

Abstract
N-linked glycosylation is an essential posttranslational modification of proteins in eukaryotes. The substrate of N-linked glycosylation, dolichol pyrophosphate (DolPP)-GlcNAc2Man9Glc3, is assembled through a complex series of ordered reactions requiring the translocation of the intermediate DolPP-GlcNAc2Man5 structure across the endoplasmic-reticulum membrane. A young patient diagnosed with a congenital disorder of glycosylation characterized by an intracellular accumulation of DolPP-GlcNAc2Man5 was found to carry a homozygous point mutation in the RFT1 gene. The c.199C→T mutation introduced the amino acid substitution p.R67C. The human RFT1 protein shares 22% identity with its yeast ortholog, which is involved in the translocation of DolPP-GlcNAc2Man5 from the cytosolic into the lumenal side of the endoplasmic reticulum. Despite the low sequence similarity between the yeast and the human RFT1 proteins, we demonstrated both their functional orthology and the pathologic effect of the human p.R67C mutation by complementation assay in Δrft1 yeast cells. The causality of the RFT1 p.R67C mutation was further established by restoration of normal glycosylation profiles in patient-derived fibroblasts after lentiviral expression of a normal RFT1 cDNA. The definition of the RFT1 defect establishes the functional conservation of the DolPP-GlcNAc2Man5 translocation process in eukaryotes. RFT1 deficiency in both yeast and human cells leads to the accumulation of incomplete DolPP-GlcNAc2Man5 and to a profound glycosylation disorder in humans.
Introduction
N-linked glycosylation is a ubiquitous posttranslational modification of proteins in eukaryotes. N-glycans convey essential signals for the folding and intracellular trafficking of glycoproteins.1 N-glycans also influence the clearance of circulating glycoproteins2 and the stability of signaling proteins at the cell surface.3 A particular feature of N-glycans is that they are first assembled in the endoplasmic reticulum (ER) as lipid-linked oligosaccharides (LLO). This assembly proceeds through the sequential addition of monosaccharides to the growing LLO. The process begins with the addition of GlcNAc monophosphate to the lipid carrier dolichol phosphate (DolP) and ends with the formation of DolPP-GlcNAc2Man9Glc3. The oligosaccharide is transferred to selected asparagine residues of nascent glycoproteins.4 The assembly of LLO requires glycosyltransferases and their respective nucleotide- and dolichol-activated monosaccharide substrates, but it also requires several proteins that regulate the complex topology of the process. For example, the MPDU1 protein makes the donor substrates DolP-Man and DolP-Glc available for completion of the LLO beyond DolPP-GlcNAc2Man5.5–7 Similarly, it was previously shown that in yeast the Rft1 protein is essential for translocation of the cytosolically oriented intermediate DolPP-GlcNAc2Man5 into the ER lumen, where LLO assembly is completed.8
The pathway of LLO assembly is strongly conserved among eukaryotes. Orthologous genes can be found from yeasts to humans for all glycosyltransferases involved. However, it is unclear whether the same degree of conservation applies to the accessory proteins. In fact, whereas MPDU1 orthologs can be found in metazoan and plant genomes, no orthologous gene can be identified in the yeast genome. In the case of RFT1, only genes with limited sequence similarity can be retrieved from the genome of higher organisms, thus casting doubt upon the functional significance of RFT1 in the assembly of LLO in general.
The identification of N-linked glycosylation disorders in humans, often referred to as congenital disorders of glycosylation (CDG), has demonstrated the conservation of the LLO-assembly pathway between yeasts and humans.9 The expression of human glycosyltransferase genes in glycosylation mutant yeasts demonstrated both the orthology of the glycosyltransferases in question and the pathological effect of the mutations identified in CDG cases.10–13
In spite of the extensive use of the yeast as a road map, many cases of CDG have remained untyped. Clinically, these cases present the symptoms typically seen in CDG patients, i.e., neurological abnormalities, failure to thrive, and varying degrees of dysmorphism.7,14 In the present study, we have identified a novel glycosylation defect in such an untyped CDG case, thereby establishing the importance of the RFT1 protein in human N-linked glycosylation.
