1-p13.3.13 Retinitis pigmentosa (RP; MIM 268000) describes a progressive constriction of the visual fields, nyctalopia, and fundus-associated changes. To date, no genes have been reported for otherwise nonsyndromic MR associated with RP, although MR and RP are present together in some syndromic forms of MR, such as several forms of Bardet-Biedl syndrome (MIM 209900).
We ascertained an ARMR family from a farming community in the district of Mianwali, Punjab province, Pakistan. The pedigree structure indicates a high degree of consanguinity (Figure 1), with first-cousin marriages among the parents of all the affected individuals and the presence of MR among the male and female offspring in three branches of the family. Institutional research ethics approval was obtained, and written informed consent was obtained for all participants in the study. Height, weight, and occipitofrontal circumference were all within the normal range. The older three patients, who are either adult or adolescent, have retinitis pigmentosa and astigmatism; the adult male patient also has early cataract. The youngest patient (currently age 3 years) does not have retinitis pigmentosa but does have astigmatism. All the patients have nystagmus. RP was not present in other members of the family. There are two difficulties in assessment of level of MR in these individuals: lack of measuring instruments with normative values for the local population and problems with administration of the instruments because of visual impairment. The patients and a number of relatives were assessed by a consultant in psychiatry, M. Ayub, who specializes in learning disability/MR and trained both in Pakistan and the UK. The patients and relatives were interviewed so that information could be gathered about development in different domains (motor, language, cognitive, social, and emotional), current level of social functioning, and help and support required for activities of daily living. All this information was discussed with a clinical psychologist practicing in Lahore with over 20 years of experience of working with people with MR in that culture. They reached consensus diagnosis for all the affected individuals (WHO 1994). A diagnosis of mild to moderate MR was reached for all the affected individuals with ICD 10 criteria. Physical and neurological assessment was performed by a consultant neurologist (M. Azam) on the youngest female affected (at 20 months) as well as the male affected (at 27 years old). There were no focal neurological abnormal signs. For both, there were no dysmorphic features, no hepatosplenomegaly, no heart murmur, and no skin abnormalities; however, vision was impaired for the male. Development was globally delayed for the female infant. Structural MRI of the brain was performed for these two affected individuals; the results were generally normal, although with some suggestion of mild cerebellar atrophy in the male affected individual. Photographs of all affected individuals were assessed for dysmorphic features by an experienced clinical geneticist; however, no unusual facial features were apparent.
Figure 1.
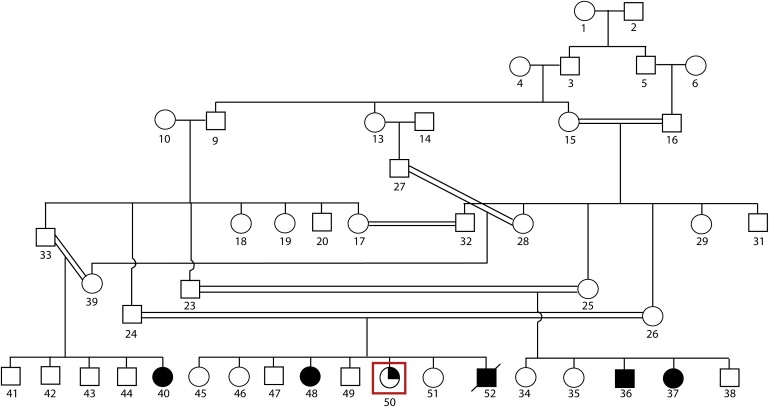
Pedigree Showing Three Branches of the Family from Mianwali District, Punjab Province, Pakistan
Filled symbols indicate individuals affected with mental retardation. The quarter-filled symbol, highlighted within a red box, indicates the patient with mental retardation but subsequently determined to have tetrasomy X.
