

Abstract
Many genetic diseases have been linked to the dysfunction of primary cilia, which occur nearly ubiquitously in the body and act as solitary cellular mechanosensory organelles. The list of clinical manifestations and affected tissues in cilia-related disorders (ciliopathies) such as nephronophthisis is broad and has been attributed to the wide expression pattern of ciliary proteins. However, little is known about the molecular mechanisms leading to this dramatic diversity of phenotypes. We recently reported hypomorphic NPHP3 mutations in children and young adults with isolated nephronophthisis and associated hepatic fibrosis or tapetoretinal degeneration. Here, we chose a combinatorial approach in mice and humans to define the phenotypic spectrum of NPHP3/Nphp3 mutations and the role of the nephrocystin-3 protein. We demonstrate that the pcy mutation generates a hypomorphic Nphp3 allele that is responsible for the cystic kidney disease phenotype, whereas complete loss of Nphp3 function results in situs inversus, congenital heart defects, and embryonic lethality in mice. In humans, we show that NPHP3 mutations can cause a broad clinical spectrum of early embryonic patterning defects comprising situs inversus, polydactyly, central nervous system malformations, structural heart defects, preauricular fistulas, and a wide range of congenital anomalies of the kidney and urinary tract (CAKUT). On the functional level, we show that nephrocystin-3 directly interacts with inversin and can inhibit like inversin canonical Wnt signaling, whereas nephrocystin-3 deficiency leads in Xenopus laevis to typical planar cell polarity defects, suggesting a role in the control of canonical and noncanonical (planar cell polarity) Wnt signaling.
Introduction
Cystic kidney diseases (CKDs) are a clinically and genetically heterogeneous group of disorders that may present in utero or be clinically silent well into adulthood.1 Recent molecular findings lead to the development of novel therapeutic strategies that are currently under investigation regarding the retardation of renal failure in CKD.2 Interestingly, most products of CKD genes, also referred to as cystoproteins, colocalize in multimeric complexes at distinct subcellular structures of the primary cilium or ciliary base comprising basal bodies and centrosomes.1 Accordingly, the term “ciliopathies” has been proposed to subsume this class of disorders, and cilia dysfunction and common pathogenic pathways may provide a molecular basis for overlapping phenotypes in ciliopathies.3,4 Consistent with this novel understanding of ciliopathies, mutations in ciliary genes have been identified in patients with a broad clinical spectrum of disorders, among others, nephronophthisis (NPHP [MIM 256100]), Joubert (JS; [MIM 213300]), and Meckel-Gruber (MKS; [MIM 249000]) syndrome.1,5–9
NPHP is a heterogeneous group of autosomal-recessive cystic kidney disorders that constitute the most frequent genetic cause of end-stage renal failure in children and young adults.1 As part of the proposed ciliary network, NPHP proteins are expressed in primary cilia, basal bodies, and/or centrosomes.1,10 We recently reported hypomorphic mutations in NPHP3 to be responsible for the adolescent type of nephronophthisis (MIM 604387).11 In some of these children and young adults, NPHP was associated with tapetoretinal degeneration called Senior-Loken syndrome (SLSN3; [MIM 606995]) and liver fibrosis. We further hypothesized that a homozygous Nphp3 missense mutation explains the phenotype in the polycystic kidney disease (pcy) mouse.11,12
In this study, we define the crucial role of nephrocystin-3 (NPHP3/Nphp3) for development in mice and man. We disrupted Nphp3 gene function by targeting of the murine Nphp3 gene. To demonstrate the pathogenic significance of the hypomorphic pcy mutation for the cystic kidney phenotype in this mouse model, we generated compound mutant Nphp3pcy/ko animals. Homozygous Nphp3-deficient (Nphp3ko/ko) animals were used for the determination of the role of nephrocystin-3 during early development and resulted in randomization of left-right body asymmetry, heterotaxia, and embryonic lethality. On the basis of our observations in mice, we investigated the role of NPHP3 for human congenital cystic kidney disease. We identified a broad spectrum of lethal and nonlethal phenotypes in patients with different NPHP3 mutations. Loss-of-function mutations were either lethal resulting in early embryonic patterning defects as seen in Meckel-Gruber-like syndrome or resulted in a severe congenital cystic kidney disease. On the basis of the clinical observation that NPHP3/Nphp3 mutations can cause situs inversus, we investigated whether nephrocystin-3 and inversin interact directly with each other and determine the role of nephrocystin-3 for canonical and noncanonical (planar cell polarity) Wnt signaling.
Material and Methods
Patients and Families
Signed and informed consent was obtained from patients and family members with protocols approved by the Institutional Ethics Review Board at the University of Aachen and collaborating institutions. In the families harboring NPHP3 mutations, DNA samples were available from all parents, healthy siblings, and all (except for the elder affected male in family F917 who died perinatally) affected individuals.
Haplotype Analysis and Genome-wide SNP Mapping
In the multiplex family F806 (second-degree consanguineous parents) linkage to MKS1-3 was excluded by haplotype analysis as described previously.13 Subsequently, genome-wide SNP mapping with the 50K Affymetrix SNP array (Affymetrix, Santa Clara, CA) and the ALLEGRO program was performed with the assumption of autosomal-recessive inheritance with complete penetrance and a disease allele frequency of 0.0001.
Mutation Analysis
NPHP3 mutation analysis was done by direct sequencing for the 27 exons encoding the 1330 amino acid (aa) nephrocystin-3 protein (GenBank: NM_153240, NP_694972; mutation numbering + 1 corresponds to the A of the ATG-translation-initiation codon). Genomic DNA from an affected individual was amplified by polymerase chain reaction (PCR) with oligonucleotide primers complementary to flanking intronic sequences (Table S1 available online). All detected splice and missense mutations were not present in 400 control chromosomes tested by denaturing high-performance liquid chromatography (DHPLC) or restriction digestion analysis.
RT-PCR
RNA was extracted from fresh blood samples from all members of family F960 with the QIAmp RNA Blood Mini Kit (QIAGEN, Hilden, Germany). The relative amount of NPHP3 messenger RNA (mRNA) was measured by reverse transcriptase (RT)-PCR and usage of the Reverse Transcription System (Promega, Mannheim, Germany) as described previously.13 For each reverse-transcriptase reaction, we used 500 ng of total RNA. The amount of PCR product was quantified by fractionation on a 2% agarose gel and measurement of ethidium bromide fluorescence. The different PCR products were gel extracted and purified with the QIAquick Gel Extraction Kit (QIAGEN) and finally sequenced as described before. Primer sequences and PCR conditions are available on request.