Material and Methods
LLO and NLO Analysis
The investigation of the patient material was approved by the Ethical Commission of the Kanton Zürich. Fibroblasts were grown in DMEM (GIBCO) containing 25 mM Glc and 10% FCS until 90% of confluence. At that point, the fibroblasts were rinsed in PBS and incubated in Glc- and FCS-free DMEM for 45 min, then labeled by addition of 150 μCi of [3H]-Man (54.0 Ci/mmol, Amersham Bioscience) for 60 min. LLO and N-linked oligosaccharides (NLO) were isolated from labeled fibroblasts by chloroform-methanol-water extraction as described previously.13 Oligosaccharides were released from LLO by mild acid hydrolysis and from NLO by N-glycosidase F (New England BioLabs) digestion,13 then subjected to HPLC.15
Mutation Analysis
Total RNA and genomic DNA were isolated from 2 × 107 fibroblasts and 5 ml blood samples, respectively, with the TRIzol LS reagent (Invitrogen) used according to the manufacturer's instructions. The human RFT1 cDNA was prepared from 2 μg of total RNA with the primer 5′-GGGCTTTTGGTCTTCACT-3′ and 2 units of Omniscript reverse transcriptase (QIAGEN). The 20 μl reaction mixtures were incubated at 37°C for 1 hr. The protein-coding region of the human RFT1 cDNA was amplified by PCR from 2 μl of RT product with the primers 5′-GGCGGCATTTCCTGGTGTCT-3′ and 5′-TGGCACTCTCTGGTGCCTCATC-3′. The exon 3 of the human RFT1 gene was amplified by PCR from 50 ng of genomic DNA with the primers 5′-GGGCAATTCAGCTTTAGG-3′ and 5′-CACCACCAGTGGTTTATG-3′. The PCR products were sequenced (Synergene Biotech, Switzerland) after removal of the unincorporated nucleotides with QIAquick columns (QIAGEN). The presence of the mutation was confirmed by detection of the Pstl site, created by the c.199C→T mutation in exon 3.
Plasmid Construction
For construction of the lentiviral expression vector, the human RFT1 cDNA was subcloned as a PCR fragment flanked by SpeI and XhoI restriction sites into the NheI and SalI sites of the pLenti6-EGFP plasmid (Invitrogen), thus yielding the pLenti6-hRFT1 vector. The yeast rft1 gene with promoter and terminator sequences was amplified from S. cerevisiae genomic DNA by PCR and ligated into YCplac3316,17 with the inserted 5′-PstI and 3′-BamHI restriction sites to generate YCplac33-ScRFT1. The pTSV30A-ScRFT116,17 plasmid was obtained by subcloning of ScRFT1 from YCplac33-ScRFT1 with SacI and BamHI restriction sites. Plasmid YCplac33-pGAL1 was constructed by amplification of the GAL1 promoter from pYES2 (Invitrogen) and ligation of the PCR product into YCplac33 with the inserted restriction sites 5′-HindIII and 3′-XbaI. The human RFT1 cDNA was obtained via the EST clone IMAGE: 6422683 (Geneservice, UK). For construction of the plasmid YCplac33 pGAL1-hRFT1, the human RFT1 ORF was amplified from the IMAGE clone by PCR and cloned into plasmid YCplac33 pGAL1 with the inserted restriction sites 5′-XbaI and 3′-SacI. The mutant RFT1[R67C] cDNA from the CDG patient was subcloned into YCplac33 pGAL1 hRFT1, resulting in YCplac33-pGAL1-hRFT1[R67C].
