

Abstract
There are two common designs for association mapping of complex diseases: case-control and family-based designs. A case-control sample is more powerful to detect genetic effects than a family-based sample that contains the same numbers of affected and unaffected persons, although additional markers may be required to control for spurious association. When family and unrelated samples are available, statistical analyses are often performed in the family and unrelated samples separately, conditioning on parental information for the former, thus resulting in reduced power. In this report, we propose a unified approach that can incorporate both family and case-control samples and, provided the additional markers are available, at the same time corrects for population stratification. We apply the principal components of a marker matrix to adjust for the effect of population stratification. This unified approach makes it unnecessary to perform a conditional analysis of the family data and is more powerful than the separate analyses of unrelated and family samples, or a meta-analysis performed by combining the results of the usual separate analyses. This property is demonstrated in both a variety of simulation models and empirical data. The proposed approach can be equally applied to the analysis of both qualitative and quantitative traits.
Introduction
Population-based association studies have been considered more powerful than family-based linkage studies in the genetic dissection of complex diseases.1,2 Such studies rely on the linkage disequilibrium (LD) between a marker variant and a disease variant in a population. LD between the alleles at two loci decays from generation to generation, depending on the distance between the two loci. As a result, strong LD can be observed only within short distances in populations. Because of the availability of dense SNPs across the genome and the reduction in high-throughput genotyping costs, association studies have become a favorite way to identify the genetic variants affecting complex traits.
The case-control design is well established in epidemiology as a reliable approach for establishing the relationship between a risk exposure and an outcome and has been widely applied in studies of the association between a genetic variant and phenotypic trait. When samples arise from different ethnic groups or an admixed population, cases and controls may have different ancestry distributions, resulting in real, but spurious, associations.3,4 This problem can be exacerbated when the sample size is large, a general requirement to obtain sufficient power to detect modest genetic effects for most complex traits.5 To overcome this problem, methods using a set of unlinked genetic markers genotyped in the same samples have been developed that control for population stratification in case-control studies.6–12 In the presence of population stratification, the chi-square (χ2) statistic of a case-control design may not follow a central chi-square distribution under the null hypothesis of biological interest. The genomic control (GC) approach simply rescales the chi-square statistic based on a set of unlinked markers,7 and this rescaled chi-square statistic is assumed to follow a chi-square distribution. An alternative approach is “structured association” (SA),9,13 which, based on a Markov Chain Monte Carlo (MCMC) method, uses a set of independent genetic markers to estimate the number of subpopulations and the ancestry probabilities of individuals from putative “unstructured” subpopulations.9 This information is then used to test for association. Satten et al.10 extended SA by applying latent-class analysis to infer the population structure while simultaneously estimating the model parameters and testing for association. When the number of subpopulations is large, the SA approach becomes computationally intensive.
A third alternative approach is to summarize the genetic background through the principal components or principal coordinate analysis of marker genotype data.6,11,12,14,15 The approach based on principal components of genetic marker data was first used for characterizing population differences.16 The principal components calculated from a matrix of genetic marker data can be further used to eliminate the effect resulting from population stratification.12 Zhang et al.11 and Chen et al.6 further modeled the relationship between the principal components and trait values through smoothing techniques. Recently, Price et al.15 presented a regression method by regressing both the phenotype and marker genotype values on the principal components for unrelated data. Association between the phenotype and marker is then tested with the residual correlation. The principal component analysis is much simpler than the MCMC-based approaches and computationally faster. Its speed depends on the singular value decomposition of a matrix with dimensions given by the number of individuals and the number of markers in a study. When the number of markers and the number of individuals are large, as in whole-genome association studies, calculating the principal components of such a matrix of genetic marker data can be extremely time consuming, requiring a huge amount of computer memory, although it can still be handled reasonably fast by modern computers.15 Bauchet et al.14 have suggested using principal coordinate analysis to summarize the genetic marker data. In fact, this approach calculates the principal components on people rather than on markers. When the number of markers is much larger than the number of individuals in a study, the principal coordinate analysis is more convenient computationally because of the smaller matrix involved.17 The first L principal components corresponding to nonzero latent roots and the first L principal coordinates are the same, because of the duality of the two analyses.17 Thus, the information used for both methods is equivalent. More recently, a simple two-step procedure has been proposed:18 first the odds of disease given the marker data is modeled by applying generalized partial least-squares, inferring the strata based on the odds of disease, then association is tested between disease and a test locus within strata. This approach is valid for testing association in the presence of population stratification and is computationally simple.18
To overcome the problem caused by population stratification, a further alternative approach is the transmission/disequilibrium test (TDT) design that utilizes family members as controls.19 The TDT compares the frequencies of genetic marker alleles that are transmitted from heterozygous parents to affected children against those that are not transmitted. In this design, the ethnic background of cases and controls is necessarily matched, and so no additional markers are required to eliminate the effect of population stratification. The TDT method has been extended to include a variety of genetic models and study designs for both qualitative traits20–24 and quantitative traits.25–30 However, compared with the case-control design, TDT-based methods require the collection of DNA samples from family members, which is more difficult than from unrelated controls, especially in the case of late-onset diseases.
The samples from case-control and family-based studies cannot be pooled naively for analysis, because of the familial correlations in the latter. As techniques advance, many whole-genome-wide association studies have started. Because of the high cost of whole-genome-wide association studies and the great amount of effort needed to genotype hundreds of thousands SNPs on each individual, the sample size of each individual study will often be limited. On the other hand, if a genetic variant contributes only a modest effect to a complex trait, a large sample size is required in order to have enough power to detect the genetic effect after correcting for the multiple tests. Collaborative studies or multistage approaches have been advocated.31 Given the availability of family samples from traditional linkage studies and the possibly better phenotypes defined in family studies than in case-control studies, and the advantages of collecting unrelated samples, we will have samples that are either in family units or unrelated. In this circumstance, it would be helpful to have access to a statistical method to analyze both family and unrelated samples simultaneously to increase the power, rather than to analyze them separately. The methods we reviewed above apply to either case-unrelated control designs or to family data. Recently, Nagelkerke et al.32 developed a method of combining the family and unrelated samples via a likelihood-based approach. Epstein et al.33 further extended this approach, relaxing the assumption of Hardy-Weinberg equilibrium (HWE) and random mating. The extended method also allows for flexible modeling and estimation of allele effects and is more powerful than methods for analyzing family and unrelated samples separately when population stratification does not play a role. However, the method requires initial testing of whether the data sources can be combined. If not, the test for association will be invalid and estimates of genotype effects can be biased. In addition, the method allows only parents-child triads and requires the rare-disease assumption if unaffected siblings are also included. Further, the information available in the family data is not fully used because only the genotype data of the parents are used. Thus, the application of this approach to pooled family and unrelated data is limited.
In this report, we describe a simple approach that can combine both family and unrelated samples without assuming a rare disease, and allowing for the inclusion of multiple affected or unaffected siblings. Our procedure uses a principal component-based approach to eliminate any effect of population stratification. Both parental phenotype and genotype data are used in the analysis. The method does not require testing whether the family and unrelated data can be combined, but does require enough markers for GC. We evaluate the performance of our approach first by using simulated data in a variety of population admixture models and then by using empirical data.