Generation of Nphp3 Mutant Mice by Gene Targeting
Nphp3 was mutated in HM1 ES cells with a replacement-type targeting vector. A murine 129/Sv genomic DNA cosmid library (obtained from the German Resource Center for Genome Research, RZPD, Berlin, Germany) was screened with a probe corresponding to exon 1 of Nphp3. One genomic clone that contained the entire Nphp3 locus (∼46 kb insert) was characterized further and used for the construction of the targeting vector. A positive selectable cassette was cloned between two homology regions (a 2.4 kb PstI fragment and a 3.7 kb SacI fragment).14 The cassette contains stop codons in all three frames, an independent ribosomal entry sequence (IRES) followed by the lacZ gene with an SV40 polyadenylation signal, and a neomycin phosphotransferase gene. By homologous recombination, the IRES-LacZ-neo cassette is inserted into exon 3 of Nphp3, and exons 4 and 5 and introns 3 and 4 are replaced (Figure S3). The Nphp3 ko allele was backcrossed to a C57BL/6 background. Nphp3pcy mice had a mixed genetic background (C57BL/6, KK/Upj-Ay/J, and CD1).
Analyses of Mutant Mice and Embryos
Mice and embryos were genotyped by PCR with genomic DNA isolated from tail biopsies or yolk sacs, respectively. In addition, lack of Nphp3 expression was confirmed by western-blot analysis (Figure S3). Adult tissues and embryos were isolated and processed for histological analyses (cryosections or paraffin embedding) according to standard procedures. Immunofluorescence stainings were performed as previously described.10 Ciliary axonemes were stained with antibodies against acetylated α-tubulin (Sigma, Taufkirchen, Germany). Secondary antibodies (Alexa Fluor 488) were from Molecular Probes (Invitrogen, Karlsruhe, Germany). Nuclei were stained with Hoechst33342 (Sigma). A Zeiss laser-scanning microscope (Axiovert 200 LSM510, Carl Zeiss, Jena, Germany) was used for confocal imaging. For cilia-length determination, the measurement and line-drawing tools in the overlay mode of the LSM image browser software (Zeiss) was used. For the determination of possible significance, a Student's t test was performed.
Coimmunoprecipitation
Coimmunoprecipitation experiments were performed as described.15 In brief, HEK293T cells were transiently transfected with the indicated plasmids by the calcium-phosphate method. After incubation for 24 hr, cells were washed twice and lysed in a 1% Triton X-100 lysis buffer. After centrifugation (15,000 × g, 15 min, 4°C) cell lysates containing equal amounts of total protein were incubated at 4°C with anti-FLAG (M2) agarose beads for approximately 2 hr. The beads were washed extensively with lysis buffer, and bound proteins were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and visualized with enhanced chemiluminescence after incubation of the blots with the respective antibodies. Experiments were repeated four times with identical results.
Luciferase Assay
Luciferase assays were performed as described previously.16 In short, HEK293T cells were seeded into 12-well plates and transiently transfected with either a TOPFlash or FOPFlash luciferase reporter construct, a β-galactosidase expression vector, and vectors directing the expression of the desired proteins, including Dishevelled-1, NPHP2/INVS, and NPHP3. Reporter constructs TOPFlash and FOPFlash were obtained from Upstate Biotechnology (Lake Placid, NY) and Millipore (Schwalbach, Germany). The total amount of DNA was adjusted to 10 μg per well. Cells were collected after 12 hr and lysed in 100 μl of reporter lysis buffer (Applied Biosystems) for 10 min at 4°C. After centrifugation of the lysates (13.500 rpm, 5 min) for the removal of insoluble material, we determined luciferase activitiy with a commerical assay system (Applied Biosystems) and normalized it to β-galactosidase activity to correct for transfection efficiency.
Xenopus laevis Embryo Manipulations
Eggs were obtained from Xenopus females, injected with 600–800 U of human chorionic gonadotropin, fertilized in vitro, and cultured in 0.1× Marc's modified Ringer's medium as described.17 Embryonic stages were determined according to Nieuwkoop and Faber.18 Two dorsal blastomeres were injected at the four-cell stage with mRNA or Morpholino antisense oligonucleotides NPHP3-MO (5′-TAACCAGAGAAGAGGCTGTCCCCAT-3′), or standard control-Morpholino (5′-GTGACTACAGAAATGCAAATGCAACAAATTC-3′) 8–32 ng per embryo (GeneTools). Capped synthetic RNA was generated with T7 RNA polymerase by in vitro transcription (mMessage mMachine kit, Ambion) of hNPHP3-pxT7 after linearization with SalI. For tracing cell movements during gastrulation, fluorescent dextran (70 kD) was injected into one dorsal blastomere at the 64-cell stage and cultured until gastrulation. Fluorescent and bright field images were taken with a Leica MZ16F stereomicroscope. Phenotypic defects were scored representing impaired convergent extension (ICE) as follows: CEI 0 (normal), no defects; CEI 1 (mild defects), less than 25% reduction of axis extension compared with uninjected embryos and mild dorsal flexure; CEI 2 (moderate defects), 25%–50% reduction of axis elongation; and CEI 3 (severe defects), shortening of more than 50%, head fused to tail or open neural folds.19 All experiments were approved by the institutional animal committee and the Regierungspräsidium Freiburg, Baden-Württemberg, Germany.
Results
In a consanguineous Turkish multiplex family (F806, logarithm of the odds [LOD] max 2.65) with Meckel-Gruber-like syndrome, we mapped the disease-causing locus to an 11.6 Mb interval on chromosome 3q21.2-q22.3 (Figure S1). The two affected females of this family had hepatobiliary ductal plate malformation and multicystic dysplastic kidneys (Figure 1) with oligo-anhydramnios consistent with renal malfunction incompatible with life. In addition, one of the girls presented with a Dandy-Walker malformation, the other with an aortic stenosis (for further details, see Table 1). Sequencing of candidate genes NEK11 (MIM 609779), CEP63 (NM_025180), and NPHP3 (MIM 608002) located in the minimal critical region identified a homozygous obligatory NPHP3 splice mutation affecting the highly conserved canonical acceptor splice site of intron 19 (c.2694-1_2 del), which segregated with the phenotype and is predicted to result in out-of-frame transcripts with premature termination of translation (Figure 2). In an African multiplex family (F888), we also detected by haplotype analysis homozygosity at the NPHP3 locus in both affected children whose phenotypes comprised enlarged multicystic-dysplastic kidneys with oligo-anhydramnios consistent with renal malfunction, multiple cysts of the liver and pancreas, ductal plate malformation, structural cardiac defects, chorioid plexus cysts, and a hypoplastic calvarium with enlarged fontanelles (Figure 1, Table 1). Both males died shortly after birth. Sequencing revealed the novel homozygous nonsense mutation c.1729C → T (p.Arg577X) in exon 11 of the NPHP3 gene (Figure 2).
Figure 1.