Complementation of rft1Δ Yeast Mutants
Yeast strains W1536 5B (MATa, ade2Δ, ade3Δ, can1-100, his3-11,15, leu2-3, 112, trp1-1, ura3-1) and W1536 8B (isogenic MATα strain) have been described earlier.17 The diploid W1536a/α was obtained by the mating of W1536 5B and W1536 8B. W1536 5B-rft1Δ was generated by introduction of the rft1::KanMX4 gene-replacement cassette into W1536a/α and selection for geneticin resistance followed by tetrad dissection on a Singer MSM manual-dissection microscope (Singer Instrument). Haploid W1536 5B-rft1Δ mutant cells carry plasmid-borne rft1 in order to be viable. The yeast cells were grown on either rich YPD (1% yeast extract, 2% peptone, and 2% D-Glc), YPGalD (1% yeast extract, 2% peptone, 2% D-Gal, and 0.05% D-Glc), YPR (1% yeast extract, 2% peptone, and 2% D-raffinose), or synthetic complete media (Sigma or QBiogene) lacking one or more nutrients. The colony-sectoring method has been described by Bender et al.18 CPY immunoblotting was performed as described elsewhere.19
Lentiviral-Mediated RFT1 Expression
HEK293T cells (3 × 106) were transfected with 20 μg of pLenti6-hRFT1 and 36 μg of the packing-plasmid mix (Invitrogen) via calcium-phosphate precipitation. Eight hours after transfection, the medium was replaced with fresh DMEM containing 10% FCS. The cell supernatant was collected after 48 hr, and lentiviruses were harvested by centrifugation at 3000 × g for 5 min and filtration through 0.45 μm membranes (Schleicher & Schuell, Germany). CDG and healthy-control fibroblasts were infected with recombinant lentiviral particles including the human RFT1 cDNA or the EGFP gene as controls. Infected cells were selected with 5 μg/ml blasticidin (Invitrogen) for 10 days.
Results
A young girl was diagnosed with a disorder of N-linked glycosylation on the basis of the detection of abnormal isoelectric focusing of serum transferrin.20 The patient, designated by the abbreviation KS,21 showed symptoms often encountered in CDG, namely, a marked developmental delay, hypotonia, seizures, hepatomegaly, and coagulopathy.21 Phosphomannomutase and phosphomannose isomerase deficiencies were ruled out by the performance of enzymatic testing (data not shown). To determine whether the glycosylation disorder was related to a defect of LLO assembly, we analyzed the LLO composition in the healthy-control and patient fibroblasts. The LLO profile of control fibroblasts was dominated by the full-length DolPP-GlcNAc2Man9Glc3 (Figure 1A). By contrast, the profile of the CDG patient was marked by an accumulation of the intermediate LLO DolPP-GlcNAc2Man5 and a strong reduction of complete LLO DolPP-GlcNAc2Man9Glc3 (Figure 1B). The analyses of NLO produced after 1 hr labeling of healthy-control and CDG fibroblasts with [3H]Man were indistinguishable. Both profiles showed peaks corresponding to GlcNAc2Man8 and GlcNAc2Man9 oligosaccharides (Figures 1C and 1D), which are normally found on glycoproteins after the trimming of the N-linked glycans by the ER glucosidases I and II and by the ER mannosidase. The absence of GlcNAc2Man5 in the NLO profile of the CDG patient suggested that only the full-length LLO DolPP-GlcNAc2Man9Glc3 was transferred to glycoproteins. This phenotype was reminiscent of the LLO and NLO profiles described in yeast depleted for the Rft1 protein.8 In conditions with limiting RFT1 activity, DolPP-GlcNAc2Man5 accumulates at the cytosolic side of the ER membrane, whereas the small amounts of flipped oligosaccharide are extended to DolPP-GlcNAc2Man9Glc3 and transferred to proteins. This results in the underglycosylation of N-glycoproteins.
Figure 1.
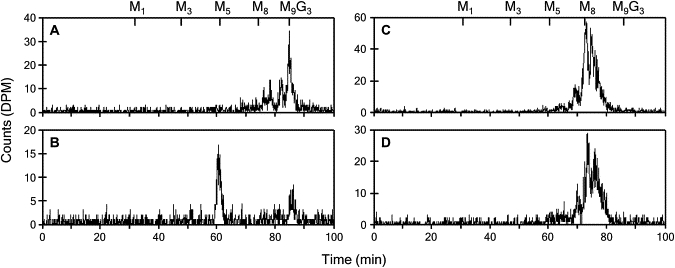
Lipid-Linked and N-Linked Oligosaccharide Profiles
The LLO isolated from healthy (A) and CDG (B) fibroblasts were separated by HPLC, demonstrating the abnormal accumulation of the LLO DolPP-GlcNAc2Man5 in the CDG sample. The NLO isolated from healthy (C) and CDG (D) fibroblasts were identical, showing that complete oligosaccharides were transferred to nascent proteins in the CDG cells. The retention times of DolPP-GlcNAc2Man1 (M1) to DolPP-GlcNAc2Man9Glc3 (M9G3) derived from a yeast standard LLO are marked at the top of the profiles.