Material and Methods
We previously suggested using the principal components of marker data to represent the genetic background of unrelated individuals.6,11,12 We now consider samples that include both family and unrelated individuals. For simplicity, we consider only nuclear families. We assume our data include Nf nuclear families. The ith family has ki members, with the first two (j = 1 or 2) being the father and mother. In addition to these families, we have Nd unrelated cases and Nc unrelated controls. The total number of individuals is thus . To simplify, we assume there are N families, i = 1, 2, …, N, with ki = 1 when i > Nf. Thus, we define each unrelated case or control as a separate family of size one. In other words, we have N = Nf + Nd + Nc. Let yij, which may be either quantitative or binary, be the trait value of the jth individual in the ith family. For a binary trait, yij takes on the value 0 or 1, indicating unaffected or affected, respectively. We do not consider any covariates, although incorporating them is straightforward. Let gij be the marker genotypic value of the jth individual in the ith family, coded according to an additive, recessive, or dominant mode of inheritance. M diallelic markers are genotyped. Let Xij = (xij1, xij2, …, xijM)T be a column vector representing the marker genotypic values for the jth individual in the ith family, where xijl is 0, 1, or 2, corresponding to a homozygote, heterozygote, and the other homozygote, l = 1, 2, …, M. We perform a principal component analysis to summarize the marker data. Because our data include both family and unrelated individuals, a naive principal component analysis with all available data will result in biased directions of maximum variability for the data. This is because the directions of maximum variability will favor the correlated data points in the marker space. Thus, the principal component analysis is applied to only the unrelated individuals, i.e, the parents in each family and the unrelated cases and controls. Let denote the variance-covariance matrix of the marker data for these unrelated individuals in our data, where is the overall mean of X. Let e1 be the lth eigenvector corresponding to the lth largest eigenvalue of Σ, l = 1, 2, …, M. Thus the eigenvectors e1, e2, …, eM represent new orthogonal axes corresponding to decreasing variability of the marker data. We calculate the lth principal component for individual j of family i by , where i = 1, 2, …, N, j = 1, 2, …, ki, and l = 1, 2, …, L. We do not incorporate the disease status in the calculation of the principal components, although such incorporation is not difficult. The disease status might be important in the analysis because of sample ascertainment; however, our simulation studies (see later) suggest that it is not critical. Here we consider only the first L principal components, assuming that the marker data can be well represented by them. In this study, we use only the first 10 principal components, which perform reasonably well in our simulation studies.
Because the principal components represent the genetic background information, we adjust both the trait and test marker values for this background by applying linear regression, as suggested by Price et al.15 However, we perform linear regression only on the unrelated individuals. That is,
and
where i = 1, 2, …, N, j = 1, 2 if i ≤ Nf and otherwise j = 1 and ɛij and τij are random errors. Let , be the least-squares estimators of β0, β1, …, βL, α0, α1, …, αL, respectively. Because the principal components are orthogonal, , can be easily calculated by
and
The residual for each individual, including the children, is calculated by
and
where i = 1, 2, …, N, j = 1, 2, …, ki.
We can view the residuals and as the projections of the phenotypic and genotypic values in the space orthogonal to the space spanned by the L principal components. Define . Under the null hypothesis of no association between the trait and test marker, and are independent and we have . We define the test statistic
| (1) |
which follows a chi-square distribution with 1 degree of freedom under the null hypothesis.
In the appendix we show that Var(T) can be calculated under the null hypothesis by
where is the correlation of the random variable between individuals j1 and j2 within a family. The variances and correlations can be estimated from the data. In view of the different ascertainments between families and unrelated cases and controls, we suggest the variances be separately estimated in families and unrelated cases and controls. When all the individuals are unrelated, (1) is the same as the test statistic proposed by Price et al.15 Note that the variance of T, when calculated from the data as given above, accounts for all residual correlations.
Simulations
Simulation 1. Discrete Model with Two Ancestral Populations
The first simulation aims to illustrate that the principal component analysis is able to cluster a mixture population of two discrete populations, by using selected ancestrally informative markers, to eliminate the effect of population stratification and to retain power when both family and unrelated data are analyzed together. In order to have samples from two different populations, we simulated 140 nuclear families, 140 unrelated cases, and 100 unrelated controls sampled from an African population and 60 nuclear families, 60 unrelated cases, and 100 unrelated controls sampled from a European population. To do this, we accessed the panel of SNPs that are informative for admixture mapping across the genome reported by Smith et al.34 The allele frequencies of the SNPs and the marker map for both the African and European populations were downloaded from the website of the American Journal of Human Genetics. We first generated 50,000 African nuclear families. The parental marker genotypes were generated according to the African SNP allele frequencies assuming the SNPs are in linkage equilibrium. We then simulated the offspring marker genotypes according to the parental genotypes and the marker map. The number of children produced by each marriage was assumed to follow a Poisson distribution with mean size 2. We assumed that an African individual has a 30% chance of being affected and this probability was used to assign an individual's disease status. We then sampled 140 families with at least one child affected. From the children in the rest of the families, we randomly selected 140 unrelated cases and 100 unrelated controls, that is, only one child was sampled per family. With the same method but European SNP allele frequencies, we generated 60 nuclear families, 60 unrelated cases, and 100 controls. We assumed that the disease prevalence in the European population is 10%. We simulated a two-allele candidate marker with susceptibility allele frequency 0.6 and 0.1 in the African and European populations, respectively. Thus, confounding resulting from population stratification was created when the samples came from the two populations with different disease prevalence.
To simulate the samples under the alternative hypothesis, we applied the same method but assigned an individual's disease status according to the penetrance of a test marker genotype under different modes of inheritance: additive, multiplicative, recessive, and dominant.
Simulation 2. Admixed Model with Two Ancestral Populations
This simulation aims to illustrate that principal component analysis is still able to eliminate the effect of population stratification and to retain power when samples are drawn from an admixed population such as the African-American population. In order to simulate 200 nuclear families, 200 unrelated cases, and 200 unrelated controls from an admixed population, we used the generalized continuous gene-flow model described in Zhu et al.35 We used the same marker panel as we did for the discrete model above. In brief, at the first generation, the marker genotypes of 50,000 unrelated African persons were simulated according to the African SNP allele frequencies. An admixed population was then formed by taking a proportion λ randomly selected from the African population to marry with people generated according to European marker allele frequencies, with the remaining proportion 1 − λ randomly mating among themselves. We let λ vary at each generation, generating it from a uniform distribution U(0, 0.06). The number of children produced by each marriage was again assumed to follow a Poisson distribution with mean size 2. We repeated this process 10 times to simulate the current families, resulting in a mixture of approximately 80%/20% of African and European ancestry in the current population. We also simulated a two-allele candidate marker with susceptibility allele frequency 0.6 and 0.1 in the African and European ancestral populations, respectively. We assigned an individual's disease status with probability equal to his African ancestry. We then sampled 200 families with at least one child affected. From the children in the rest of the families, we randomly selected 200 unrelated cases and 200 unrelated controls, i.e., only one child per family was sampled. Thus, confounding resulting from population stratification was created for testing association between a marker and disease status.
To simulate the samples under the alternative hypothesis, we applied the same method but assigned an individual's disease status according to the penetrance of a test marker genotype under different modes of inheritance: additive, multiplicative, recessive, and dominant.
Simulation 3. Discrete Model with Three Ancestral Populations
This simulation aims to illustrate the performance of principal component analysis with randomly chosen markers when samples are from three discrete populations. We simulated samples with three ancestral populations with the haplotype data released by the HapMap project.36 The HapMap project consists of three populations: 120 European chromosomes (CEU), 120 African chromosomes (Yoruba), and 178 East Asian chromosomes (90 Han Chinese and 88 Japanese). In these simulations, we used only the haplotype data on chromosome 22. To generate the genotypes of unrelated individuals in a large population, we first generated a number of crossovers across the chromosome by a Poisson process, with an average of 6 crossovers per Morgan, in order to create more independent chromosomes than in the original HapMap data. The crossover locations were generated according to a uniform distribution. Then, starting at one end of the chromosome, a random choice was made from the haplotypes of HapMap chromosomes between two successive crossovers. The offspring genotypes were generated by randomly transmitting one of the two haplotypes of the father and the mother with the crossovers occurring according to the genetic map. To simulate an individual's disease status, we set the population disease prevalence to be 25%, 15%, and 10% in African, European, and Asian populations, respectively. We then sampled 100, 60, and 40 nuclear families with at least one affected offspring from the African, European, and Asian populations, respectively. We further selected 100 unrelated cases and 66 unrelated controls from the African population, 60 cases and 67 controls from the European population, and 40 cases and 67 controls from the East Asian populations. Thus, our analysis sample included a total of 200 nuclear families, 200 unrelated cases, and 200 unrelated controls from the three discrete populations, no marker being associated with the disease status. 10,000 randomly selected SNPs on chromosome 22 were used in the analyses for calculating the principal components. The LD pattern across a chromosome is generally preserved for the SNPs that are closely located.
The samples under the alternative hypothesis were simulated according to the penetrance of a test marker genotype under different modes of inheritance: additive, multiplicative, recessive, and dominant. The test marker was chosen to be one of the 10,000 SNPs.