Blood samples were collected from five affected and 12 unaffected members of the family. Genomic DNA was extracted by standard methods. Lymphoblast cell lines were also successfully established for several family members (Figure 1, individuals 23, 24, 25, 26, 37, and 50), RNA was extracted, and first-strand cDNA was synthesized by standard methods. DNA from the five affected and one unaffected individuals were analyzed with the Affymetrix GeneChip Mapping 500K array NspI chip, which allowed us to genotype ∼260,000 SNPs. The arrays were scanned with a GeneChip Scanner, and the data were processed with GeneChip Operating Software (GCOS) and GeneChip Genotyping Analysis Software (GTYPE) software (ver. 3.0.2) for generation of SNP allele calls. Analysis revealed a common ∼11.2 Mb homozygous and haploidentical region on the short arm of chromosome 4 (4p15.2-p15.33; Figure 2) in four affected individuals but not in the normal individual or the fifth affected individual. This was the only large (>0.5 Mb) homozygous region present in four out of five affected individuals, and no such regions were present in all five. We confirmed the boundaries of the 11.2 Mb critical region containing ∼39 genes (UCSC Genome Browser, March 2006) by sequencing the flanking SNPs, rs2191685 and rs7664104 (at 14.001 and 25.203 Mb from the p telomere, respectively), in order to confirm their heterozygous status.
Figure 2.
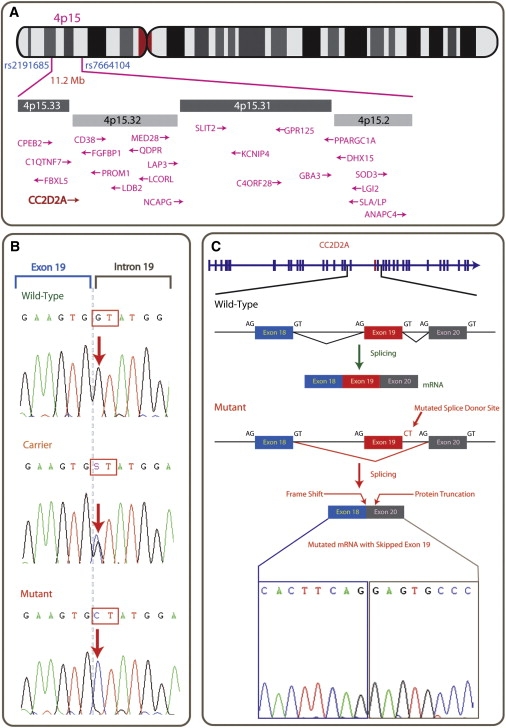
Ideogrammatic Representation of the Mutation Region on 4p15.33
(A) Position of the 11.2 Mb common homozygous stretch on 4p15. and the organization of various known genes within this region.
(B) The intron 19 donor splice-site mutation sequence, with wild-type, followed by carrier (heterozygote) sequence and the homozygous mutation from affected individuals.
(C) The genomic organization of CC2D2A and the consequence of the mutation, namely the splicing out of exon 19, which results in a shift of reading frame leading to 13 nonsense amino acids followed by a stop codon, thus prematurely truncating the protein at this point. Sequence analysis was performed with BigDye Terminator v3.1 Cycle Sequencing (Applied Biosystems).
Because homozygosity mapping did not reveal any common region in all five affected individuals, we further hypothesized that there might be a chromosomal deletion or duplication segregating with phenotype. Copy-number variations (CNVs) were inferred by comparative analysis of hybridization intensities with dChip analyzer.14–16 After normalization, we used the hidden Markov model (HMM) to infer the DNA copy number. Interestingly, copy-number analysis indicated a gain across the entire X chromosome in the fifth affected individual, who did not share the 4p15.3 haplotype with the other four affected members of the family. Subsequent cytogenetic analysis indicated the karyotype 48,XXXX. This rare tetrasomy is almost always associated with mental retardation, with an IQ range of 30 to 75 (mean IQ of 60),17 and thus suggests that this female is a phenocopy. No other CNVs of interest were identified among the affected individuals.
We confirmed linkage to the locus on chromosome 4p15.33-p15.2 by genotyping 17 members of the family with the 12 microsatellite markers across the 4p region (Table 1) and using an ABI 3730xl DNA analyzer and Genemapper software (Applied Biosystems). Parametric linkage analysis was performed with MLINK (2-point) and LINKMAP (shifting three-point).18,19 Linkage analysis revealed a maximum two-point logarithm of odds (LOD) score of 3.59 (theta = 0.0) at marker D4S419 (Table 1), whereas a maximum LOD score of 4.1 was obtained by multipoint analysis. A shared disease haplotype was identified for the microsatellite markers D4S3048, D4S1525, D4S419, D4S1546, and D4S425 (Figure 3).
Table 1.