The search for a human ortholog to the yeast Rft1 protein pointed to a single gene (GenBank, NM_052859) that encodes a protein of 541 amino acids sharing 22% identity with yeast Rft1. Sequencing of the NM_052859 cDNA in the CDG fibroblasts revealed a C-to-T transition at nucleotide position 199 (Figure 2A). This point mutation led to the amino acid substitution p.R67C in the human RFT1 protein ortholog. Analysis of the NM_052859 gene in the DNA of the CDG patient's parents confirmed the heterozygosity (Figure 2B). The c.199C→T mutation was not seen in 210 control chromosomes of European origin, thus excluding this mutation as a single-nucleotide polymorphism. The p.R67C mutation was localized to a 50-amino-acid-long hydrophilic stretch in the overall hydrophobic Rft1 protein ortholog (Figure 2C).
Figure 2.
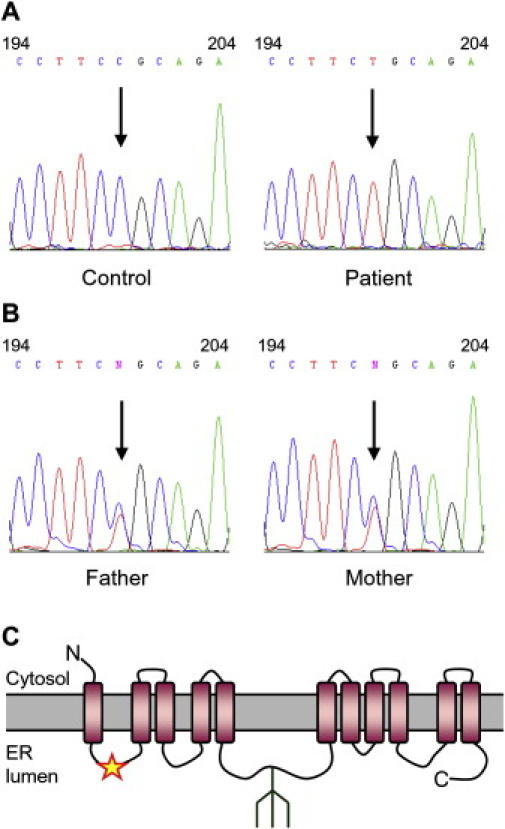
Mutation Analysis of Human RFT1 in CDG
(A) Electropherograms of RFT1 cDNA surrounding nucleotide position 199, marked by an arrow, sequenced from healthy-control and patient cDNA.
(B) The same region was sequenced from the genomic DNA of the CDG patient's parents, showing the heterozygozity for the c.199C→T mutation.
(C) The resulting p.R67C substitution, marked with a star, was predicted to be localized in a hydrophilic loop within the highly hydrophobic RFT1 protein. A potential N-glycosylation site detected at position N227 is shown schematically.
Genes encoding proteins with varying degrees of similarity to the human RFT1 protein can be retrieved from all eukaryote genomes analyzed, for which the sequence identity ranges from 87% for the mouse ortholog down to 17% for fungal orthologs (Figure 3). Although the overall protein sequence identity is limited, some regions of RFT1 are strongly conserved across species. It is noteworthy that the region composing the p.R67C mutation was strongly conserved and that several arginine residues, including R67 in the human protein, were found in all sequences analyzed (Figure 3), suggesting the importance of these amino acids for proper functionality.
Figure 3.

RFT1 Protein-Sequence Comparison
Amino acid sequences of proteins derived from Homo sapiens, Mus musculus, Danio rerio, and Caenorhabditis elegans showing similarity to the Saccharomyces cerevisiae RFT1 protein as performed by ClustalW analysis.30 Residues conserved in all five species are shown in black. The R residue at position 67 in the human RFT1 protein (see arrow) is conserved in the five species. Total amino acid identity with the human sequence ranged from 87% for the mouse protein down to 22% for the S. cerevisiae Rft1 protein.