Simulation 4. Admixed Model with Three Ancestral Populations
This simulation aims to illustrate the performance of principal component analysis when randomly chosen markers are used for samples from a population admixed by three ancestral populations. Again, we simulated samples based on the chromosome 22 data of the HapMap project. We first generated haplotype exchange points on the chromosome among the populations by using a Poisson process, with an average of 6 crossovers per Morgan. This is equivalent to a population that has been admixed for an average of 6 generations. In each region between two exchange points, we determined which ancestral population a haplotype came from based on a distribution of admixture proportions of Africans, Europeans, and East Asians, which we set to (0.7, 0.2, 0.1). We then applied the same method as for Simulation 3 to generate a person's genotypes from the selected ancestral population. The method in Simulation 3 for generating offspring genotypes was also applied. To simulate an individual's disease status, we assumed that the probability of persons becoming affected is dependent on their own admixture proportions. Letting a person's African, European, and East Asian admixture proportions be (λYRI, λCEU, λEA), the probability of being affected for the person was 0.5λYRI + 0.2λCEU + 0.1λEA. We then generated 200 nuclear families with at least one offspring affected, 200 unrelated cases, and 200 unrelated controls. 10,000 randomly selected SNPs on chromosome 22 were used in the analyses. Again, the LD pattern across a chromosome is in this way preserved when two SNPs are closely located.
As before, the samples under the alternative hypothesis were simulated according to the penetrance of a test marker genotype, chosen from the 10,000 SNPs, under different modes of inheritance: additive, multiplicative, recessive, and dominant.
Results
The Performance of Principal Component Analysis
Figure 1 presents the principal component analysis for the samples generated according to Simulation 1. Only the first two principal components were plotted for 200, 400, and 800 SNPs. People from African and European populations can apparently be correctly grouped. The children's genotypes were not used to obtain the principal components although their principal component values were obtained through the eigenvectors obtained from the genotypes of their parents and the unrelated cases and controls. We observed that the children can also be correctly grouped. Even the first principal component alone can cluster individuals into correct groups when samples are from a population consisting of two discrete subpopulations, consistent with the results in Zhu et al.12 We then standardized the principal components over the whole sample and estimated their standard deviations for Africans and Europeans separately. The standard deviation of the first principal component within populations is substantially smaller than that of the second principal component (Figure 1). The within-population standard deviation is reduced as the number of SNPs increases for the first principal component, but not for the second one. ANOVA suggests that 99.5% to 99.9% of the total variance can be expressed by the clusters using the first principal component alone for 200 to 800 SNPs, but almost no variation was expressed for the second principal component. The results also hold for the children, although we did not use their genotype information for calculating the eigenvectors. The results indicate that the principal components can well capture the variation of an individual's ancestry and that a child's ancestry can also be estimated through the prediction of the principal components obtained from the unrelated individuals' principal components. The results for the next eight principal components were similar to those for the second principal component.
Figure 1.
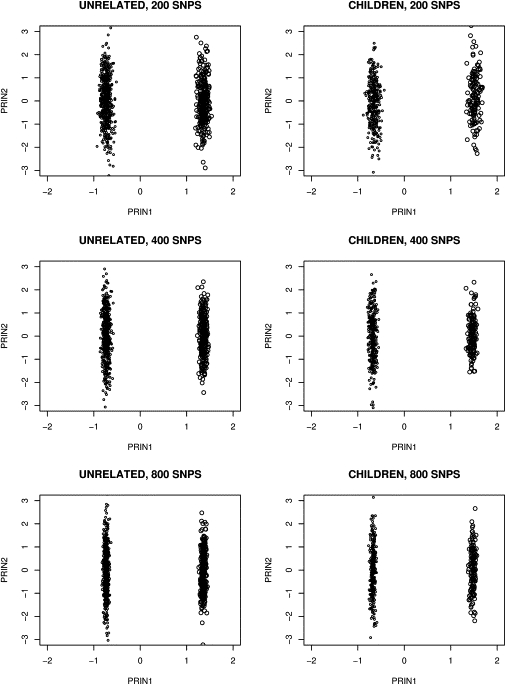
Plot of the First Two Principal Components When Samples Were Generated in Simulation 1, Where Samples Were Drawn from Two Discrete Populations
200, 400, and 800 informative SNPs obtained from Smith et al.34 were generated with no LD between SNPs in two subpopulations. Left and right dots represent individuals from African and European populations, respectively. The children's principal components were calculated by projection to the axes obtained from the independent samples. It can be observed that the first principal component can distinguish individuals from two subpopulations for both independent samples and children.
Simulation 1 generated samples comprising only a discrete mixture of subpopulations. For the samples from an admixed population generated by Simulation 2, we did not observe a clear picture when we plotted the first two principal components (Figure 2). This is because the population has been substantially mixed after 10 generations and each person carries a portion of African and European ancestries. We then plotted each person's true ancestry against the first two principal components (Figure 3). The true ancestry is calculated here as the proportion of alleles from the ancestral African population, standardized by the sample mean and standard deviation. We observed that the first principal component is highly correlated with the true ancestry defined this way, with the correlation coefficients ranging from 0.97 to 0.99 for 200 SNPs to 800 SNPs. In comparison, the correlation between the second principal component and the true ancestry is less than 0.14. The results suggest that the principal components are able to capture the ancestry variation even for data generated through a generalized continuous gene flow model, such as an African-American population. The results also hold for the children, although we did not use their genotype information for calculating the eigenvectors.
Figure 2.
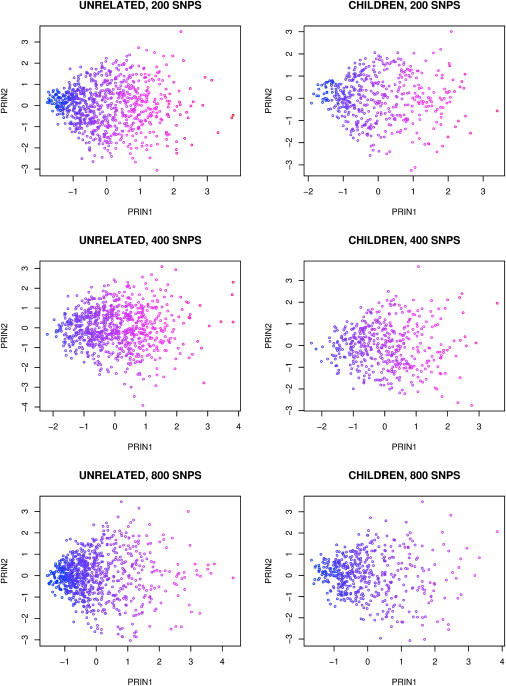
Plot of the First Two Principal Components When Samples Were Generated in Simulation 2, Where Samples Were Drawn from an Admixed Population of Two Ancestral Populations
200, 400, and 800 informative SNPs obtained from Smith et al.34 were generated with no LD between SNPs in two ancestral populations. Blue and red colors indicate that an individual has more African and European ancestral alleles, respectively. The children's principal components were calculated by projection to the axes obtained from the independent samples. Because each individual carries a portion of SNPs from each ancestral population, we cannot observe clean clusters as in Figure 1.
Figure 3.
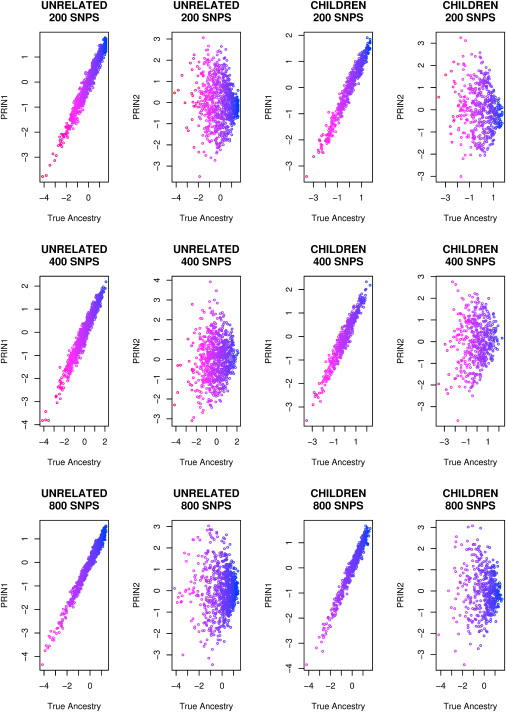
Plot of the First Two Principal Components against the True Ancestry for the Same Data as in Figure 2
We observe that the first principal component, but not the second, is highly correlated with the true ancestry.