Two-Point Linkage Analysis of Microsatellite Markers across the 4p Region
| Marker | Physical Position (Mb) | Genetic Distance Female (cM) | Genetic Distance Male (cM) | Sex Averaged Genetic Distance (cM) | LOD |
|---|---|---|---|---|---|
| D4S1599 | 10.404 | 25.5 | 20.9 | 23.2 | −0.98979 |
| D4S3036 | 11.728 | 26.5 | 22.3 | 24.4 | −0.662288 |
| D4S403 | 13.259 | 28.7 | 23.4 | 26.05 | −1.479108 |
| D4S3048 | 15.516 | 29.7 | 28.8 | 29.25 | 2.304191 |
| D4S1525 | 16.092 | 29.7 | 29.9 | 29.8 | 2.208764 |
| D4S419 | 18.357 | 36.2 | 30.9 | 33.55 | 3.597317 |
| D4S1546 | 20.275 | 37.3 | 33.1 | 35.2 | 2.007724 |
| D4S425 | 23.165 | 38.3 | 34.1 | 36.2 | 2.822141 |
| D4S391 | 27.121 | 46.9 | 40.6 | 43.75 | −5.677216 |
| D4S2408 | 30.813 | 50.1 | 41.9 | 46 | −1.635551 |
| D4S405 | 39.947 | 69.4 | 44.8 | 57.1 | −1000 |
| D4S1592 | 57.276 | 91.6 | 48 | 69.8 | −1000 |
We assumed an autosomal-recessive model with complete penetrance and a disease allele frequency of 0.0001. We assumed that marker-allele frequencies were equal. Markers shaded gray are within the homozygous critical region shared by the four affected individuals. Positive LOD scores are highlighted in italics.
Figure 3.
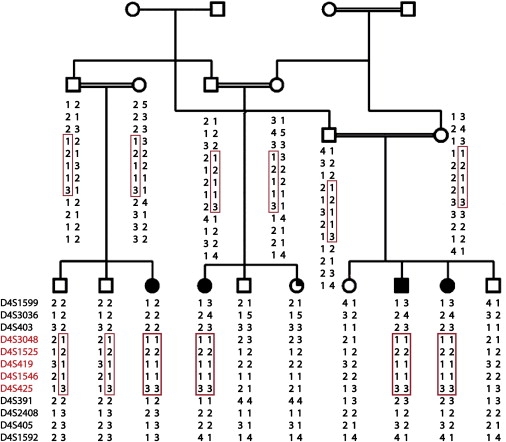
Haplotype Analysis
Markers are shown in red, and haplotypes within a red box indicate the disease haplotype shared between affected individuals (homozygous) and carriers (heterozygous).
The 11.2 Mb critical region, as defined by the homozygosity data, contains ∼39 annotated genes (UCSC 2006). First, we excluded QDPR, the gene for phenylketonuria type 2 (PKU2; MIM +262630), which lies within the critical region, by sequence analysis and biochemical analysis of patients' blood. We then sequenced all other known genes in the region and identified a splice-donor-site mutation (IVS19+1:G→C) in exon 19 of a gene, coiled-coil and C2 domain 2A, CC2D2A (Figure 2). This homozygous mutation segregates with the phenotype in the Mianwali family. CC2D2A (NM_001080522; also known as KIAA1345) has an mRNA ∼5 kb in length and is found in at least 13 different isoforms with up to 37 exons spanning ∼131.5 kb of genomic DNA (from 15,080,760–15,212,278 bp; UCSC March 2006) on 4p15.33. The CC2D2A protein contains up to 1620 amino acids (depending on exon usage), and a C2 domain is predicted from residue 1042-1202. RT-PCR amplification followed by sequence analysis with lymphoblast-derived cDNA from the affected individuals indicated that the splice mutation results in the skipping of exon 19. The splicing of exon 18 to exon 20 causes a frameshift, resulting in the addition of 13 nonsense amino acids added (p.V728EfsX741; NP_001073991) beyond exon 18 before a premature stop codon truncates the protein after amino acid 740, thus abolishing the C2 domain (Figure 2). This mutation was not present in 460 chromosomes of healthy Pakistani controls.