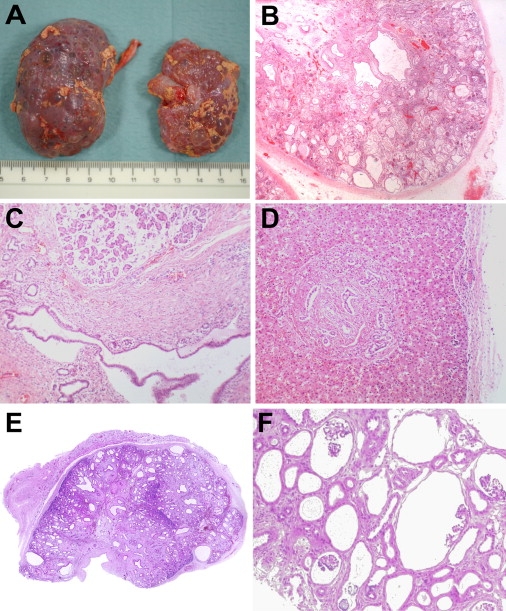
Pathology Findings in Patients with Loss-of-Function NPHP3 Mutations
(A–D) Autopsy findings in the second affected patient of family 888.
(A) Multicystic dysplastic kidneys. The smaller right kidney had a dilated pelvis due to ureteric atresia in utero. The left kidney had a patent, but thinned ureter and reduced urinary output.
(B) Higher-magnification histology (×20) shows a picture with renal cysts of variable size and location as well as interstitial fibrosis consistent with multicystic-dysplastic kidney disease.
(C) Congenital pancreatic disease with smaller and larger cysts and fibrosis. Pancreatic acini have normal appearance (×40).
(D) Liver histology reveals ductal plate malformation with hyperplastic biliary ducts and congenital hepatic fibrosis (×40).
(E) Low-magnification histology of a renal cross-section (×2.5) from the first affected patient of family 806 that is consistent with multicystic dysplastic kidney disease.
(F) Renal biopsy specimen of the proposita of family 960 displaying diffuse glomerulocystic kidney disease (×40). Please note glomerular structures within the cysts.
Table 1.
Phenotypes and Genotypes of Novel Patients Included in This Study
| Family | Origin | NPHP3 Mutations | Patient | Age or Death | CNS | Kidneys | Liver | Pancreas | Heart | Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|
| 806 (C) | Turkey | c.2694-1_2 del (P) | Female | US/TOP 23rd gw, postmortem | Dandy-Walker malformation | Enlarged multicystic dysplastic kidneys, oligohydramnios | Ductal plate malformation | Normal on autopsy | Normal on autopsy | Single cyst anterior part of bursa omentalis |
| c.2694-1_2 del (M) | ||||||||||
| 806 (C) | Turkey | c.2694-1_2 del (P) | Female | Perinatal death, respiratory insufficiency, kidney-liver biopsy | n.d. | Enlarged multicystic dysplastic kidneys, oligohydramnios | Ductal plate malformation | n.d. | Aortic stenosis | |
| c.2694-1_2 del (M) | ||||||||||
| 888 | Cameroon | c.1729C → T (p.Arg577X)(P) | Male | Death at 49 days | Neurologically very abnormal, muscular hypertonia, small subdural collections, cyst on floor of right ventricle, bilateral chorioid plexus cysts | Enlarged multicystic dysplastic kidneys | Liver cysts | n.d. | Atrial septal defect, persistent ductus arteriosus, right ventricular hypertrophy | |
| c.1729C → T (p.Arg577X) (M) | ||||||||||
| 888 | Cameroon | c.1729C → T (p.Arg577X)(P) | Male | Perinatal death, postmortem | Hypoplastic calvarium, extremely enlarged fontanelles | Enlarged multicystic dysplastic kidneys, anhydramnios | Ductal plate malformation | Cystic pancreas | PDA, nodular dysplasia of valve cusps | Atresia of right ureter, high forehead, small, deep set, abnormally folded ears different from typical Potter's sequence |
| c.1729C → T (p.Arg577X) (M) | ||||||||||
| 917 | Switzerland | c.2918G → A (p.Arg973Gln) (P) | Male | Perinatal death, respiratory insufficiency, postmortem | n.d. | Enlarged multicystic dysplastic kidneys, oligohydramnios | Ductal plate malformation | Normal on autopsy | Normal on autopsy | |
| c.3340C → T (p.Gln1114X) (M) | ||||||||||
| 917 | Switzerland | c.2918G → A (p.Arg973Gln) (P) | Male | 17 years | Normal neurological and cognitive assessment | Multicystic kidneys | Ductal plate malformation, biliary cirrhosis | Increased echogenicity | Normal | Hepatosplenomegaly, end-stage renal failure and peritoneal dialysis at 14 months, progressive liver cirrhosis and liver failure, combined liver-kidney transplantation at age 3 years |
| c.3340C → T (p.Gln1114X) (M) | ||||||||||
| 960 | Vietnam | c.1985+5G → A (P) | Female | 8 years | Normal neurological and cognitive assessment | Glomerulocystic kidney disease | Hepatopathy with cholestasis, cirrhosis and portal hypertension | Pancreatic amylase constantly increased, normal endocrine and exocrine function | Normal | Situs inversus totalis, bilateral preauricular fistulas, postaxial polydactyly left foot, bilateral inguinal hernia, end-stage renal failure and peritoneal dialysis at 3 months, combined liver-kidney transplantation at age 4 years |
| c.1985+5G → A (M) |
GenBank NM_153240; nucleotide +1 is the A of the ATG-translation initiation codon. The following abbreviations are used: parental consanguinity known (C), paternally inherited allele (P), maternally inherited allele (M), not determined (n.d.), gestational week (gw), ultrasound findings with subsequent termination of pregnancy (US/TOP), and congenital hepatic fibrosis with hyperplastic biliary ducts (ductal plate malformation).
Figure 2.
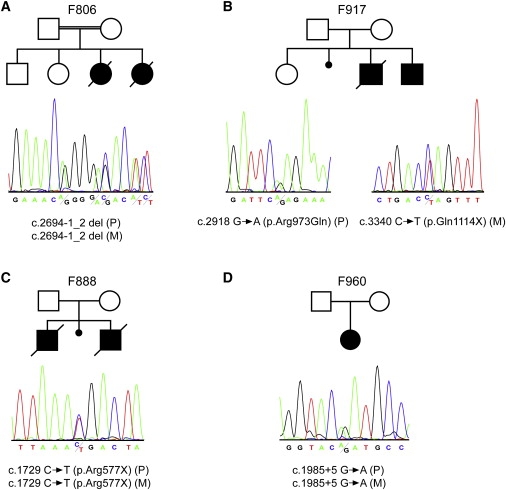
NPHP3 Mutations in Four Families
The upper panel shows the pedigree structure of the families, and the lower panel shows sequence chromatograms illustrating the predicted NPHP3 loss-of-function mutations in the described families. Mutations segregated with the disease status in each family (P indicates paternally inherited allele; M indicates maternally inherited allele). In case of a homozygous mutation, a heterozygous electropherogram of one of the parents is shown for reasons of clarity.
