

Abstract
Epigenetic misregulation is consistent with various non-Mendelian features of schizophrenia and bipolar disorder. To date, however, few studies have investigated the role of DNA methylation in major psychosis, and none have taken a genome-wide epigenomic approach. In this study we used CpG-island microarrays to identify DNA-methylation changes in the frontal cortex and germline associated with schizophrenia and bipolar disorder. In the frontal cortex we find evidence for psychosis-associated DNA-methylation differences in numerous loci, including several involved in glutamatergic and GABAergic neurotransmission, brain development, and other processes functionally linked to disease etiology. DNA-methylation changes in a significant proportion of these loci correspond to reported changes of steady-state mRNA level associated with psychosis. Gene-ontology analysis highlighted epigenetic disruption to loci involved in mitochondrial function, brain development, and stress response. Methylome network analysis uncovered decreased epigenetic modularity in both the brain and the germline of affected individuals, suggesting that systemic epigenetic dysfunction may be associated with major psychosis. We also report evidence for a strong correlation between DNA methylation in the MEK1 gene promoter region and lifetime antipsychotic use in schizophrenia patients. Finally, we observe that frontal-cortex DNA methylation in the BDNF gene is correlated with genotype at a nearby nonsynonymous SNP that has been previously associated with major psychosis. Our data are consistent with the epigenetic theory of major psychosis and suggest that DNA-methylation changes are important to the etiology of schizophrenia and bipolar disorder.
Introduction
Schizophrenia (SZ) [MIM 181500] and bipolar disorder (BD) [MIM 125480] are etiologically related psychiatric conditions,1 together termed “major psychosis.” Studies of major psychosis have focused primarily on the interplay between genetic and environmental risk factors. Twin and adoption studies highlight a clear inherited component to both disorders,2 but whereas replicated findings exist for a number of genes, association studies are characterized by nonreplication, small effect sizes, and significant heterogeneity.3 Several epidemiological, clinical, and molecular peculiarities associated with major psychosis are difficult to explain with traditional gene- and environment-based approaches. Such peculiarities include the noncomplete concordance between monozygotic twins for both SZ (41%–65%) and BD (∼60%),2,4 which cannot be accounted for by only environmental factors.3,5 Other complexities of major psychosis include a fluctuating disease course with periods of remission and relapse, sexual dimorphism, peaks of susceptibility to disease coinciding with major hormonal rearrangements, and parent-of-origin effects.3 These observations have led to speculation about the importance of epigenetic factors in mediating susceptibility to both SZ and BD.3
Epigenetics refers to the heritable, but reversible, regulation of various genetic functions, including gene expression, mediated through modifications of DNA and histones.6 Epigenetic processes are essential for normal cellular development and differentiation, and they allow the regulation of gene function through nonmutagenic mechanisms. The impact of DNA methylation on gene activity has been explained by two proven mechanisms. The “critical site” model puts an emphasis on the methylation of specific cytosines in transcription-factor binding sites, responsible for reducing binding affinity and thus the transcription of mRNA.7 The “methylation density” model suggests that the proportion of methylated cytosines across a region, rather than at any specific position, controls chromatin conformation and thus the transcriptional potential of the gene.7
The epigenetic model of major psychosis is based upon three general principles.3 First, like the DNA sequence, the epigenetic profile of somatic cells is mitotically inherited, but unlike the DNA sequence, epigenetic signals are dynamic. The epigenetic status of the genome is tissue-specific, developmentally regulated, and influenced by both stochastic and environmental factors. Second, because epigenetic processes regulate various genetic and genomic functions, epigenetic factors can have profound phenotypic effects. Genes, even those containing no mutations or disease-predisposing polymorphisms, can be harmful if not expressed in the appropriate amount, at the correct time of the cell cycle, or in the correct compartment of the nucleus. Third, some epigenetic signals, rather than being reset and erased during gametogenesis, could be transmitted meiotically across generations.8 This has obvious ramifications for the identification of the molecular substrate of inherited predisposition, in which heritable phenotypic variation is assumed to result exclusively from DNA-sequence variants.