RT-PCR analysis indicates that CC2D2A is expressed in many adult tissues (Figure 4). In a panel of cDNAs from 12 tissues, expression was detected in each tissue, albeit at varying levels, with maximum expression seen in the prostate, pancreas, kidney, lung, and liver, and lower expression in the spleen, small intestine, colon, skeletal muscle, ovary, thymus, and heart. Brain expression with human fetal brain Marathon Ready cDNA (Clontech) was also strong (not shown). To study cellular localization of the CC2D2A protein, we overexpressed a CC2D2A-GFP fusion protein in Cos7 cells. Localization appeared to be almost exclusively cytoplasmic, despite the predicted presence of potential nuclear localization signals (data not shown).
Figure 4.
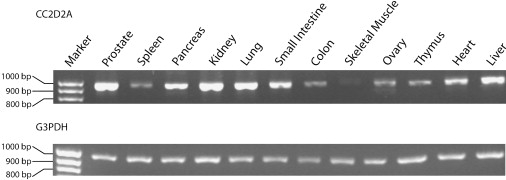
Tissue Transcription Analysis of CC2D2A
RT-PCR expression analysis with a multiple-tissue panel of normalized first-strand cDNA (Clontech) for CC2DA (top) and housekeeping gene G3PDH, which was run as a control (bottom). For RT-PCR amplification, we used a forward primer from exon 18 (5′-ACAGTCAGTCGGCCACTAGG-3′) and a reverse primer from exon 25 (5′- GTTCTGCCAGCTTGAAAAGG-3′) with PCR product size of 963 bp. A 100 bp size ladder is shown to the left.
Genomic organization of CC2D2A on 4p15.33 is shown in Figure 2. Analysis of the sequence at and around the CC2D2A gene with the PromoterInspector program indicated the presence of a putative promoter sequence of 773 bp from 15,080,088–15,080,860 bp (from the 4p telomere; UCSC March 2006). Analysis of the protein sequence using SMART indicated the presence of a C2 domain from residue 1042 to 1202 (in the 1620 amino acid isoform). The C2 domain, also known as protein kinase C conserved region 2 (CalB), is a Ca2+-dependent membrane-targeting module present in many proteins involved in membrane trafficking or signal transduction. Transmembrane (TM) prediction with TMpred suggested two possibilities: either one strong TM helix (from residues 1278 to 1297) or no clear TM domains. Coiled-coil analysis with COILSv.2.1 predicted three stretches of the protein with >90% probability of coiled-coil structure: amino acid residues 442–463, 472–492, and 533–580 (with a 21-residue window). SOSUI predicts the protein to be soluble, with average hydrophobicity of −0.655. Secondary structure is predicted to be mainly alpha helix and random coil, with some extended strand (network protein sequence analysis). No signal peptides were detected. In addition, initial analysis indicates that CC2D2A appears to have a number of potential CaMKII recognition sites (9 RXXS/T and 3 S/TXD sites), as well as numerous putative PKC phosphorylation sites. Two putative nuclear localization signals were also detected with the PredictProtein suite (QRAKKKKRK at residue 587 and RPRRK at 1021). Protein-alignment analysis indicates that CC2D2B has no homology with either CC2D1A or CC2D1B. There is 34% homology between CC2D2A and CC2D2B (NM_001001732; 10q23), but only toward the C-terminal end (residues 1250–1620; Figure 5B) of CC2D2A. CC2D2B has neither coiled-coil nor C2 domains.
Figure 5.

Comparative Protein Sequence Analysis of CC2D2A
Boxshade alignments for (A) a section of the C2 domain (residues 1099 to 1202 of the human protein) and (B) a section of the C-terminal region (1280–1463). A selection of sequences from 11 representative species (out of 18 studied) across the evolutionary spectrum are shown. Alignment with the C-terminal region of the human paralogue, CC2D2B, is also shown; however, no homology was detected with the C2 region. The upstream half of the C2 region is not shown because sections of this region are not present in some lower-order vertebrates and nonvertebrates. Also, only a portion of the C-terminal region is shown, for the sake of succinctness. Residues that were conserved across all 18 species analyzed (including those not included in the figure: chimp, horse, cow, rat, tetraodon, zebrafish, and Drosophila) are marked with an asterisk. Accession numbers for sequences used are as follows in parentheses: chimpanzee (XM_526530), rhesus monkey (XM_001118936), horse (XM_001500638), cow (UPI0000F333EB), dog (UPI0000EB4478), mouse (NM_172274), rat (XM_001055728), opossum (XM_001369737), chicken (XM_420777), Xenopus (BC077972), zebrafish (XM_693709), fugu (NEWSINFRUT00000150241), tetraodon (GSTENT00032796001), sea urchin (XM_775975), fruitfly (NM_137386), mosquito (XM_001658220), and nematode (NM_059625).