(A) Sequence chromatogram depicting the deletion of the canonical acceptor splice site nucleotides at position −1_2 of the intron 19 splice acceptor site predicted to result in out-of-frame transcripts and a premature stop.
(B) On the left, a sequence chromatograph of the affected individual of family 917 showing a heterozygous G → A exchange which predicts an p.Arg973Gln amino acid substitution is displayed. On the right, a heterozygous C → T exchange predicting a stop codon (p.Gln1114X) is shown.
(C) Sequence chromatograph showing a heterozygous C → T exchange that predicts a stop codon (p.Arg577X).
(D) Sequence chromatogram illustrating the heterozygous G → A exchange at position +5 of the intron 13 splice donor site resulting in skipping of exon 13 and an out-of-frame transcript that leads to premature termination of translation (Figure S2).
In contrast to these NPHP3 null alleles that are obviously incompatible with longer survival, we recently reported hypomorphic NPHP3 mutations in children and young adults with isolated adolescent nephronophthisis and associated hepatic fibrosis or tapetoretinal degeneration.11 To further delineate genotype-phenotype correlations for NPHP3, we performed sequence analyses in patients with overlapping phenotypes and identified mutations in two other families. Clinical data of a nonconsanguineous Swiss multiplex family (F917) with renal-hepatic-pancreatic dysplasia has been previously described in detail (Table 1).20 Paternally, in exon 20, the novel missense mutation c.2918G → A (p.Arg973Gln) that affects an evolutionarily highly conserved arginine residue and was not present among 400 control chromosomes was identified. Maternally, the novel nonsense mutation c.3340C → T (p.Gln1114X) was detected in exon 24 (Figure 2). In a Vietnamese family, we identified the novel homozygous splice mutation c.1985+5G → A that affects the donor splice site of intron 13 and was absent in 400 control chromosomes (Figure 2). Experiments on the RNA level confirmed the pathogenicity of this splice mutation revealing out-of-frame transcripts in the patient (Figure S2). The phenotype of the 8-year-old proposita is of particular interest because she demonstrates a wide range of early embryonic patterning/laterality defects with situs inversus totalis and postaxial polydactyly, as well as branchial arch maldevelopment with bilateral preauricular fistulas (Table 1). Moreover, she developed early renal failure and was on peritoneal dialysis since her third month of life. A combined kidney-liver transplantation was performed at the age of 4 years because of renal-hepatic-pancreatic dysplasia (Figure 2).
To validate and further delineate our data obtained in men, we generated an Nphp3 null allele by knockout technology (Figure S3). Heterozygous mice did not show any obvious morphological defects or behavioral abnormalities. Reproduction and life span were also within the normal range, thus ruling out any heterozygosity effect (the oldest animals analyzed for morphological defects by visual examination and cystic kidney disease by hematoxylin-eosin (HE) staining of cryosections were as follows: 21 weeks, n = 2; 31 weeks n = 2; 35 weeks n = 1; and 42 weeks n = 2). This is in line with our data in humans (parents of affected children) with NPHP3 heterozygosity that neither showed any abnormality. We then crossed these knockout mice with our Nphp3pcy/pcy mice harboring the hypomorphic missense variant I614S that leads to a renal-exclusive nephronophthisis phenotype. We considered the I614S variant a hypomorphic allele because the mouse phenotype is less severe than that observed in Nphp3-deficient mice. In addition, we demonstrate by western-blot analysis that nephrocystin-3 is still expressed in Nphp3pcy/pcy mice (data not shown). A total of 14 Nphp3pcy/wt and 14 Nphp3pcy/ko mice were analyzed (ten 4-week-old animals and four 12-week-old animals in each group). In contrast to wild-type, Nphp3ko/wt, and Nphp3pcy/wt mice, all 14 (seven females, seven males) compound-heterozygous animals (Nphp3pcy/ko) developed cystic kidneys. This is in line with our argument that Nphp3pcy indeed represents a hypomorphic Nphp3 allele that underlies the cystic kidney phenotype in these mice (Figure 3). Cysts in these animals were evident as early as 4 weeks after birth, and cyst size and number progressively increased with age. We did not compare severity of the cystic kidney disease phenotypes between Nphp3pcy/ko and Nphp3pcy/pcy mice because both alleles are not on identical genetic backgrounds, so modifier effects cannot be ruled out concerning disease severity.
Figure 3.
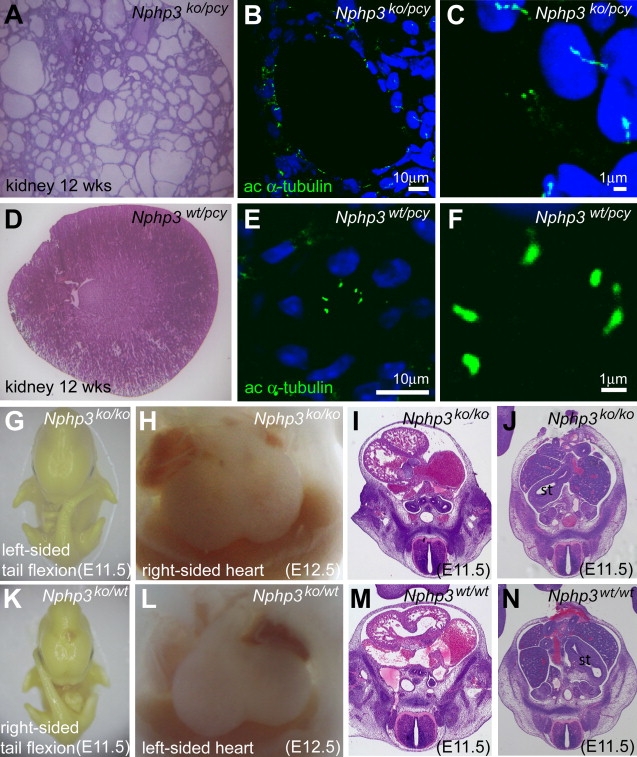
Cystic Kidney Disease in Compound-Heterozygous Nphp3pcy/ko Mice and Situs Inversus Totalis in Nphp3ko/ko Embryos
Compound-heterozygous Nphp3pcy/ko mice (A–C) develop cystic kidney disease, whereas control (data not shown) and monoallelic Nphp3pcy/ko (D–F) mice did not exhibit an obvious renal phenotype. Hematoxylin-eosin stainings of a cryosectioned kidney from a Nphp3pcy/ko mouse (A) and a Nphp3pcy/wt mouse (D) are shown. Immunofluorescence staining with antibodies against acetylated α-tubulin (green) as a cilia marker (nuclei, blue) of kidney cryosections from Nphp3pcy/ko mice (B) demonstrates that epithelial cells of renal cysts carry monocilia that are longer than renal monocilia from control mice (E). (C) and (F) show higher magnifications of the images shown in (B) and (E). Homozygous Nphp3ko/ko embryos (G–J) have situs inversus and die during embryonic development. Situs inversus in Nphp3ko/ko embryos is indicated by left-sided tail flexion (G) and mirror-image arrangement of the heart (H). Histological examination of Nphp3ko/ko embryos confirms dextrocardia (I) and position of the stomach on the right side (J), consistent with situs inversus totalis. Normal situs arrangement was noted in heterozygous Nphp3ko/wt and control embryos (K–N).