Figure 4 presents the principal component analysis for the samples generated according to Simulation 3, where we have a mixture of three populations and 10,000 random SNPs were simulated. The unrelated individuals, as well as the children, from the three populations can apparently be correctly grouped when using a large number of random SNPs, rather than SNPs designed for admixture mapping. The first two principal components are sufficient to cluster individuals into correct groups when the samples come from a population consisting of three discrete populations. We then standardized the principal components over the whole sample and estimated the standard deviations for Africans, Europeans, and Asians separately. The standard deviations of the first two principal components within populations is substantially smaller than those of the third principal components (Figure 4). We then performed linear regression analysis, regressing each of the true population-specific ancestries on the first three principal components. The R-square values were 0.986, 0.987, and 0.995 for European, Asian, and African ancestries, respectively, suggesting that the principal components can capture the individual ancestry variation. The results for children were similar even though we did not use their genotype information for calculating the eigenvectors.
Figure 4.
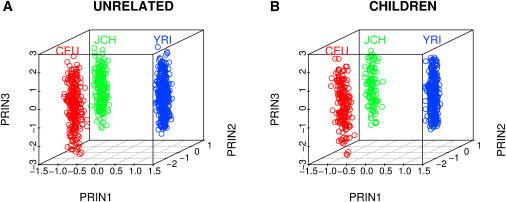
The First Three Principal Components for Data from Simulation 3
Plot of the first three principal components when samples were generated in Simulation 3, where samples were drawn from three discrete populations simulated with the data on chromosome 22 of YRI, CEU, and Japanese and Chinese (JCH) from the HapMap project. 10,000 randomly selected SNPs were generated and the LD between SNPs was preserved as in the HapMap data. The children's principal components were calculated by projection on to the axes obtained from the independent samples. Red, green, and blue represent individuals who were from CEU, JCH, and YRI, respectively. It can be observed that the first two principal components can distinguish individuals from three subpopulations for both independent samples and children.
(A) Independent samples.
(B) Children samples.
When the samples came from the admixed population with three ancestral populations generated in Simulation 4, we did not observe a clear picture on plotting the first three principal components (Figure 5), because each person carried a portion of African, European, and Asian ancestry. When compared with the true ancestries, however, a substantial correlation can be observed. We performed linear regression analysis by regressing the true ancestries on the first three principal components and obtained the R-square values of 0.239, 0.596, and 0.951 for European, Asian, and African ancestries, respectively, indicating that the first three principal components are not enough to capture all the ancestry variation. When using the first 10 principal components in the regression model, the R-square values increased to 0.808, 0.902, and 0.962 for European, Asian, and African ancestries, respectively. We noticed that some of the top principal components may express less variation of the true ancestry than lower ones, as demonstrated by the cumulative R-square values (Table 1). The results for children were similar, though we did not use their genotype information for calculating the eigenvectors.
Figure 5.
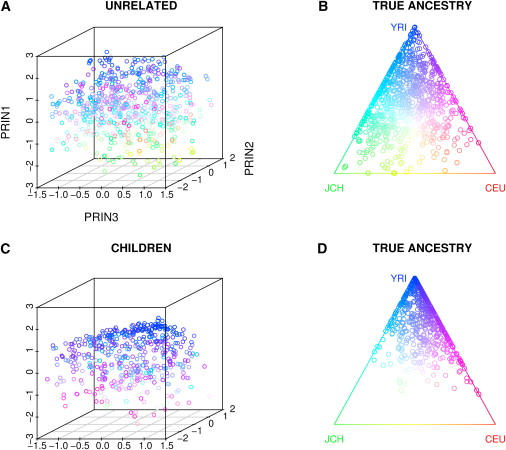
The First Three Principal Components for Data from Simulation 4
Plot of the first three principal components when samples were generated in Simulation 4, where samples were drawn from an admixed population simulated with the data on chromosome 22 of YRI, CEU, and Japanese and Chinese (JCH) from the HapMap project. The individual true ancestry is also presented. 10,000 randomly selected SNPs were generated and the LD between SNPs was preserved as in HapMap data. The children's principal components were calculated by projection on to the axes obtained from the independent samples. Because each individual carries a portion of SNPs from each ancestral population, we can not observe distinct clusters as in Figure 4. Color designates an individual's ancestral proportion, as seen in the right panel.
(A) Three principal components of independent individuals.
(B) True independent individual ancestry.
(C) Three principal components of children.
(D) True ancestry of children.
Table 1.
T Test Statistic Values and Cumulative R2 in Regression Analysis of True Ancestry on Each of the First 10 Principal Components for the Data Generated in Simulation 4
| Unrelated |
Children |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CEU |
JCH |
YRI |
CEU |
JCH |
YRI |
|||||||
| T | R2 | T | R2 | T | R2 | T | R2 | T | R2 | T | R2 | |
| PRIN1 | 20.6 | 0.10 | 69.2 | 0.59 | −136.1 | 0.89 | 15.4 | 0.12 | 55.7 | 0.57 | −107.7 | 0.87 |
| PRIN2 | 0.7 | 0.10 | 4.3 | 0.59 | −7.4 | 0.89 | 1.8 | 0.13 | 3.9 | 0.57 | −8.9 | 0.88 |
| PRIN3 | −23.7 | 0.24 | 3.9 | 0.60 | 35.8 | 0.95 | −20.8 | 0.29 | 3.4 | 0.57 | 31.4 | 0.95 |
| PRIN4 | −45.7 | 0.75 | 47.4 | 0.87 | 11.1 | 0.96 | −36.9 | 0.75 | 37.3 | 0.85 | 10.3 | 0.95 |
| PRIN5 | 2.2 | 0.75 | −3.0 | 0.87 | 0.55 | 0.96 | 1.3 | 0.75 | −2.7 | 0.85 | 1.7 | 0.95 |
| PRIN6 | −2.4 | 0.75 | 4.5 | 0.88 | −2.4 | 0.96 | −1.6 | 0.75 | 2.9 | 0.86 | −2.2 | 0.95 |
| PRIN7 | −5.4 | 0.76 | 5.6 | 0.88 | 1.4 | 0.96 | −6.1 | 0.76 | 5.6 | 0.86 | 2.7 | 0.95 |
| PRIN8 | 7.2 | 0.77 | −2.6 | 0.88 | −8.9 | 0.96 | 5.5 | 0.77 | −2.8 | 0.86 | −5.6 | 0.96 |
| PRIN9 | 8.0 | 0.78 | −6.2 | 0.89 | −5.0 | 0.96 | 5.3 | 0.78 | −4.0 | 0.87 | −3.5 | 0.96 |
| PRIN10 | −10.1 | 0.81 | 11.3 | 0.90 | 1.2 | 0.96 | −8.0 | 0.80 | 8.8 | 0.88 | 1.2 | 0.96 |
Type I Error for Association Analysis
The main purpose of this report is to focus on developing a statistical method for combining family and unrelated samples. We thus examined the type I error of the proposed statistic S2 that can combine both family and unrelated samples with data generated from the four simulation scenarios. To illustrate how much power can be gained by using both parents' genotype and phenotype information, we also compared the proposed method with the transmission/disequilibrium test (TDT) with the parent-affected-child trio data only.19 We further compared S2 with Fisher's meta-analysis method37 of combining the p values of the family and unrelated case-control tests, resulting in a statistic that follows a chi-square distribution with 4 degrees of freedom. When only parent-affected-child trios are available, we also created an unaffected pseudo-child having the two alleles not transmitted from the parents to the affected child at each marker locus. We then analyzed the data with the proposed method and compared the type I error and power when using parent-affected-child data only. Table 2 presents the type I error for these test statistics when data were generated from the four simulation scenarios. The proposed test S2 has reasonable type I error for all the scenarios: with parent-affected-child trios only (S2), adding to each family an unaffected pseudo-child whose alleles are not transmitted when combining family and unrelated samples (S2∗), and separate analyses of family and unrelated samples. The TDT statistic also has reasonable type I error, as well as Fisher's method of combining p values with the p values obtained by the proposed method for family and unrelated data separately. We noticed that Fisher's method leads to significant inflation of the type I error rate for the Simulation 2 data with 800 markers in the principal component analysis. A possible reason for this may be that the number of markers in the principal component analysis is still not adequate.