Comparative analysis of the protein sequence with ClustalW across 18 species shows a high degree of conservation across vertebrate evolution (Figure 5). The human sequence is 99.4% identical to that of chimp, 96.9% identical to that of rhesus monkey, 88.8% identical to that of horse, 84.8% identical to that of mouse and rat, 75.7% identical to that of opossum, 70.7% identical to that of chicken, 60% identical to that of Xenopus, 59.6% identical to that of zebrafish, and 44.7% identical to that of sea urchin. The human protein is 21.6% identical to C. elegans protein K07G5.3 (NP_492026) and 29.9% identical to Drosophila protein CG18631 (NP_611230), with the strongest overlap and homology occurring toward the C-terminal end (from amino acid 1301 in human, 894 in C. elegans, and 200 in Drosophila, to the carboxyl terminus), suggesting conserved functionality to this region in addition to the C2 domain. The WormBase information on the C. elegans orthologous gene K07G5.3 indicates that it is expressed in the nervous system, including head and tail neurons (as well as other unidentified cells in the head and tail) during both adult and larval stages. Knockdown of K07G5.3 through RNAi was nonlethal.
We studied rates of evolutionary change for CC2D2A gene and for domain-encoding regions within CC2D2A, with pairwise comparisons between more closely related species. We calculated Ka and Ks values20–22 to examine the level of amino acid changing and silent nucleotide divergence between gene pairs. Ka is the number of nonsynonymous substitutions per nonsynonymous site, and Ks is the number of synonymous substitutions per synonymous site. Comparing the Ka/Ks ratio can provide insight into the evolutionary profile of a gene sequence because a Ka/Ks ratio significantly greater than 1.0 is strong evidence of positive selection, whereas a Ka/Ks ratio of less than 0.45 indicates negative selection. Interestingly, the C2 domain appears to have a different evolutionary profile in the primate lineage than the rodent lineage. In the primates, the C2 domain has higher Ka and Ks than either the full-length sequence (Table 2, rows 1–3) or the regions flanking the C2 domain, FR1 and FR2 (Table 3, upper section). In the rodent lineage, however, based on Ka values, the C2 domain is more conserved at the amino acid level than the full-length sequence (Table 2, row 6) or at FR1 and FR2 (Table 3, upper section). This suggests that the C2 domain in the primate lineage may be more permissive of amino-acid-changing substitutions than the orthologous region in the rodent lineage. This may suggest a primate-specific role for this subdomain in neuronal development because affected individuals in this study express a truncated CC2D2A lacking the C2 domain. Additional mapping of point mutations in this region may help to confirm this hypothesis. For the C-terminal region, in all pairwise comparisons, the Ka/Ks ratios were significantly lower than for the N terminal region (Table 3, lower section), providing additional evidence that the C-terminal region is also well conserved across species.
Table 2.
Pairwise Analysis of Synonymous versus Nonsynonymous Nucleotide Substitutions for the Full-Length CC2D2A Sequence and for the C2 Domain
| Species | cDNA Full-Length |
C2 Domain |
||||
|---|---|---|---|---|---|---|
| Ka | Ks | Ka/Ks | Ka | Ks | Ka/Ks | |
| Human-Chimp | 0.0029 | 0.0074 | 0.3937 | 0.0087 | 0.0184 | 0.4755 |
| Human-Rhesus | 0.0155 | 0.0558 | 0.2775 | 0.0182 | 0.0732 | 0.2493 |
| Chimp-Rhesus | 0.0155 | 0.0521 | 0.2983 | 0.0221 | 0.0675 | 0.3272 |
| Human-Mouse | 0.0813 | 0.5117 | 0.1588 | 0.0443 | 0.5567 | 0.0796 |
| Human-Rat | 0.0814 | 0.5249 | 0.1551 | 0.0503 | 0.6294 | 0.0799 |
| Mouse-Rat | 0.0258 | 0.1689 | 0.1527 | 0.0056 | 0.2466 | 0.0227 |
The Ka is the number of nonsynonymous substitutions per nonsynonymous site, and the Ks is the number of synonymous substitutions per synonymous site.