Growth of the yeast strain W1536-5B-rft1Δ relies on the presence of the yeast rft1 gene on the plasmid pTSV30A-ScRFT1. Cells of this strain background carrying this plasmid develop a red pigment, due to the presence of the ADE3 gene on pTSV30A, whereas loss of this plasmid results in white cells.18 Accordingly, W1536-5B-rft1Δ/pTSV30A-ScRFT1 forms colonies that are uniformly red. This strain was transformed with the additional plasmid YCplac33pGAL1-hRFT1 that leads to expression of the human RFT1 cDNA controlled by the galactose-inducible GAL1 promoter. Such cells formed colonies that contain white sectors, due to the fact that the pTSV30A-ScRFT1 plasmid was no longer essential for growth and the plasmid could be lost, as visualized by the white sectors (Figure 4A). This color change indicated functional complementation of the rft1 defect by expression of the human RFT1 cDNA. This experiment demonstrated that the investigated human cDNA does indeed encode the orthologous protein to yeast Rft1. The same experiment performed with a plasmid expressing the CDG RFT1[R67C] allele did not yield any sectoring colonies (Figure 4A), thus demonstrating that the p.R67C mutation led to reduced function even in the yeast system. As shown previously,8 Rft1 depletion in yeast leads to the underglycosylation of the vacuolar N-linked glycoprotein carboxypeptidase Y. When W1536 5B-rft1Δ yeasts were complemented with YCplac33 pGAL1-hRFT1, a normal carboxypeptidase Y glycosylation profile was restored (Figure 4B), thus showing that the effect of the human RFT1 on rft1Δ yeast was indeed related to N-glycosylation.
Figure 4.
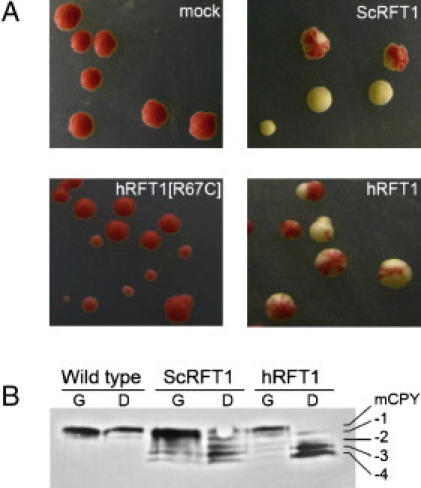
Complementation of rft1Δ Yeasts
(A) W1536 5B rft1Δ/pTSV30A-ScRFT1 cells transformed with a plasmid carrying a complementing RFT1 variant will form sectored colonies as they are allowed to lose the red pigment-inducing pTSV30A -ScRFT1 plasmid. Cells were transformed with: YCplac33 (mock), YCp33 GAL ScRFT1, YCp33 GAL hRFT1[R67C], and YCp33 GAL hRFT1.
(B) Immunoblotting analysis of the yeast strain W1536 rftΔ transformed with yeast and human RFT1 expressed from the yeast GAL1 promoter showed near wild-type levels of carboxypeptidase Y glycosylation when grown on 2% Gal [G] and an accumulation of underglycosylated isoforms when RFT1 expression was repressed by 4% Glc [D]. Carbon source did not affect carboxypeptidase Y glycosylation in wild-type yeast.
Finally, we introduced a normal human RFT1 cDNA in the fibroblasts of the CDG patient to demonstrate that the glycosylation disorder was the consequence solely of the identified RFT1 mutation. Healthy-control and CDG fibroblasts were infected with recombinant lentiviruses expressing either the normal RFT1 cDNA or EGFP as a negative control. The analysis of LLO profiles in the infected fibroblasts showed an increased formation of the full-length DolPP-GlcNAc2Man9Glc3 and a reduced presence of DolPP-GlcNAc2Man5 (Figure 5A), whereas the EGFP expression control had no effect on the LLO profile (Figure 5B). The expression of either normal human RFT1 or EGFP in healthy control fibroblasts had no effect on the LLO profiles (data not shown).
Figure 5.