On the basis of our findings in patients with NPHP3 null alleles, we also hypothesized a much more severe phenotype in mice homozygous for Nphp3 loss-of-function mutations. Genotypic analyses of 70 mice (32 females, 38 males) derived from 11 intercrosses of heterozygous Nphp3wt/ko mice revealed a ratio of approximately 1:2:0 for wild-type (n = 20; 12 females, 8 males) to heterozygous (n = 50; 20 females, 30 males) to homozygous littermates at birth, consistent with embryonic lethality of Nphp3ko/ko mice. We then analyzed embryos (n = 308; derived from 39 pregnant females obtained by intercrosses of Nphp3wt/ko mice) at different stages of development to determine the time point of lethality and its underlying causes. In E9.5–E12.5 embryos (n = 221), we found left-sided tail flexion in approximately 17% (n = 37) of all analyzed specimens, indicating that embryonic turning is possibly inverted or randomized in Nphp3-deficient embryos (Figure 3). Genotype analyses of E9.5–E13.5 embryos (n = 148; including 36 Nphp3wt/wt and 83 Nphp3wt/ko embryos) revealed that Nphp3ko/ko embryos (n = 29) either had situs solitus (n = 8), situs reversal (n = 15),or inconsistent situs (n = 6) arrangements. In addition, some embryos could not be assessed because of intrauterine death and resorption or complex developmental defects. Comparison of histological stainings of serially sectioned E11.5–E12.5 Nphp3ko/ko embryos (n = 17) with wild-type and heterozygous littermates (n = 7) confirmed situs inversus totalis (dextroposition of the heart and dextroposition of the stomach; n = 7), situs solitus (n = 5) and heterotaxia (n = 5) as well as complex cardiac defects in Nphp3-deficient embryos (Figure 3). Thus, our results indicate that homozygous deletion of Nphp3 regularly results in randomization of left-right asymmetry and midgestational embryonic lethality, probably caused by associated complex cardiac defects.
The crucial role NPHP3/Nphp3 plays throughout development from early embryonic stages to maintenance of morphologic integrity in later life is illustrated by the wide range of renal changes in patients and mice with NPHP3/Nphp3 mutations. Hypomorphic NPHP3 alleles in individuals with late-onset (adolescent) nephronophthisis resulted in shrunken kidneys with only few cysts confined to the corticomedullary junction. Histologically, tubular dilatation and atrophy, interstitial fibrosis, and tubular basement membrane changes were present.11,12 In this study, we had the opportunity to analyze in detail the effects of truncating NPHP3 mutations on renal morphology. Surprisingly, we found a range of gross congenital renal abnormalities, including glomerulocystic kidney disease (Figure 1F) and enlarged multicystic-dysplastic kidneys with early embryonic renal failure (Figures 1B and 1E) (Table 1). Histologically, cysts were of manifold morphology and located in all segments of the nephron. In accordance with early embryonic maldevelopment, ureteric atresia was observed in one of our patients with predicted homozygous NPHP3 loss-of-function mutations.
Our previously reported in situ hybridization analyses are consistent with the reported broad spectrum of disease phenotypes demonstrating specific Nphp3 expression in embryonic cells of the node, brain, kidney tubules, biliary tract, and liver, all known to carry monocilia.11
To investigate whether cyst formation in Nphppcy/ko mice is caused by lack of cilia generation, we stained kidneys of 12-week-old control and mutant animals with antibodies directed against acetylated α-tubulin (Figure 3). High-resolution immunofluorescence image analyses demonstrated presence of renal monocilia in epithelial cells of normal and dilated renal tubules, thus ruling out a ciliogenesis defect in Nphppcy/ko mice. Interestingly, renal monocilia (n = 93; average length 4.87 μm; standard deviation [SD] 2.35) in the dilated kidney tubules of Nphppcy/ko mutant animals (n = 3) were longer than monocilia (n = 86; average length 1.62 μm; SD 0.57) of Nphppcy/wt control mice (n = 2) (Figure 3, p < 0.005), which may indicate a possible defect in cilia-length control.
We previously have demonstrated that nephrocystin (NPHP1) directly interacts with inversin (NPHP2) and nephrocystin-3 (NPHP3).11,21 Both NPHP proteins nephrocystin and inversin have been sublocalized to cilia and/or the ciliary base,10,21,22 suggesting a similar localization for nephrocystin-3. Close physical and functional interrelations between nephrocystin-3 and inversin are also suggested by overlapping disease phenotypes in patients and mice with NPHP3/Nphp3 and INVS/Invs mutations, respectively.21 This phenotypic similarity becomes even more evident by the fact that both inv and Nphp3ko/ko mice cause situs inversus and that both genes can result in hepatopancreatic and renal disease. To investigate whether inversin and nephrocystin-3 directly interact, we coexpressed different truncations of nephrocystin-3 with epitope-tagged inversin in HEK293T cells. V5-tagged inversin coprecipitated with nephrocystin-3 but not with control proteins (Figure 4). Truncations of nephrocystin-3 could possibly map the interaction to aa 1 to 603 (data not shown).
Figure 4.
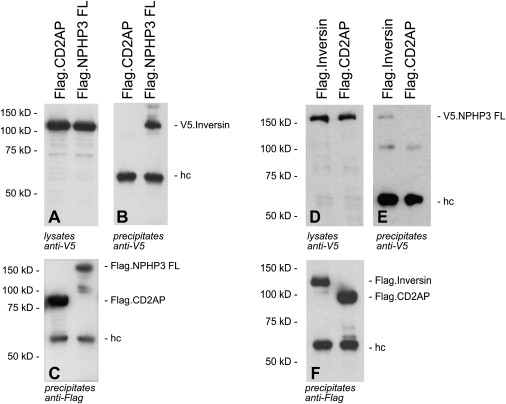
Interaction of Nephrocystin-3 and Inversin
(A–C) V5-tagged inversin was coexpressed with N-terminally FLAG-tagged full-length nephrocystin-3 (Flag.NPHP3 FL) or FLAG-tagged CD2AP protein (Flag.CD2AP) as a negative control. Expression of V5-tagged inversin in cellular lysates was confirmed by immunoblotting with V5 antibodies (A). After immunoprecipitation with FLAG antibody, coprecipitating V5-tagged inversin was detected with V5-specific antiserum (B). As control, FLAG-specific antibodies detected precipitated Flag.Inversin and Flag.CD2AP (C).