Table 3.
Analysis of Rates of Evolutionary Change for CC2D2A
| Species | FR1 |
FR2 |
||||
|---|---|---|---|---|---|---|
| Ka | Ks | Ka/Ks | Ka | Ks | Ka/Ks | |
| Human-Chimp | 0.0000 | 0.0000 | n/a | 0.0031 | 0.0000 | n/a |
| Human-Rhesus | 0.0060 | 0.0487 | 0.1232 | 0.0031 | 0.0251 | 0.1235 |
| Chimp-Rhesus | 0.0060 | 0.0487 | 0.1232 | 0.0061 | 0.0252 | 0.2421 |
| Human-Mouse | 0.0500 | 0.3178 | 0.1573 | 0.0533 | 0.5594 | 0.0953 |
| Human-Rat | 0.0479 | 0.2821 | 0.1698 | 0.0485 | 0.5551 | 0.0874 |
| Mouse-Rat | 0.0141 | 0.0925 | 0.1524 | 0.0103 | 0.1907 | 0.0540 |
| Species |
N Terminal |
C-Terminal |
||||
| Ka | Ks | Ka/Ks | Ka | Ks | Ka/Ks | |
| Human-Chimp | 0.0027 | 0.0027 | 0.9853 | 0.0011 | 0.0000 | na |
| Human-Rhesus | 0.0254 | 0.0515 | 0.4940 | 0.0038 | 0.0275 | 0.1384 |
| Chimp-Rhesus | 0.0254 | 0.0485 | 0.5245 | 0.0027 | 0.0275 | 0.0989 |
| Human-Mouse | 0.1803 | 0.4834 | 0.3730 | 0.0486 | 0.4130 | 0.1176 |
| Human-Rat | 0.1847 | 0.5643 | 0.3273 | 0.0439 | 0.3870 | 0.1134 |
| Mouse-Rat | 0.0608 | 0.1870 | 0.3254 | 0.0099 | 0.1985 | 0.0499 |
Pairwise comparison of synonymous versus nonsynonymous nucleotide substitutions for flanking regions FR1 and FR2 500 bp upstream and downstream of C2, respectively (upper section), and for the N-terminal region (the first 1115 nucleotides at the N terminus in human) and C-terminal region (encompassing the last 1113 nucleotides of the coding sequence in the human) (lower section).
CC2D2A lies on 4p15.3 within a region involved in proximal interstitial deletions (proximal to the Wolf-Hirschhorn syndrome (WHS; MIM #194190) and Pitt-Rogers-Dank syndrome (PRDS; MIM #262350) deletion regions. To date, more than 20 cases have been reported, and the clinical features include facies and other dysmorphologcal features as well as varying degrees of mental retardation (discussed in Tonk et al.23); however, RP is not a known feature.
CC2D1A, one of the four previously identified ARMR genes,8 also encodes a single C2 domain toward the C-terminal end of the protein in addition to coiled-coil regions, and thus we speculate that these two proteins, CC2D1A and CC2D2A, may have similar functions and may be components of the same or parallel pathways that are important components for neuronal development, disruption of which leads to developmental delay. CC2D1A is also known as Freud-1 (five prime repressor under dual repression binding protein-1). A deletion mutation was identified that spans 3.6 kb, deleting exons 14 to 16, thereby resulting in a truncated protein lacking the C2 domain. The mouse homolog of CC2D1A/Freud-1 was initially identified as a transcription factor involved in the negative regulation of the serotonin 1A receptor gene (HTR1A).24,25 Serotonin 1A receptors are expressed in the cortex, limbic system, and hypothalamus and are associated with depression, anxiety, sexual activity, stress response, and learning.26 CC2D1A/Freud-1 is colocalized with HTR1A receptors in both presynaptic and postsynaptic serotonin system neurons.25 Recent studies have shown that CC2D1A/Freud-1 also regulates transcription of the dopamine receptor DRD2.27
Freud-1 is highly conserved through evolution, with orthologs in invertebrate species such as C. elegans. Freud-1 contains four DM14 domains (D. melanogaster 14; unknown function), one helix-loop-helix (HLH) domain, one C2 domain, a proline-rich domain believed to be involved in protein binding,28 coiled-coil oligomerization motifs,29 putative phosphorylation sites for protein kinases A and C, and two calcium calmodulin-dependent protein kinase (CaMK) II/IV sites. Our preliminary examination of CC2D2A protein indicates the presence of the coiled-coil domains and the C2 domain, as well as PKC and CaMKII sites. However, homology analysis indicates that this protein is not a member of the Freud protein family. A recent study aimed at screening the human proteome for calmodulin (CaM) protein binding partners with the mRNA display technique has identified CC2D2A as a potential CaM-binding protein.30 CaM is the most widespread and well studied of Ca2+ sensors in eukaryotic cells. This adds further support to a role for CC2D2A in Ca2+-regulated signaling pathways.