Table 2.
Type I Error in Percent of the Test Statistics at the Nominal 5% and 1% Significance Levels When the Samples Were from Simulations 1 to 4
| No. of Markers |
S2 |
|
TDT |
|
|
|
Fisher's χ2 |
S2 |
|
TDT |
|
|
|
Fisher's χ2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5% | 1% | |||||||||||||||||||||
| Discrete, Simulation 1a | ||||||||||||||||||||||
| 200 | 5.3 | 5.80 | 5.65 | 5.4 | 5.65 | 5.60 | 5.25 | 1.5 | 1.45 | 1.2 | 0.85 | 0.95 | 1.0 | 1.2 | ||||||||
| 400 | 5.6 | 5.35 | 5.8 | 5.55 | 5.3 | 5.30 | 5.3 | 0.85 | 1.20 | 1.15 | 1.3 | 0.95 | 0.95 | 1.3 | ||||||||
| 800 | 5.46 | 5.83 | 5.29 | 5.21 | 5.21 | 5.13 | 5.5 | 1.08 | 1.17 | 0.79 | 1.04 | 1.21 | 0.92 | 1.13 | ||||||||
| Admixed, Simulation 2a | ||||||||||||||||||||||
| 200 | 5.65 | 4.90 | 5.35 | 6.0 | 5.45 | 4.25 | 5.65 | 1.05 | 0.75 | 0.95 | 1.2 | 1.05 | 1.1 | 1.0 | ||||||||
| 400 | 6.9 | 5.65 | 4.6 | 6.05 | 5.85 | 5.85 | 6.15 | 1.15 | 1.60 | 0.95 | 1.25 | 1.55 | 0.95 | 1.05 | ||||||||
| 800 | 5.67 | 4.83 | 6.08 | 5.63 | 5.88 | 5.46 | 7.77 | 1.17 | 1.25 | 1.12 | 1.54 | 1.58 | 1.13 | 1.63 | ||||||||
| Discrete, Simulation 3b | ||||||||||||||||||||||
| 10,000 | 5.47 | 5.13 | 4.94 | 6.61 | 4.52 | 4.61 | 5.73 | 1.25 | 0.99 | 1.02 | 1.28 | 0.85 | 0.77 | 1.28 | ||||||||
| Admixed, Simulation 4b | ||||||||||||||||||||||
| 10,000 | 4.34 | 4.34 | 5.33 | 4.35 | 5.24 | 5.28 | 4.73 | 0.84 | 0.81 | 0.95 | 0.75 | 1.04 | 0.90 | 0.80 | ||||||||
200 parent-affected-child trios, 200 cases, and 200 controls. Abbreviations: S2, the proposed method is applied to both family and unrelated data; S2∗, the proposed method is applied to both family and unrelated data, each family being augmented by an unaffected pseudo-child whose alleles were not transmitted; , the proposed method is applied to case-control data only; , the proposed method is applied to family data only; , the proposed method is applied to family data only, but each family being augmented by an unaffected pseudo-child whose alleles were not transmitted.
Type I error is calculated based on 1000 replications. Fisher's χ2 was calculated based on the S2 statistics for unrelated and families, respectively.
Type I error is calculated as the percentage of 10,000 SNPs reaching the nominal significance level.
Power Analysis
We also performed power analyses for the data generated from the four simulation scenarios. Table 3 presents the power of the test statistics when data were generated by Simulation 1 under multiplicative, additive, recessive, and dominant modes of inheritance. We used 200 SNPs to control for the effect of population stratification for Simulations 1 and 2. The proposed test gains substantial power when compared with separate analyses and better power than analyses combined by Fisher's method. The power of using the unaffected pseudo-children is slightly better than without using them. Interestingly, the proposed method improves the power substantially over the TDT method, indicating that parental phenotype information does contribute information in association analysis. The results for the data generated in Simulation 2 are similar (Table 4).
Table 3.
Power for Test Statistics at the Nominal Significance Levels 5% and 1% When Samples Are from Simulation 1
| Significance Level | S2 | TDT | Fisher's χ2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Multiplicative: rDD = 0.225, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 96.8 | 96.8 | 72.4 | 65.5 | 84.8 | 86.2 | 94.8 | ||||
| 1% | 87.4 | 88.8 | 51.4 | 40.4 | 65.4 | 68.3 | 83.0 | ||||
| Additive: rDD = 0.2, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 91.7 | 92.6 | 63.7 | 57.5 | 79.4 | 80.3 | 89.0 | ||||
| 1% | 80.6 | 81.5 | 39.0 | 34.4 | 53.4 | 55.1 | 75.5 | ||||
| Recessive: rDD = 0.2, rDd = 0.1, rdd = 0.1 | |||||||||||
| 5% | 47.8 | 50.2 | 31.9 | 23.4 | 35.9 | 36.8 | 43.0 | ||||
| 1% | 27.2 | 28.2 | 13.8 | 9.1 | 17.4 | 18.7 | 22.1 | ||||
| Dominant: rDD = 0.2, rDd = 0.2, rdd = 0.1 | |||||||||||
| 5% | 99.0 | 99.1 | 84.9 | 83.3 | 93.9 | 94.8 | 98.6 | ||||
| 1% | 96.3 | 96.8 | 65.5 | 62.7 | 81.4 | 83.2 | 94.7 | ||||
Power was calculated based on 1000 replications. 200 parent-affected-child trios, 200 cases, and 200 controls. 200 SNPs were used. Disease allele frequency is 0.3 and 0.2 in Africans and Europeans, respectively. 200 SNPs were simulated to correct for population stratification. Abbreviations: S2, the proposed method is applied to both family and unrelated data; S2∗, the proposed method is applied to both family and unrelated data, each family being augmented by an unaffected pseudo-child whose alleles are not transmitted; , the proposed method is applied to case-control data only; , the proposed method is applied to family data only; , the proposed method is applied to family data only, but each family being augmented by an unaffected pseudo-child whose alleles were not transmitted.
Table 4.
Power for Test Statistics at the Nominal Significance Levels 5% and 1% When Samples Are from Simulation 2
| Significance Level | S2 | TDT | Fisher's χ2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Multiplicative: rDD = 0.225, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 99.0 | 99.2 | 78.4 | 78.6 | 91.4 | 91.4 | 98.2 | ||||
| 1% | 95.9 | 96.4 | 56.1 | 55.2 | 76.5 | 78.6 | 92.7 | ||||
| Additive: rDD = 0.2, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 97.4 | 97.6 | 67.6 | 68.9 | 84.7 | 85.6 | 95.3 | ||||
| 1% | 90.7 | 92.4 | 44.6 | 45.4 | 65.4 | 67.0 | 85.3 | ||||
| Recessive: rDD = 0.2, rDd = 0.1, rdd = 0.1 | |||||||||||
| 5% | 70.3 | 70.0 | 38.0 | 36.5 | 49.0 | 50.7 | 61.8 | ||||
| 1% | 47.1 | 49.3 | 18.6 | 16.2 | 24.4 | 26.1 | 37.8 | ||||
| Dominant: rDD = 0.2, rDd = 0.2, rdd = 0.1 | |||||||||||
| 5% | 100 | 100 | 86.4 | 89.4 | 97.1 | 97.6 | 99.7 | ||||
| 1% | 99.4 | 99.5 | 64.2 | 71.4 | 90.1 | 90.7 | 98.3 | ||||
For details and abbreviations, see legend to Table 3.
We next compared the power for the samples generated by Simulations 3 and 4, which consist of admixtures of three populations. The results are also similar to those of data generated from Simulations 1 and 2 (Tables 5 and 6).
Table 5.