To date, few studies have investigated the role of epigenetic factors in major psychosis, and none has taken a genome-wide epigenomic approach. DNA-methylation differences have been reported in the vicinity of both catechol-O-methyltransferase (COMT)9 and reelin (RELN),10 although these findings were not confirmed using fully quantitative methylation-profiling methods.11,12 In this article we report findings from a comprehensive epigenomic study of major psychosis. Using DNA from the frontal cortex—a region previously implicated in the etiology of major psychosis13—derived from individuals with SZ, BD, and from matched controls (CTRL), we examined DNA methylation by utilizing two complementary approaches. First, we performed a microarray-based epigenomic scan of major psychosis using CpG-island microarrays after enrichment of the unmethylated fraction of brain DNA. Second, we performed a hypothesis-driven analysis of DNA methylation across candidate genes for which a priori evidence for a role in the etiology of major psychosis exists. In addition, to investigate whether epigenetic differences could be observed in the germline, we also used CpG-island microarrays to profile germline DNA methylation in BD patients and controls. Consistent with the epigenetic theory of major psychosis, we find considerable evidence for epigenetic changes associated with schizophrenia and bipolar disorder.
Material and Methods
Samples
Frontal-cortex postmortem brain tissue from individuals with DSM-IV diagnosed SZ (n = 35), BD (n = 35), and matched controls (n = 35) was provided by the Stanley Medical Research Institute brain-array collection (courtesy of Drs. Michael B. Knable, E. Fuller Torrey, Maree J. Webster, and Robert H. Yolken). The samples consisted of frozen tissue sections, which were stored at −80°C prior to DNA extraction. Demographic data associated with these samples are summarized in Table 1. In addition, germline-DNA samples were obtained from mature spermatozoa of BD patients (n = 20) and unaffected controls (n = 20) from an ongoing study at the Centre for Addiction and Mental Health (Toronto, Canada). These individuals were matched for age (BD mean age = 44.2; CTRL mean age = 41.3) and ethnic background (all individuals of European ancestry). Extraction of all DNA was performed with a standard phenol-chloroform extraction method. The quality and quantity of DNA was assessed by spectrophotometry and agarose-gel analysis, and DNA was subsequently stored at −20°C until further use. Samples for which good-quality, nondegraded DNA was not available were excluded from subsequent analyses. The project has been fully approved by the Ethics Committee of the Centre for Addiction and Mental Health, Toronto.
Table 1.
Demographic Data Associated with Frontal-Cortex Brain-Tissue Samples Utilized in this Study
| CTRL | SZ | BD | |
|---|---|---|---|
| Number of Tissue Samples | 35 | 35 | 35 |
| Number Hybridized | 28 | 35 | 32 |
| Mean Age and Range | 44.1 (31-59) | 42.6 (19-59) | 45.3 (19-64) |
| Race | 35 white | 35 White | 33 White 1 Black 1 Native American |
| Sex | 26M, 9F | 26M, 9F | 17M, 18F |
| Diagnosis | no axis I | 27 undifferentiated 7 paranoid 1 disorganized | 26 BP I 4 BP II 4 BP NOS 1 BP-SA |
| Psychotic Features | 0 | 35+ | 20+, 11−, 4 unclear |
| Cause of Death | 32 cardiac 3 other medical | 14 cardiac 13 other medical 1 accident 7 suicide | 12 cardiac 4 other medical 4 accidents 15 suicide |
| Postmortem Interval | Male: 26.9 (11.7) Female: 35.50 (9.6) | Male: 31.4 (17.0) Female: 30.88 (9.1) | Male: 37.8 (20.6) Female: 39.59 (17.7) |
| Refrigerator Interval | Male: 2.9 (1.6) Female: 2.83 (0.4) | Male: 5.67 (4.3)∗ Female: 6.25 (4.1) | Male: 7.80 (7.4)∗ Female: 13.18 (12.7)∗ |
| Brain pH | Male: 6.65 (0.26) Female: 6.45 (0.30) | Male: 6.44 (0.25)∗ Female: 6.61 (0.17) | Male: 6.46 (0.26)∗ Female: 6.44 (0.29) |