(D–F) In the reverse experiment, V5-tagged full-length nephrocystin-3 (V.5NPHP3 FL) was coexpressed with Flag-tagged inversin or FLAG-tagged CD2AP. The additional approximately 60 kDa band visible in all lanes (B, C, E, and F) represents the heavy chain. The 100 kDa bands that are visible in (E) are nonspecific bands that also appeared upon long exposure time in (B).
Additionally, both inversin and nephrocystin-3 were able to inhibit Dishevelled-1-induced canonical Wnt-signaling activity (Figure 5A) and demonstrated a synergistic effect if coexpressed in HEK293T cells. Changes observed upon morpholino-oligonucleotide knockdown of nephrocystin-3 in Xenopus laevis embryos were similar to those of inversin knockdown.19 We observed defective morphogenetic cell movements during gastrulation by following fluorescently labeled clones of cells. These cells did not intercalate and converge to the medial midline, leading to a severe delay of gastroporus closure (Figure 5B). At lower doses (16 ng), the neural folds remained open (Figure 5C), and shortened body axes (Figure 5D) were observed in a dose-depended manner at later stages (Figure 5E). Specificity of the knockdown was demonstrated by amelioration of the phenotype by overexpression of human nephrocystin-3 RNA (Figure 5E). These hallmark phenotypes of defective convergent extension movements suggest a role for NPHP3 in noncanonical PCP Wnt-pathway activation.
Figure 5.
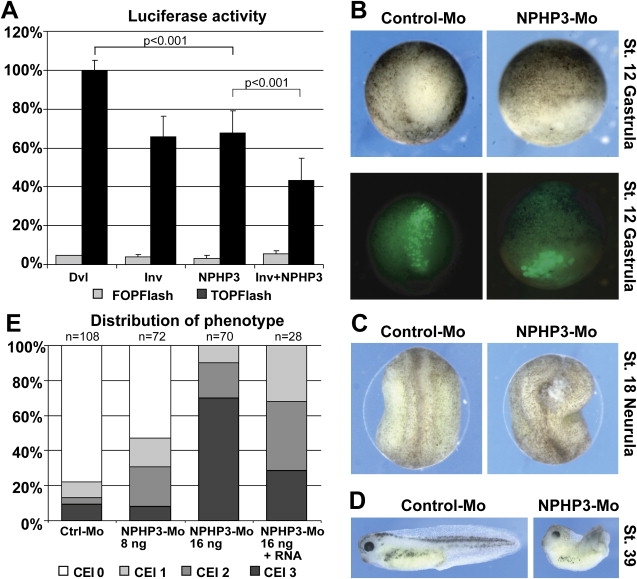
Inversin and Nephrocystin-3 Inhibit Dishevelled-1-Induced Canonical Wnt-Signaling Activity
(A) Both inversin and NPHP3 inhibit Dvl1-induced activation of a TCF/LEF-1-dependent luciferase reporter construct (TOPFlash) in HEK293T cells. A combined expression of both NPHP proteins leads to a further suppression of the luciferase activity. A luciferase reporter construct with mutated TCF/LEF-1 binding sites (FOPFlash) showed no significant background stimulation. At least four independent experiments were done, each in triplicate. Dishevelled-induced stimulation was always above 20-fold over background. Data was normalized to β-galactosidase expression. p values were calculated with the two-tailed Student's t test.
(B–E) Morphogenetic cell movements during gastrulation and neurulation are defective upon NPHP3 knockdown in Xenopus laevis embryos and suggest a role in the PCP Wnt pathway. Gastrulation movements are delayed after NPHP3 knockdown by Morpholino-antisense oligonucleotide injected into two dorsal blastomeres (B). A clone of cells was labeled in the 64-cell stage with fluorescent dextrane, and the cell movements were followed by fluorescent microscopy at stage 12. Upper and lower panels show bright-field and fluorescent images of identical embryos at gastrulation. Incomplete closure of neural folds (C) and phenotypes of shortened body axis and dorsal bending (D) were observed at later stages, suggestive of disrupted PCP/Wnt signaling. This phenotype was dose-dependent and could be partially rescued by coinjection of NPHP3-RNA (E). The phenotypic changes were scored with the convergent extension index (CEI).
Discussion
In this study, we chose a combinatorial approach in patients and mice to define the phenotypic spectrum of NPHP3/Nphp3 mutations and the role of its encoded protein nephrocystin-3. The wide range of phenotypes observed indicates a prominent functional role for nephrocystin-3 throughout development from early embryonic stages to maintenance of morphologic integrity in later life. This is impressively illustrated by the wide spectrum of renal and urinary tract anomalies ranging from shrunken kidneys with interstitial fibrosis and only very few cysts confined to the corticomedullary junction to glomerulocystic kidney disease and grossly enlarged multicystic-dysplastic kidneys with early embryonic renal failure. Cysts in those latter patients were of great number, manifold morphology, and located in all segments of the nephron. Furthermore, ureteric atresia was additionally observed in a severely affected patient harboring a homozygous NPHP3 nonsense mutation. Congenital anomalies of the kidney and urinary tract (CAKUT) are in accordance with early embryonic maldevelopment and indicate that seemingly unrelated distinct cystic renal phenotypes can be caused by very similar molecular mechanisms. Interestingly, one of our patients with early renal failure, situs inversus totalis, and postaxial polydactyly was further characterized by bilateral preauricular fistulas and thus phenotypically overlaps with branchiootorenal (BOR) syndrome that may link ciliopathies to branchial arch maldevelopment.
The pcy mouse has been used for novel therapeutic strategies for cystic kidney diseases. However, so far, the pathogenic character of the pcy missense variant I614S was only speculated to be causative for the nephronophthisis phenotype of these mice. Our experimental setup does not completely rule out digenic disease mechanisms postulating the rare event that a second gene in close vicinity of the Nphp3 gene locus is mutated in Nphp3pcy mice. However, our data obtained in compound heterozygous Nphp3ko/pcy are consistent with the interpretation that the pcy mutation indeed generates a hypomorphic allele responsible for the cystic kidney disease phenotype. These findings are of major clinical importance for future therapeutic approaches in patients afflicted with nephronophthisis and other ciliopathies, because several pharmacological interventional studies have demonstrated that the renal phenotype in pcy mice can be considerably ameliorated.23
In contrast to mice bearing the hypomorphic pcy allele, Nphp3 mice with complete loss-of-function alleles presented with situs inversus, congenital heart defects, and embryonic lethality. Genotype-phenotype correlations based on the mutational type have been previously described for tg737 and Rpgrip1l (Ftm) mutant mice. Tg737 mice harbor a mutation in the intraflagellar transport protein IFT88 (polaris) that is important for intraciliary trafficking.24,25 Rpgrip1l/RPGRIP1L was recently shown to encode a basal-body protein necessary for cilium-related Hedgehog signaling and developmental processes such as the establishment of left-right asymmetry and patterning of the neural tube and the limbs.6,7,26 In accordance with our Nphp3 mutational data, hypomorphic Tg737 and Rpgrip1l alleles result in similar, viable phenotypes, whereas predicted loss-of-function mutations result in embryonic-lethal laterality defects observed in Nphp3-deficient animals. Another animal model that resembles Nphp3 mutants and supports a cilia-related disease mechanism is the Ofd1-deficient mouse, which displays laterality defects and renal cysts due to lack of generation of primary cilia.27 Interestingly, we also report here a defect in cilia generation. However, it is distinct from lack of cilia generation and is best described as defect in cilia-length control (Figure 3).