Power for Test Statistics at Significance Levels 5% and 1% When Samples Are from Simulation 3
| Significance Level | S2 | TDT | Fisher's χ2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Multiplicative: rDD = 0.225, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 99.5 | 99.4 | 79.0 | 88.8 | 94.1 | 99.8 | 99.3 | ||||
| 1% | 98.2 | 98.3 | 55.8 | 71.3 | 85.2 | 85.4 | 97.1 | ||||
| Additive: rDD = 0.2, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 98.5 | 98.8 | 66.3 | 75.6 | 84.8 | 85.6 | 75.7 | ||||
| 1% | 91.9 | 92.2 | 39.6 | 51.6 | 67.6 | 68.1 | 87.1 | ||||
| Recessive: rDD = 0.2, rDd = 0.1, rdd = 0.1 | |||||||||||
| 5% | 98.5 | 98.2 | 71.6 | 78.1 | 86.8 | 88.0 | 96.4 | ||||
| 1% | 92.6 | 93.0 | 49.1 | 55.8 | 68.6 | 69.9 | 87.7 | ||||
| Dominant: rDD = 0.2, rDd = 0.2, rdd = 0.1 | |||||||||||
| 5% | 96.8 | 96.9 | 58.0 | 73.8 | 82.1 | 84.5 | 94.7 | ||||
| 1% | 89.6 | 91.0 | 32.1 | 49.2 | 61.5 | 66.0 | 83.5 | ||||
For details and abbreviations, see legend to Table 3.
Table 6.
Power for Test Statistics at Significance Levels 5% and 1% When Samples Are from Simulation 4
| Significance Level | S2 | TDT | Fisher's χ2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Multiplicative: rDD = 0.225, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 99.9 | 99.8 | 80.7 | 91.3 | 94.8 | 96.0 | 99.6 | ||||
| 1% | 99.2 | 99.0 | 58.4 | 77.5 | 85.9 | 87.1 | 98.7 | ||||
| Additive: rDD = 0.2, rDd = 0.15, rdd = 0.1 | |||||||||||
| 5% | 98.7 | 98.9 | 66.5 | 79.7 | 85.1 | 86.3 | 97.8 | ||||
| 1% | 95.6 | 95.5 | 42.8 | 57.5 | 69.2 | 69.9 | 90.3 | ||||
| Recessive: rDD = 0.2, rDd = 0.1, rdd = 0.1 | |||||||||||
| 5% | 99.8 | 99.8 | 77.7 | 87.1 | 92.3 | 93.3 | 99.4 | ||||
| 1% | 98.5 | 98.3 | 55.3 | 67.1 | 78.6 | 78.8 | 96.8 | ||||
| Dominant: rDD = 0.2, rDd = 0.2, rdd = 0.1 | |||||||||||
| 5% | 96.6 | 97.1 | 57.6 | 73.3 | 82.2 | 83.9 | 94.6 | ||||
| 1% | 90.9 | 92.6 | 33.0 | 51.4 | 62.0 | 63.9 | 83.8 | ||||
For details and abbreviations, see legend to Table 3.
Application to Angiotensin I-Converting Enzyme Data
The rennin-angiotensin system (RAS) plays a key role in blood-pressure regulation. The angiotensin I-converting enzyme (ACE [MIM 106180]) is a key component of the RAS because it catalyzes the conversion of angiotensin I to angiotensin II, a potent vasoconstrictor that leads to the constriction of blood vessels and retention of salt and water. The ACE gene polymorphism has been extensively studied,38–40 although a causative relationship between the ACE gene and hypertension is still not established. Bouzekri et al.41 described the association between 13 variants in the ACE gene at an average distance of 2 kb apart and the ACE plasma level in three population samples, from Nigeria, Jamaica, and an African-American community in the US. Several polymorphisms have been shown to be significantly associated with plasma ACE level, with ACE8 being the most significant one. A portion of the Nigerian and US samples have also been genotyped with microsatellite markers by the Mammalian Genotyping Service in Marshfield, WI.42,43 To illustrate the application of our method, we tested whether the association evidence of these 13 SNPs can be improved on combining the Nigerian and US samples, by comparing with FBAT,21 which applies only to family data.
The data consist of 312 Nigerian and 312 US families, respectively. We were able to identify 428 individuals from 119 Nigerian nuclear families, 66 unrelated Nigerians, and 32 unrelated US individuals, who have available 13 polymorphisms in the ACE gene and 269 overlapping microsatellite markers across the genome. The missing genotyping rate of each individual is less than 15%. We recoded a Nigerian as affected if his/her ACE level is greater than 715 and unaffected otherwise. Similarly, a US individual is considered as affected if his/her ACE level is greater than 634 and unaffected otherwise. These thresholds are calculated by adding one standard deviation to the population mean in the corresponding populations.21 For simplicity, we dichotomized the 269 microsatellite markers based on the mean of the marker values, i.e, we recorded a microsatellite marker allele as 1 if it is less than its average and 2 otherwise. Any missing marker value was imputed by using the marker mean. We performed the principal component analysis with all the parents from each of the families and the unrelated Nigerian and Maywood individuals, for a total 324 persons. Our analysis based on these samples, including both the family and unrelated individuals, clearly demonstrated that the proposed method has increased power over that of FBAT with only the Nigerian family data (Table 7). We also observed that 7.4% of the microsatellite markers reach the 5% significance level. Although this rate is relatively high, which could be due to some association or incomplete control of population stratification, because of the relatively small number of microsatellites, this value is within the 95% confidence interval for a true 5% value.
Table 7.
The Association of the ACE Polymorphisms and the ACE Plasma Level Analyzed by Proposed Method and FBAT
|
S2 |
FBAT |
|
|---|---|---|
| Marker | p Value | p Value |
| ACE1 | 0.153 | 0.734 |
| ACE2 | 0.102 | 0.804 |
| ACE3 | 0.127 | 0.796 |
| ACE4 | 0.036 | 0.121 |
| ACE5 | 0.224 | - |
| ACEs12 | 0.779 | 0.438 |
| ACEs11 | 0.012 | 0.675 |
| ACE6 | 0.950 | 0.317 |
| NewACE6 | 0.594 | - |
| ACE7 | 0.404 | 0.416 |
| I/D | 0.573 | 0.962 |
| ACE8 | 1.06 × 10−5 | 0.017 |
| ACE9 | 0.178 | 0.431 |
FBAT did not calculate the p value because of rare allele frequency. S2 is calculated based on both family and unrelated individuals, for a total of 526 individuals. FBAT is calculated based on the family data only, with a sample size of 428 individuals.
Discussion
We present a new method to combine family and unrelated samples, while avoiding the effects of population stratification. Unlike the method developed by Nagelkerke et al.32 and extended by Epstein et al.,33 the proposed method does not require us to test whether the data from different studies can be combined. In fact, the proposed method is able to integrate the data from either different or admixed populations and is therefore more general than the methods of Nagelkerke et al.32 and Epstein et al.33 Simulation studies suggest that the proposed new method is robust to population stratification and more powerful than the usual way of analyzing family and unrelated samples separately, and of then using Fisher's method of combining p values from the separate data sets. In addition, the proposed method uses both parental phenotype and genotype information and allows for multiple siblings. Compared to the TDT method, the proposed method improves power substantially, although the TDT does not require additional markers to be genotyped. Thus, we suggest that the proposed method should be used for a family study when data on many markers are available across the genome. When only parent-affected-child trios are available, our simulation studies suggest that using unaffected pseudo-children may slightly improve the power. However, further studies should be conducted in order to understand how much power can be gained for different population admixture models. The gain in power is mainly due to the effectively increased sample size available when the analysis of the family data is not conditional on parental information, and due to being a one degree of freedom test when compared to Fisher's method, which is a four degrees of freedom test. The methods of Nagelkerke et al.32 and Epstein et al.33 are sensitive to population stratification and require the assumption that the controls are from the same population as that of the parents. Because our method focuses on integrated samples from family and unrelated data, while correcting for population stratification, we did not directly compare it with the methods of Nagelkerke et al. and Epstein et al., who focus on testing whether the samples can be combined. Our simulated samples are not valid for the methods proposed by Nagelkerke et al. and Epstein et al. When the samples of family or unrelated persons are from the same population, our method should be expected to have more power than those of Nagelkerke et al. and Epstein et al., which use the conditional likelihood approach of the TDT and are less powerful than an association method that uses all the phenotype and genotype information in family data.30
The application of our method to the ACE data also demonstrates that combining family and unrelated data has an advantage over the method of using family data only. However, the type I error based on the 269 microsatellite markers is slightly high, although it is within the 95% confidence interval for a true 5% value. In general, a large number of markers are necessary to well control the effect of population stratification.