Given the various commonalities between CC2D1A/Freud-1 and CC2D2A in terms of protein structure and the phenotypic consequences of truncation mutation, we hypothesize that they may function in similar pathways or mechanisms, i.e., transcriptional regulation and signaling, which may be crucial for correct neuronal development. Study of a common pathway may help identify other genes involved in NSMR (also possibly in RP), as well as potential therapeutic targets for developmental delay.
Acknowledgments
We would like to express our deepest gratitude to the family from Mianwali for their participation in this study. We also thank The Centre for Applied Genomics at The Hospital for Sick Children, and Ciara Fahey in particular for the cytogenetic work, and Dr. Bridget Fernandez at Memorial University of Newfoundland Health Sciences Centre, St. John's, Newfoundland, for consultation on possible dysmorphic features in the Mianwali family. Funding support for this work was through Genome Canada/Ontario Genomics Institute and through an Early Researcher Award from the Ontario provincial government to J.B.V.
Web Resources
The URLs for data presented herein are as follows:
ICD 10, http://www.who.int/classifications/apps/icd/icd10online/
Network Protein Sequence analysis, http://npsa-pbil.ibcp.fr
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
PredictProtein, http://www.predictprotein.org/newwebsite/
PromoterInspector, http://www.genomatix.de
SOSUI, http://sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html
UCSC, http://genome.ucsc.edu
Worm Base, http://www.wormbase.org/
References
- 1.Leonard H., Wen X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 2.Murphy C.C., Boyle C., Schendel D., Decouflé P., Yeargin-Allsopp M. Epidemiology of mental retardation in children. Ment. Retard. Dev. Disabil. Res. Rev. 1998;4:6–13. [Google Scholar]
- 3.American Psychiatric Association . Fourth Edition, Text Revision. DSM-IV; Washington, DC: 1994. The Diagnostic & Statistical Manual of Mental Disorders. [Google Scholar]
- 4.Shea S.E. Mental retardation in children ages 6–16. Seminars in Pediatric Neurology. 2006;13:262–270. doi: 10.1016/j.spen.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Curry C.J. Rational evaluation of the adolescent with mental retardation. Adolesc. Med. 2002;13:331–343. [PubMed] [Google Scholar]
- 6.Chelly J., Khelfaoui M., Francis F., Cherif B., Bienvenu T. Genetics and pathophysiology of mental retardation. Eur. J. Hum. Genet. 2006;14:701–713. doi: 10.1038/sj.ejhg.5201595. [DOI] [PubMed] [Google Scholar]
- 7.Basel-Vanagaite L. Genetics of autosomal recessive non-syndromic mental retardation: Recent advances. Clin. Genet. 2007;72:167–174. doi: 10.1111/j.1399-0004.2007.00881.x. [DOI] [PubMed] [Google Scholar]
- 8.Basel-Vanagaite L., Attia R., Yahav M., Ferland R.J., Anteki L., Walsh C.A., Olender T., Straussberg R., Magal N., Taub E. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J. Med. Genet. 2006;43:203–210. doi: 10.1136/jmg.2005.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molinari F., Rio M., Meskenaite V., Encha-Razavi F., Auge J., Bacq D., Briault S., Vekemans M., Munnich A., Attie-Bitach T. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–1781. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- 10.Higgins J.J., Pucilowska J., Lombardi R.Q., Rooney J.P. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology. 2004;63:1927–1931. doi: 10.1212/01.wnl.0000146196.01316.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motazacker M.M., Rost B.R., Hucho T., Garshasbi M., Kahrizi K., Ullmann R., Abedini S.S., Nieh S.E., Amini S.H., Goswami C. A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am. J. Hum. Genet. 2007;81:792–798. doi: 10.1086/521275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Najmabadi H., Motazacker M.M., Garshasbi M., Kahrizi K., Tzschach A., Chen W., Behjati F., Hadavi V., Nieh S.E., Abedini S.S. Homozygosity mapping in consanguineous families reveals extreme heterogeneity of non-syndromic autosomal recessive mental retardation and identifies 8 novel gene loci. Hum. Genet. 2007;121:43–48. doi: 10.1007/s00439-006-0292-0. [DOI] [PubMed] [Google Scholar]