Overall, our mutational findings add to the increasing body of evidence that ciliary genes/proteins display pleiotropic effects with phenotypic overlap between related disorders and seemingly unrelated clinical entities.3 Nevertheless, it is still a matter of debate and poorly understood how comparable or even identical mutations in the same gene can cause very different phenotypes.28–30 Given the postulated network of cystoproteins, it might be legitimate to claim modifiers and epistatic effects by proteins interacting with each other to be causative for some of the observed phenotypic variability.31 Recently, Badano et al. elegantly dissected the genetic basis for Bardet-Biedl syndrome (BBS), one of the prime examples of pleiotropic and oligogenic inheritance.32 They found that the MGC1203/CCDC28B gene (NM_024296.3), which encodes a pericentriolar protein that colocalizes and interacts with BBS proteins, contributes epistatic alleles to BBS. Some evidence for possible digenic and oligogenic inheritance was recently also reported for nephronophthisis.33 Digenic and oligogenic inheritance patterns are also well conceivable among other plausible explanations (e.g., lack of mutation detection in case of intronic sequence variations or large deletions) for our patients with only one identified NPHP3 mutation.11 Changes in other ciliary genes may also account for some of the observed intrafamiliar phenotypic variability among affected siblings that was also present to a minor extent in our families. In accordance, Tory et al. suggested that epistatic effects provided by heterozygous CEP290 (NM_025114.3) and AHI1 (NM_017651.3) mutations and variants may contribute to the aggravation of phenotype from nonsyndromic nephronophthisis to Joubert syndrome with occurrence of extrarenal features in patients harboring NPHP1 mutations.34 Identification of the mechanisms that explain pleiotropic gene effects will require further studies but will most probably give valuable insights into disease pathogenesis and may also provide a general concept for the modification of disease expression.
Given that virtually all NPHP proteins are expressed in primary cilia, basal bodies, and/or centrosomes, it is now widely accepted that defective cilia function contributes to cyst formation in nephronophthisis.1,10,21,22,35 We next aimed to better characterize the functional role of nephrocystin-3. The similar phenotypes in humans and mice with NPHP2/INVS (Nphp2/Invs) or NPHP3 (Nphp3) mutations prompted us to investigate whether nephrocystin-3 and inversin exert similar functions. In line with this notion/hypothesis, we demonstrated direct interaction of nephrocystin-3 with inversin. Studies in HEK cells confirmed that nephrocystin-3 like inversin can inhibit the canonical Wnt-signaling pathway.19 In addition, analyses in Xenopus laevis with nephrocystin-3 knockdown identified phenotypes of defective convergent extension, suggesting a role in noncanonical PCP Wnt-pathway activation. Taken together with the demonstrated effect on canonical Wnt signaling, a similar role for both inversin and nephrocystin-3 in the control of the switch between canonical and noncanonical (planar cell polarity) Wnt signaling can be postulated.19 Interestingly, both nephrocystins enhance the inhibiting activity on canonical Wnt signaling of the respective other. This observation leads us to believe that both proteins do not function up- or downstream from each other but in parallel, converging on the same downstream targets when modulating canonical and noncanonical Wnt signaling (Figure 5). Altered Wnt signaling was not only found in nephronophthisis-related disorders but also in Bardet-Biedl syndrome.36,37
In this study, we have demonstrated in patients and mice that NPHP3/Nphp3 mutations cause a broad clinical spectrum of early embryonic patterning defects comprising situs inversus, polydactyly, central nervous system malformations, structural heart defects, preauricular fistulas, and a wide range of renal and urinary tract anomalies (CAKUT). Conclusively, NPHP3/Nphp3 mutations can result in isolated nephronophthisis, Senior-Loken syndrome, renal-hepatic-pancreatic dysplasia, Meckel-Gruber-like syndrome, and embryonic lethality.
Supplemental Data
Three figures and one table are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer 3, http://www.broad.mit.edu/cgi-bin/primer/primer3_www.cgi/
University of California, Santa Cruz Genome Bioinformatics, http://genome.ucsc.edu/
Acknowledgments
We thank the patients and families for their participation in this study, E. Golz-Staggemeyer, E. von Heel, E. Bünger, T. Knöbl-Deledezma, C. Tessmer, S. Franz, U. Ackermann, C. Böhm, J. Kalnitski, C. Reinhardt, K. Sutter, and C. Kopp for technical assistance, and H. U. Völker and C. Monoranu for microscopic photographs. We thank O. Strauch, N. Klemm, N. Störmer, D. Guhl, K. Thumm, J. Herbst, M. Bohrer, E. Klimczak, J. Christophel, and V. Skude for excellent animal handling and R. Nitschke and S. Haxelmans for support with confocal imaging. This work was supported by the German Kidney Foundation (Deutsche Nierenstiftung) (C.B.), the START program of the medical faculty of the Rheinisch-Westfälische Technische Hochschule (RWTH) Aachen University (C.B.), and the Deutsche Forschungsgemeinschaft (K.Z. and C.B., H.O. [SFB592]).