Our method can be easily applied to the association analysis of quantitative traits. However, a more powerful way may be based on using a multivariate regression framework in which a quantitative trait is assumed to be multivariate normally distributed. Although our method can be theoretically extended in an obvious manner to the analysis of large pedigrees, there are then many more familial correlations to be estimated and those based on pair types that are infrequent in the data set will not be accurately estimated. One possibility is to assume that all the familial correlations are simple functions of a single parameter, such as heritability.44 This idea was later extended to include the estimation of three more parameters (variance components that allow for extra sibling, marital, and/or nuclear family correlations),45 and this is implemented in ASSOC, part of the program package S.A.G.E. If the necessary markers are available, our method of using principal components to summarize the genetic data for inferring population structure from unrelated samples to family samples provides a way of making the association analysis implemented in ASSOC robust to stratification with more power than is afforded by the use of a transmitted allele indicator,26 which yields a TDT type of analysis. The use of the principal components calculated from marker genotype data has already been established for unrelated samples.6,11,12 The principal component analysis assumes that the samples are independent and calculating principal components will result in bias if applied naively to data with lager family sizes. Thus, we propose to calculate the principal components for the children through the eigenvectors calculated from the independent samples in the available data. We made the assumption that the axes of the principal components can be well represented by just the independent samples in the data. This is a reasonable assumption when the parental genotype data are available, because the children carry half of both parental genomes. When parental DNA is not available, we can randomly choose one of the siblings for the principal component analysis. This should not be a major impediment, provided that the average proportion from each ancestral population in the sample is not too small,46 so that the SNPs from each ancestral population are well represented.
When a study involves hundreds of thousands of markers, such as in a whole-genome association study, calculating the principal components is computationally intensive but less intensive than the MCMC approach.9 Because the principal components can be calculated through the singular value decomposition of the matrix of marker data, and the computation time is dependent on the singular value decomposition, Price et al.15 suggested that this calculation is rather fast. In fact, we found that calculating the principal components for 800 individuals and 10,000 SNPs took 3.5 min on the Intel Xeon 1.6 Ghz cluster. When the number of SNPs is extremely large, such as is the case when more than a million SNPs are available, an alternative approach for calculating principal components is a two-stage approach. First, we divide the markers into nonoverlapping subsets/chromosomes and calculate the principal components on every subset/chromosome. Then, the first L principal components on each subset/chromosome are used to calculate new principal components and these principal components are used to control the effect of population stratification. We compared this two-stage approach to directly calculating the principal components and found that very little information is lost. For example, we divided the 10,000 SNPs into 20 subsets each of 500 SNPs for the data generated in Simulations 3 and 4 and then calculated the principal components by the two-stage approach. First, we calculated the top 10 principal components in each of the 20 subsets, and we then calculated the top 10 principal components from the top 10 principal components from each of the 20 subsets. We found that the variation of true ancestry can be captured just as well by the first 10 principal components calculated this way as when calculated in one step (data not shown). The multistage approach can be fast and requires less computer memory in dealing with large data sets. Recently, Bauchet et al.14 suggested using principal coordinate analysis in which the principal components are calculated on people. However, with this method it is not feasible to infer the children's principal components.
In our analysis, we used the first 10 principal components for controlling the population stratification. This number was also suggested by Price et al.15 When hundreds of thousands of SNPs are available, even subtle population admixture can be detected by principal component analysis. However, using only the first 10 principal components may not be sufficient when a population is admixed with several ancestral populations. Thus, the method developed by Patterson et al.46 might be useful to find out how many principal components are necessary.
It should be noted that our regression of both the trait and test marker on the principal components is based on unrelated subjects only, while the test statistic is calculated on both related and unrelated subjects. This may bring additional variability into the denominator of the test statistic T, resulting in either E(g∗) or E(y∗) not being 0 under the null hypothesis of no association. However, our simulation studies suggested that both E(g∗) and E(y∗) = 0 under the null hypothesis of no association. We argue that the effect resulting from the population structure for the family members can be well predicted when a large number of markers across the genome are available. Therefore, E(g∗) and E(y∗) will still be close to 0, even if they are estimated based on both unrelated and related individuals. In the case of large pedigrees, we believe that, as long as the founders' genotype and phenotype values are available, our method should work well. However, in the case that many founders' genotype and phenotype information is missing, which is the case for many family studies, our method may result in too few individuals from which to obtain good estimates. In this case, the specific parts of the large pedigrees should be chosen for the purpose of estimating the regression coefficients. However, details of how to accomplish this will require further research. An alternative approach could be the mixed-model method developed by Yu et al.47 However, this method requires that population assignments be obtained from other methods, such as STRUCTURE.48
It should also be noted that pedigrees collected for linkage analyses may be selected differently from the subjects collected for a case-control study. For example, if the pedigree data are collected based on prevalent cases whereas the case-control study is based on incident cases, this could lead to survival bias. Clearly, this is an issue that should be considered in designing an appropriate case-control study when following up on a linkage study.
In summary, we developed a simple method to integrate the data from family-based studies and unrelated samples while correcting for population stratification. This method can be applied to both qualitative and quantitative traits and is more powerful than a method that analyzes family and unrelated samples separately, the former by a conditional approach and the latter by GC. This method should be useful for current association studies when different groups use different study designs. The program FamCC, to combine family and unrelated samples, will be available online and incorporated into the S.A.G.E. program package.
Appendix A
Under the null hypothesis, genotype value and phenotype value are independent.
where is the correlation of the random variable between individuals j1 and j2 within a family. The variances and correlations can be estimated from the data. Because of the different ascertainments between families and unrelated cases and controls, we suggest that the variances be estimated in families and unrelated cases and controls separately.
Web Resources
The URLs for data presented herein are as follows:
American Journal of Human Genetics, http://www.ajhg.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Statistical Analysis for Genetic Epidemiology (SAGE), http://darwin.cwru.edu/sage/
Acknowledgments
We thank two referees for their constructive comments. We also think Dr. S.L. Zhang for providing a C program to generate admixed samples based on the general continuous gene flow model. This work was supported in part by U.S. Public Health Service Research grants: HG003054 from the National Human Genome Research Institute, HL074166 from the National Heart, Lung, and Blood Institute, GM28356 from the National Institute of General Medical Sciences, and Cancer Center Support Grant P30CAD43703 from the National Cancer Institute. The 3-dimensional plots in Figures 4 and 5 were drawn by scatterplot3d.R.