- 13.Uyguner O., Kayserili H., Li Y., Karaman B., Nurnberg G., Hennies H., Becker C., Nurnberg P., Basaran S., Apak M.Y. A new locus for autosomal recessive non-syndromic mental retardation maps to 1p21.1-p13.3. Clin. Genet. 2007;71:212–219. doi: 10.1111/j.1399-0004.2007.00762.x. [DOI] [PubMed] [Google Scholar]
- 14.Li C., Wong W.H. DNA-chip analyzer (dChip) In: Parmigiani G., Garrett E.S., Irizarry R., Zeger S.L., editors. The Analysis of Gene Expression Data: Methods and Software. Springer; New York: 2003. [Google Scholar]
- 15.Zhao X., Li C., Paez J.G., Chin K., Janne P.A., Chen T.H., Girard L., Minna J., Christiani D., Leo C. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X., Weir B.A., LaFramboise T., Lin M., Beroukhim R., Garraway L., Beheshti J., Lee J.C., Naoki K., Richards W.G. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–5570. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 17.Linden M.G., Bender B.G., Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96:672–682. [PubMed] [Google Scholar]
- 18.Cottingham R.W., Jr., Idury R.M., Schaffer A.A. Faster sequential genetic linkage computations. Am. J. Hum. Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 19.Schaffer A.A., Gupta S.K., Shriram K., Cottingham R.W., Jr. Avoiding recomputation in linkage analysis. Hum. Hered. 1994;44:225–237. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]
- 20.Comeron J.M. A method for estimating the numbers of synonymous and nonsynonymous substitutions per site. J. Mol. Evol. 1995;41:1152–1159. doi: 10.1007/BF00173196. [DOI] [PubMed] [Google Scholar]
- 21.Li W.H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- 22.Pamilo P., Bianchi N.O. Evolution of the Zfx and Zfy genes: Rates and interdependence between the genes. Mol. Biol. Evol. 1993;10:271–281. doi: 10.1093/oxfordjournals.molbev.a040003. [DOI] [PubMed] [Google Scholar]
- 23.Tonk V.S., Jalal S.M., Gonzalez J., Kennedy A., Velagaleti G.V. Familial interstitial deletion of chromosome 4 (p15.2p16.1) Ann. Genet. 2003;46:453–458. doi: 10.1016/s0003-3995(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 24.Ou X.M., Jafar-Nejad H., Storring J.M., Meng J.H., Lemonde S., Albert P.R. Novel dual repressor elements for neuronal cell-specific transcription of the rat 5–HT1A receptor gene. J. Biol. Chem. 2000;275:8161–8168. doi: 10.1074/jbc.275.11.8161. [DOI] [PubMed] [Google Scholar]
- 25.Ou X.M., Lemonde S., Jafar-Nejad H., Brown C.D., Goto A., Rogaeva A., Albert P.R. Freud-1: A neuronal calcium-regulated repressor of the 5–HT1A receptor gene. J. Neurosci. 2003;23:7415–7425. doi: 10.1523/JNEUROSCI.23-19-07415.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barnes N.M., Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 27.Rogaevea A., Ou X.M., Jafar-Nejad H., Lemonde S., Albert P.R. Differential repression by Fred-1/CC2D1A at a polymorphic site in the dopamine-D2 receptor gene. J. Biol. Chem. 2007;282:20897–20905. doi: 10.1074/jbc.M610038200. [DOI] [PubMed] [Google Scholar]
- 28.Williamson M.P. The structure and function of proline rich-regions in proteins. Biochem. J. 1994;297:249–260. doi: 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burkhard P., Stetfeld J., Strelkov S.J. Coiled-coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 30.Shen X., Valencia C.A., Szostak J.W., Dong B., Liu R. Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. USA. 2007;102:5969–5974. doi: 10.1073/pnas.0407928102. [DOI] [PMC free article] [PubMed] [Google Scholar]