References
- 1.Hildebrandt F., Otto E. Cilia and centrosomes: A unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 2.Harris P.C., Torres V.E. Understanding pathogenic mechanisms in polycystic kidney disease provides clues for therapy. Curr. Opin. Nephrol. Hypertens. 2006;15:456–463. doi: 10.1097/01.mnh.0000232888.65895.e7. [DOI] [PubMed] [Google Scholar]
- 3.Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 4.Mykytyn K. Clinical variability in ciliary disorders. Nat. Genet. 2007;39:818–819. doi: 10.1038/ng0707-818. [DOI] [PubMed] [Google Scholar]
- 5.Baala L., Romano S., Khaddour R., Saunier S., Smith U.M., Audollent S., Ozilou C., Faivre L., Laurent N., Foliguet B. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 7.Arts H.H., Doherty D., van Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Marker T., Voesenek K., Kartono A. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 8.Baala L., Audollent S., Martinovic J., Ozilou C., Babron M.C., Sivanandamoorthy S., Saunier S., Salomon R., Gonzales M., Rattenberry E. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel Syndrome. Am. J. Hum. Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank V., den Hollander A.I., Ortiz Brüchle N., Zonneveld M.N., Nürnberg G., Becker C., du Bois G., Kendziorra H., Roosing S., Senderek J. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat. 2008;29:45–52. doi: 10.1002/humu.20614. [DOI] [PubMed] [Google Scholar]
- 10.Fliegauf M., Horvath J., von Schnakenburg C., Olbrich H., Muller D., Thumfart J., Schermer B., Pazour G.J., Neumann H.P., Zentgraf H. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J. Am. Soc. Nephrol. 2006;17:2424–2433. doi: 10.1681/ASN.2005121351. [DOI] [PubMed] [Google Scholar]
- 11.Olbrich H., Fliegauf M., Hoefele J., Kispert A., Otto E., Volz A., Wolf M.T., Sasmaz G., Trauer U., Reinhardt R. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 12.Omran H., Haffner K., Burth S., Fernandez C., Fargier B., Villaquiran A., Nothwang H.G., Schnittger S., Lehrach H., Woo D. Human adolescent nephronophthisis: Gene locus synteny with polycystic kidney disease in pcy mice. J. Am. Soc. Nephrol. 2001;12:107–113. doi: 10.1681/ASN.V121107. [DOI] [PubMed] [Google Scholar]
- 13.Frank V., Ortiz Brüchle N., Mager S., Senderek J., Frints S.G.M., Bohring A., du Bois G., Debatin I., Seidel H., Besbas N. Aberrant splicing is a common mutational mechanism in MKS1, a key player in Meckel Gruber syndrome. Hum. Mutat. 2007;28:638–639. doi: 10.1002/humu.9496. [DOI] [PubMed] [Google Scholar]
- 14.Levanon D., Bettoun D., Harris-Cerruti C., Woolf E., Negreanu V., Eilam R., Bernstein Y., Goldenberg D., Xiao C., Fliegauf M. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J. 2002;21:3454–3463. doi: 10.1093/emboj/cdf370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benzing T., Gerke P., Hopker K., Hildebrandt F., Kim E., Walz G. Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers phosphorylation of Pyk2. Proc. Natl. Acad. Sci. USA. 2001;98:9784–9789. doi: 10.1073/pnas.171269898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim E., Arnould T., Sellin L.K., Benzing T., Fan M.J., Grüning W., Sokol S.Y., Drummond I., Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J. Biol. Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 17.Newport J., Kirschner M. Regulation of the cell cycle during early Xenopus development. Cell. 1982;3:675–686. doi: 10.1016/0092-8674(84)90409-4. [DOI] [PubMed] [Google Scholar]
- 18.Nieuwkoop P.D., Faber J. North Holland Publishing Company; Amsterdam, Holland: 1967. Normal Table of Xenopus laevis (Daudin) [Google Scholar]
- 19.Simons M., Gloy J., Ganner A., Bullerkotte A., Bashkurov M., Kronig C., Schermer B., Benzing T., Cabello O.A., Jenny A. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neuhaus T.J., Sennhauser F., Briner J., van Damme B., Leumann E.P. Renal-hepatic-pancreatic dysplasia: An autosomal recessive disorder with renal and hepatic failure. Eur. J. Pediatr. 1996;155:791–795. doi: 10.1007/BF02002909. [DOI] [PubMed] [Google Scholar]
- 21.Otto E.A., Schermer B., Obara T., O'Toole J.F., Hiller K.S., Mueller A.M., Ruf R.G., Hoefele J., Beekmann F., Landau D. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mollet G., Silbermann F., Delous M., Salomon R., Antignac C., Saunier S. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum. Mol. Genet. 2005;14:645–656. doi: 10.1093/hmg/ddi061. [DOI] [PubMed] [Google Scholar]
- 23.Torres V.E., Harris P.C. Mechanisms of disease: Autosomal dominant and recessive polycystic kidney diseases. Nat. Clin. Pract. Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 24.Moyer J.H., Lee-Tischler M.J., Kwon H.Y., Schrick J.J., Avner E.D., Sweeney W.E., Godfrey V.L., Cacheiro N.L., Wilkinson J.E., Woychik R.P. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science. 1994;264:1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- 25.Murcia N.S., Richards W.G., Yoder B.K., Mucenski M.L., Dunlap J.R., Woychik R.P. The Oak Ridge polycystic kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127:2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- 26.Vierkotten J., Dildrop R., Peters T., Wang B., Rüther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–2577. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- 27.Ferrante M.I., Zullo A., Barra A., Bimonte S., Messaddeq N., Studer M., Dolle P., Franco B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006;38:112–117. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- 28.Jaeken J., Carchon H. Congenital disorders of glycosylation: A booming chapter of pediatrics. Curr. Opin. Pediatr. 2004;16:434–439. doi: 10.1097/01.mop.0000133636.56790.4a. [DOI] [PubMed] [Google Scholar]
- 29.Chiang A.P., Beck J.S., Yen H.J., Tayeh M.K., Scheetz T.E., Swiderski R.E., Nishimura D.Y., Braun T.A., Kim K.Y., Huang J. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11) Proc. Natl. Acad. Sci. USA. 2006;103:6287–6292. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rankin J., Ellard S. The laminopathies: A clinical review. Clin. Genet. 2006;70:261–274. doi: 10.1111/j.1399-0004.2006.00677.x. [DOI] [PubMed] [Google Scholar]
- 31.Woo D.D., Nguyen D.K., Khatibi N., Olsen P. Genetic identification of two major modifier loci of polycystic kidney disease progression in pcy mice. J. Clin. Invest. 1997;100:1934–1940. doi: 10.1172/JCI119724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Badano J.L., Leitch C.C., Ansley S.J., May-Simera H., Lawson S., Lewis R.A., Beales P.L., Dietz H.C., Fisher S., Katsanis N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- 33.Hoefele J., Wolf M.T., O'Toole J.F., Otto E.A., Schultheiss U., Dêschenes G., Attanasio M., Utsch B., Antignac C., Hildebrandt F. Evidence of oligogenic inheritance in nephronophthisis. J. Am. Soc. Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- 34.Tory K., Lacoste T., Burglen L., Moriniere V., Boddaert N., Macher M.A., Llanas B., Nivet H., Bensman A., Niaudet P. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: Potential epistatic effect of NPHP6 and AHI1 ,utations in patients with NPHP1 mutations. J. Am. Soc. Nephrol. 2007;18:1566–1575. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- 35.Christensen S.T., Pedersen L.B., Schneider L., Satir P. Sensory cilia and integration of signal transduction in human health and disease. Traffic. 2007;8:97–109. doi: 10.1111/j.1600-0854.2006.00516.x. [DOI] [PubMed] [Google Scholar]
- 36.Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Chapple J.P., Munro P.M., Fisher S. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 37.Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.