References
- 1.Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 2.Risch N.J. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–856. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- 3.Knowler W.C., Williams R.C., Pettitt D.J., Steinberg A.G. Gm3;5,13,14 and type 2 diabetes mellitus: an association in American Indians with genetic admixture. Am. J. Hum. Genet. 1988;43:520–526. [PMC free article] [PubMed] [Google Scholar]
- 4.Lander E.S., Schork N.J. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 5.Marchini J., Cardon L.R., Phillips M.S., Donnelly P. The effects of human population structure on large genetic association studies. Nat. Genet. 2004;36:512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 6.Chen H.S., Zhu X., Zhao H., Zhang S. Qualitative semi-parametric test for genetic associations in case-control designs under structured populations. Ann. Hum. Genet. 2003;67:250–264. doi: 10.1046/j.1469-1809.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- 7.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 8.Hoggart C.J., Parra E.J., Shriver M.D., Bonilla C., Kittles R.A., Clayton D.G., McKeigue P.M. Control of confounding of genetic associations in stratified populations. Am. J. Hum. Genet. 2003;72:1492–1504. doi: 10.1086/375613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritchard J.K., Stephens M., Rosenberg N.A., Donnelly P. Association mapping in structured populations. Am. J. Hum. Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satten G.A., Flanders W.D., Yang Q. Accounting for unmeasured population substructure in case-control studies of genetic association using a novel latent-class model. Am. J. Hum. Genet. 2001;68:466–477. doi: 10.1086/318195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S., Zhu X., Zhao H. On a semiparametric test to detect associations between quantitative traits and candidate genes using unrelated individuals. Genet. Epidemiol. 2003;24:44–56. doi: 10.1002/gepi.10196. [DOI] [PubMed] [Google Scholar]
- 12.Zhu X., Zhang S., Zhao H., Cooper R.S. Association mapping, using a mixture model for complex traits. Genet. Epidemiol. 2002;23:181–196. doi: 10.1002/gepi.210. [DOI] [PubMed] [Google Scholar]
- 13.Pritchard J.K., Rosenberg N.A. Use of unlinked genetic markers to detect population stratification in association studies. Am. J. Hum. Genet. 1999;65:220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauchet M., McEvoy B., Pearson L.N., Quillen E.E., Sarkisian T., Hovhannesyan K., Deka R., Bradley D.G., Shriver M.D. Measuring European population stratification with microarray genotype data. Am. J. Hum. Genet. 2007;80:948–956. doi: 10.1086/513477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 16.Cavalli-Sforza L.L., Menozzi P., Piazza A. Princeton University Press; Princeton, NJ: 1994. The History and Geography of Human Genes. [Google Scholar]
- 17.Gower J.C. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika. 1966;53:325–338. [Google Scholar]
- 18.Epstein M.P., Allen A.S., Satten G.A. A simple and improved correction for population stratification in case-control studies. Am. J. Hum. Genet. 2007;80:921–930. doi: 10.1086/516842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spielman R.S., McGinnis R.E., Ewens W.J. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am. J. Hum. Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- 20.Boehnke M., Langefeld C.D. Genetic association mapping based on discordant sib pairs: the discordant-alleles test. Am. J. Hum. Genet. 1998;62:950–961. doi: 10.1086/301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laird N.M., Horvath S., Xu X. Implementing a unified approach to family-based tests of association. Genet. Epidemiol. 2000;19(Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 22.Schaid D.J. General score tests for associations of genetic markers with disease using cases and their parents. Genet. Epidemiol. 1996;13:423–449. doi: 10.1002/(SICI)1098-2272(1996)13:5<423::AID-GEPI1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Spielman R.S., Ewens W.J. A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am. J. Hum. Genet. 1998;62:450–458. doi: 10.1086/301714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao H., Zhang S., Merikangas K.R., Trixler M., Wildenauer D.B., Sun F., Kidd K.K. Transmission/disequilibrium tests using multiple tightly linked markers. Am. J. Hum. Genet. 2000;67:936–946. doi: 10.1086/303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allison D.B. Transmission-disequilibrium tests for quantitative traits. Am. J. Hum. Genet. 1997;60:676–690. [PMC free article] [PubMed] [Google Scholar]
- 26.George V., Tiwari H.K., Zhu X., Elston R.C. A test of transmission/disequilibrium for quantitative traits in pedigree data, by multiple regression. Am. J. Hum. Genet. 1999;65:236–245. doi: 10.1086/302444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rabinowitz D. A transmission disequilibrium test for quantitative trait loci. Hum. Hered. 1997;47:342–350. doi: 10.1159/000154433. [DOI] [PubMed] [Google Scholar]
- 28.Xiong M.M., Krushkal J., Boerwinkle E. TDT statistics for mapping quantitative trait loci. Ann. Hum. Genet. 1998;62:431–452. doi: 10.1046/j.1469-1809.1998.6250431.x. [DOI] [PubMed] [Google Scholar]
- 29.Zhu X., Elston R.C., Cooper R.S. Testing quantitative traits for association and linkage in the presence or absence of parental data. Hum. Hered. 2001;51:183–191. doi: 10.1159/000053341. [DOI] [PubMed] [Google Scholar]
- 30.Zhu X., Elston R.C. Transmission/disequilibrium tests for quantitative traits. Genet. Epidemiol. 2001;20:57–74. doi: 10.1002/1098-2272(200101)20:1<57::AID-GEPI6>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 31.Hirschhorn J.N., Daly M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 32.Nagelkerke N.J., Hoebee B., Teunis P., Kimman T.G. Combining the transmission disequilibrium test and case-control methodology using generalized logistic regression. Eur. J. Hum. Genet. 2004;12:964–970. doi: 10.1038/sj.ejhg.5201255. [DOI] [PubMed] [Google Scholar]
- 33.Epstein M.P., Veal C.D., Trembath R.C., Barker J.N., Li C., Satten G.A. Genetic association analysis using data from triads and unrelated subjects. Am. J. Hum. Genet. 2005;76:592–608. doi: 10.1086/429225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith M.W., Patterson N., Lautenberger J.A., Truelove A.L., McDonald G.J., Waliszewska A., Kessing B.D., Malasky M.J., Scafe C., Le E. A high-density admixture map for disease gene discovery in african americans. Am. J. Hum. Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu X., Zhang S., Tang H., Cooper R. A classical likelihood based approach for admixture mapping using EM algorithm. Hum. Genet. 2006;120:431–445. doi: 10.1007/s00439-006-0224-z. [DOI] [PubMed] [Google Scholar]
- 36.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher R.A. Thirteenth Edition. Oliver & Loyd; London: 1925. Statistical Methods for Research Workers. [Google Scholar]
- 38.Zhu X., McKenzie C.A., Forrester T., Nickerson D.A., Broeckel U., Schunkert H., Doering A., Jacob H.J., Cooper R.S., Rieder M.J. Localization of a small genomic region associated with elevated ACE. Am. J. Hum. Genet. 2000;67:1144–1153. doi: 10.1016/s0002-9297(07)62945-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu X., Bouzekri N., Southam L., Cooper R.S., Adeyemo A., McKenzie C.A., Luke A., Chen G., Elston R.C., Ward R. Linkage and association analysis of angiotensin I-converting enzyme (ACE)-gene polymorphisms with ACE concentration and blood pressure. Am. J. Hum. Genet. 2001;68:1139–1148. doi: 10.1086/320104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox R., Bouzekri N., Martin S., Southam L., Hugill A., Golamaully M., Cooper R., Adeyemo A., Soubrier F., Ward R. Angiotensin-1-converting enzyme (ACE) plasma concentration is influenced by multiple ACE-linked quantitative trait nucleotides. Hum. Mol. Genet. 2002;11:2969–2977. doi: 10.1093/hmg/11.23.2969. [DOI] [PubMed] [Google Scholar]
- 41.Bouzekri N., Zhu X., Jiang Y., McKenzie C.A., Luke A., Forrester T., Adeyemo A., Kan D., Farrall M., Anderson S. Angiotensin I-converting enzyme polymorphisms, ACE level and blood pressure among Nigerians, Jamaicans and African-Americans. Eur. J. Hum. Genet. 2004;12:460–468. doi: 10.1038/sj.ejhg.5201166. [DOI] [PubMed] [Google Scholar]
- 42.Cooper R.S., Luke A., Zhu X., Kan D., Adeyemo A., Rotimi C., Bouzekri N., Ward R. Genome scan among Nigerians linking blood pressure to chromosomes 2, 3, and 19. Hypertension. 2002;40:629–633. doi: 10.1161/01.hyp.0000035708.02789.39. [DOI] [PubMed] [Google Scholar]
- 43.Zhu X., Cooper R.S., Luke A., Chen G., Wu X., Kan D., Chakravarti A., Weder A. A genome-wide scan for obesity in African-Americans. Diabetes. 2002;51:541–544. doi: 10.2337/diabetes.51.2.541. [DOI] [PubMed] [Google Scholar]
- 44.George V.T., Elston R.C. Testing the association between polymorphic markers and quantitative traits in pedigrees. Genet. Epidemiol. 1987;4:193–201. doi: 10.1002/gepi.1370040304. [DOI] [PubMed] [Google Scholar]
- 45.Elston R.C., George V.T., Severtson F. The Elston-Stewart algorithm for continuous genotypes and environmental factors. Hum. Hered. 1992;42:16–27. doi: 10.1159/000154043. [DOI] [PubMed] [Google Scholar]
- 46.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J., Pressoir G., Briggs W.H., Vroh B., Yamasaki I., Doebley M., Mc J.F., Mullen M.D., Gaut B.S., Nielsen D.M. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- 48.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]