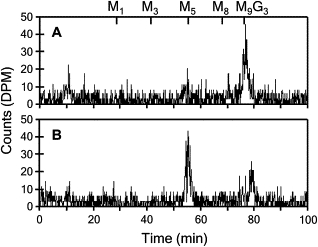
Lentiviral-Mediated Complementation in CDG Fibroblasts
The LLO profiles of fibroblasts infected with recombinant lentiviruses expressing either human RFT1 (A) or EGFP as negative control (B) were analyzed. The profiles show that the expression of the normal RFT1 cDNA in CDG fibroblasts restored the synthesis of the complete LLO DolPP-GlcNAc2Man9Glc3. The expression of EGFP in CDG fibroblasts had no effect on the pathologic profile characterized by the accumulation of DolPP-GlcNAc2Man5. The retention times of DolPP-GlcNAc2Man1 (M1) to DolPP-GlcNAc2Man9Glc3 (M9G3) are marked at the top of the profiles.
Discussion
The identification of a human glycosylation disorder associated with an RFT1 defect underlines the functional conservation of the RFT1 protein in eukaryotes. In spite of a limited sequence similarity between the yeast and the human RFT1 proteins, RFT1 deficiency led to identical biochemical phenotypes in both species, i.e., to an accumulation of the LLO DolPP-GlcNAc2Man5 and to a severe protein underglycosylation. Clinically, the RFT1-deficient CDG patient presented with symptoms frequently encountered in CDG patients, namely, failure to thrive, psychomotor retardation, seizures, hypotonia, and coagulopathy. Among the various types of CDG, the clinical severity of the RFT1 deficiency resembled the diseases caused by ALG3 and DPM1 deficiencies, also known as CDG-Id22 and CDG-Ie,23,24 respectively. The ALG3 gene encodes the DolP-Man-dependent mannosyltransferase that catalyzes the elongation of LLO from DolPP-GlcNAc2Man5 to DolPP-GlcNAc2Man6.25 The DPM1 protein is the catalytic subunit of the DolP-Man synthase complex.26 In CDG-Ie, the shortage in DolP-Man impairs the elongation of the LLO DolPP-GlcNAc2Man5 in the ER lumen. Although DPM1, ALG3, and RFT1 defects all lead to the accumulation of the LLO DolPP-GlcNAc2Man5, their impact on protein N-glycosylation is expected to be different. In fact, an RFT1 defect yields complete LLO structures for the transfer to proteins, yet in limited amount, whereas ALG3 and DPM1 defects yield low amounts of complete LLO combined with the lumenal accumulation of the DolPP-GlcNAc2Man5. The clinical similarities among these three glycosylation defects suggest that the limited availability of complete LLO alone dictates the extent of the clinical manifestations. In the case of DPM1 deficiency, the decreased DolP-Man availability is expected to also affect O-mannosylation and GPI-anchor formation. Yet, the comparison between the clinical features of DPM1 deficiency and those of ALG3 and RFT1 deficiencies suggests that the majority of the symptoms are due to abnormal N-glycosylation.
The functional conservation of the yeast and human RFT1 proteins emphasizes the essential role of RFT1 in lower and higher eukaryotes. However, the function of the RFT1 protein still remains unclear. The contribution of RFT1 to the specific translocation of DolPP-GlcNAc2Man5 suggests that it may function as a flippase,8 although such an activity could not be confirmed in vitro. A similar uncertainty relates to the function of the MPDU1 protein,5 which is involved in making DolP-Man and DolP-Glc substrates available to ER lumenal mannosyl- and glucosyltransferases. It is presently unclear whether MPDU1 affects the flipping of these substrates across the ER membrane or whether MPDU1 affects their local concentration by a different mechanism. It is noteworthy that a deficiency of MPDU1 in humans leads to CDG, which is associated with the parallel accumulation of LLO DolPP-GlcNAc2Man5 and DolPP-GlcNAc2Man9.6,7
To date, 21 forms of CDG have been divided into 13 types of CDG-I and eight types of CDG-II on the basis of the nature of the glycosylation defects.27–29 In accordance with the nomenclature guidelines established previously,27 we propose to name the RFT1 deficiency CDG-In.
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We thank Peter Clayton for the clinical assessment of the CDG patient. F.M.P. and A.J.K. would like to thank Kalervo Hiltunen for his help with the project. This work was supported by the Körber Foundation and by grants from the Swiss National Science Foundation to T.H. (PP00A-106756) and to M.A. (3100A0-105541), as well as by grants from the Academy of Finland and the Sigrid Juselius Foundation to F.M.P. and A.J.K.
References
- 1.Helenius A., Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 2.Mi Y., Shapiro S.D., Baenziger J.U. Regulation of lutropin circulatory half-life by the mannose/N-acetylgalactosamine-4-SO4 receptor is critical for implantation in vivo. J. Clin. Invest. 2002;109:269–276. doi: 10.1172/JCI13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chantret I., Dancourt J., Dupre T., Delenda C., Bucher S., Vuillaumier-Barrot S., Ogier de Baulny H., Peletan C., Danos O., Seta N. A deficiency in dolichyl-P-glucose:Glc1Man9GlcNAc2-PP-dolichyl alpha3-glucosyltransferase defines a new subtype of congenital disorders of glycosylation. J. Biol. Chem. 2003;278:9962–9971. doi: 10.1074/jbc.M211950200. [DOI] [PubMed] [Google Scholar]
- 4.Burda P., Aebi M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta. 1999;1426:239–257. doi: 10.1016/s0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- 5.Anand M., Rush J.S., Ray S., Doucey M.A., Weik J., Ware F.E., Hofsteenge J., Waechter C.J., Lehrman M.A. Requirement of the Lec35 gene for all known classes of monosaccharide-P-dolichol-dependent glycosyltransferase reactions in mammals. Mol. Biol. Cell. 2001;12:487–501. doi: 10.1091/mbc.12.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kranz C., Denecke J., Lehrman M.A., Ray S., Kienz P., Kreissel G., Sagi D., Peter-Katalinic J., Freeze H.H., Schmid T. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If) J. Clin. Invest. 2001;108:1613–1619. doi: 10.1172/JCI13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schenk B., Imbach T., Frank C.G., Grubenmann C.E., Raymond G.V., Hurvitz H., Korn-Lubetzki I., Revel-Vik S., Raas-Rotschild A., Luder A.S. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J. Clin. Invest. 2001;108:1687–1695. doi: 10.1172/JCI13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helenius J., Ng D.T., Marolda C.L., Walter P., Valvano M.A., Aebi M. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature. 2002;415:447–450. doi: 10.1038/415447a. [DOI] [PubMed] [Google Scholar]
- 9.Aebi M., Hennet T. Congenital disorders of glycosylation: Genetic model systems lead the way. Trends Cell Biol. 2001;11:136–141. doi: 10.1016/s0962-8924(01)01925-0. [DOI] [PubMed] [Google Scholar]
- 10.Imbach T., Burda P., Kuhnert P., Wevers R.A., Aebi M., Berger E.G., Hennet T. A mutation in the human ortholog of the Saccharomyces cerevisiae ALG6 gene causes carbohydrate-deficient glycoprotein syndrome type-Ic. Proc. Natl. Acad. Sci. USA. 1999;96:6982–6987. doi: 10.1073/pnas.96.12.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grubenmann C.E., Frank C.G., Kjaergaard S., Berger E.G., Aebi M., Hennet T. ALG12 mannosyltransferase defect in congenital disorder of glycosylation type lg. Hum. Mol. Genet. 2002;11:2331–2339. doi: 10.1093/hmg/11.19.2331. [DOI] [PubMed] [Google Scholar]
- 12.Frank C.G., Grubenmann C.E., Eyaid W., Berger E.G., Aebi M., Hennet T. Identification and functional analysis of a defect in the human ALG9 gene: Definition of congenital disorder of glycosylation type-IL. Am. J. Hum. Genet. 2004;75:146–150. doi: 10.1086/422367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grubenmann C.E., Frank C.G., Hülsmeier A.J., Schollen E., Matthijs G., Mayatepek E., Berger E.G., Aebi M., Hennet T. Deficiency of the first mannosylation step in the N-glycosylation pathway causes congenital disorder of glycosylation type Ik. Hum. Mol. Genet. 2004;13:535–542. doi: 10.1093/hmg/ddh050. [DOI] [PubMed] [Google Scholar]
- 14.Wu X., Rush J.S., Karaoglu D., Krasnewich D., Lubinsky M.S., Waechter C.J., Gilmore R., Freeze H.H. Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij. Hum. Mutat. 2003;22:144–150. doi: 10.1002/humu.10239. [DOI] [PubMed] [Google Scholar]
- 15.Zufferey R., Knauer R., Burda P., Stagljar I., te Heesen S., Lehle L., Aebi M. STT3, a highly conserved protein required for yeast oligosaccharyl transferase activity in vivo. EMBO J. 1995;14:4949–4960. doi: 10.1002/j.1460-2075.1995.tb00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gietz R.D., Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 17.Kastaniotis A.J., Autio K.J., Sormunen R.T., Hiltunen J.K. Htd2p/Yhr067p is a yeast 3-hydroxyacyl-ACP dehydratase essential for mitochondrial function and morphology. Mol. Microbiol. 2004;53:1407–1421. doi: 10.1111/j.1365-2958.2004.04191.x. [DOI] [PubMed] [Google Scholar]
- 18.Bender A., Pringle J.R. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burda P., te Heesen S., Brachat A., Wach A., Dusterhoft A., Aebi M. Stepwise assembly of the lipid-linked oligosaccharide in the endoplasmic reticulum of Saccharomyces cerevisiae: Identification of the ALG9 gene encoding a putative mannosyl transferase. Proc. Natl. Acad. Sci. USA. 1996;93:7160–7165. doi: 10.1073/pnas.93.14.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stibler H., Holzbach U., Kristiansson B. Isoforms and levels of transferrin, antithrombin, alpha(1)-antitrypsin and thyroxine-binding globulin in 48 patients with carbohydrate-deficient glycoprotein syndrome type I. Scand. J. Clin. Lab. Invest. 1998;58:55–61. doi: 10.1080/00365519850186832. [DOI] [PubMed] [Google Scholar]
- 21.Imtiaz F., Worthington V., Champion M., Beesley C., Charlwood J., Clayton P., Keir G., Mian N., Winchester B. Genotypes and phenotypes of patients in the UK with carbohydrate-deficient glycoprotein syndrome type 1. J. Inherit. Metab. Dis. 2000;23:162–174. doi: 10.1023/a:1005669900330. [DOI] [PubMed] [Google Scholar]
- 22.Korner C., Knauer R., Stephani U., Marquardt T., Lehle L., von Figura K. Carbohydrate deficient glycoprotein syndrome type IV: Deficiency of dolichyl-P-Man:Man(5)GlcNAc(2)-PP-dolichyl mannosyltransferase. EMBO J. 1999;18:6816–6822. doi: 10.1093/emboj/18.23.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imbach T., Schenk B., Schollen E., Burda P., Stutz A., Grunewald S., Bailie N.M., King M.D., Jaeken J., Matthijs G. Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J. Clin. Invest. 2000;105:233–239. doi: 10.1172/JCI8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S., Westphal V., Srikrishna G., Mehta D.P., Peterson S., Filiano J., Karnes P.S., Patterson M.C., Freeze H.H. Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie) J. Clin. Invest. 2000;105:191–198. doi: 10.1172/JCI7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aebi M., Gassenhuber J., Domdey H., te Heesen S. Cloning and characterization of the ALG3 gene of Saccharomyces cerevisiae. Glycobiology. 1996;6:439–444. doi: 10.1093/glycob/6.4.439. [DOI] [PubMed] [Google Scholar]
- 26.Maeda Y., Tanaka S., Hino J., Kangawa K., Kinoshita T. Human dolichol-phosphate-mannose synthase consists of three subunits, DPM1, DPM2 and DPM3. EMBO J. 2000;19:2475–2482. doi: 10.1093/emboj/19.11.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aebi M., Helenius A., Schenk B., Barone R., Fiumara A., Berger E.G., Hennet T., Imbach T., Stutz A., Bjursell C. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: An updated nomenclature for CDG. First International Workshop on CDGS. Glycoconj J. 1999;16:669–671. doi: 10.1023/a:1017249723165. [DOI] [PubMed] [Google Scholar]
- 28.Freeze H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006;7:537–551. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 29.Jaeken J., Matthijs G. Congenital Disorders of Glycosylation: A Rapidly Expanding Disease Family. Annu. Rev. Genomics Hum. Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- 30.Thompson J.D., Higgins D.G., Gibson T.J. Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]