| Brain Weight | Male: 1486.4 (137.9) Female: 1314.2 (106.3) | Male: 1444.6 (104.8) Female: 1418.1 (128.8) | Male: 1467.6 (112.9) Female: 1316.2 (111.4) |
| Lifetime Alcohol Use | Male: 0.73 (0.94) Female: 1.00 (1.55) | Male: 2.36 (2.00)∗∗ Female: 1.38 (2.00) | Male: 2.86 (1.70)∗∗ Female: 2.29 (1.86)∗ |
| Lifetime Drug Use | Male: 0.27 (0.70) Female: 0.17 (0.41) | Male: 2.08 (2.00)∗∗ Female: 0.29 (0.76) | Male: 2.93 (2.05)∗∗ Female: 1.76 (1.8)∗ |
Summary of demographic data for the brain samples obtained from the Stanley Foundation. Comparison of brain samples utilized for microarray-based epigenomic profiling. Given are the mean for each group (with SD). Affected individuals were found to have significantly lower brain pH (male SZ, male BD) and higher lifetime alcohol and drug use (male SZ, male BD, female BD) compared to unaffected controls of the same sex, reflecting data reported elsewhere. None of these demographic variables was correlated with DNA methylation.
∗denotes a t test of p < 0.05 comparing affected group with controls.
∗∗denotes a t test of p < 0.001 comparing affected group with controls.
Enrichment of Unmethylated DNA and Microarray Hybridization
We used our developed technology for enrichment of the unmethylated DNA fraction and for epigenetic profiling with microarrays, described in detail elsewhere.14 In brief, the methylation-sensitive restriction enzyme HpaII (New England Biolabs) was used to digest 1 μg of genomic DNA. DNA adaptors (annealing products of two primers, U-CG1A and U-CG1B [see Table S1]) were ligated to the cleaved DNA fragments, followed by treatment with McrBC (New England Biolabs), which digests DNA fragments containing two or more methylated cytosines, thereby further enriching the unmethylated fraction. Adaptor-PCR amplification of the ligated products, with the use of primers complementary to the adaptor sequence, consisted of 250 ng of ligated DNA, 2.5 mM MgCl2, 0.2 mM aminoallyl-dNTPs (15 mM aminoallyl–2′-deoxyuridine 5′-triphosphate, 10 mM 2′-deoxythymidine 5′-triphosphate, and 25 mM each of 2′-deoxycytidine 5′-triphosphate, 2′-deoxyguanosine 5′-triphosphate, and 2′-deoxyadenosine 5′-triphosphate), 200 pmol primer U-CG1B, and 5 U Taq polymerase (New England Biolabs) in 1 × PCR reaction buffer (Sigma), to a final volume of 100 μl. PCR conditions are adjusted in such a way that fragments < 1.5 kb (i.e., those that are digested, short, and thus unmethylated) will amplify preferentially. Cycling consisted of an initial cycle at 72°C for 5 min and 95°C for 1 min, 25 cycles at 95°C for 40 s and 68°C for 2 min 30 s, and a final extension at 72°C for 5 min. Given that the role of epigenetic effects in disease etiology could be sex-specific and that considerable differences are observed in the course and prognosis of major psychosis between males and females, we split our sample according to gender. For the brain samples, equal amounts of amplicons from CTRL male samples were mixed to form a male common-reference pool, and equal amounts of amplicons from CTRL female samples were mixed to form a female common-reference pool. Individual samples, including all CTRL samples, were then cohybridized with the relevant common-reference pool sample. For the germline samples, all samples were cohybridized with a common-reference pool made by combination of amplicons from all CTRL samples. Samples were hybridized on 12,192 CpG-island microarrays obtained from the University Health Network Microarray Facility in Toronto. For the brain samples, good-quality-DNA extraction, enrichment, and microarray hybridization was successful for 28 CTRL samples, 35 SZ samples, and 32 BD samples. Seven of the initial 35 CTRL samples and three of the initial 35 BD samples were excluded from the experiment on the basis of degraded DNA that could compromise efficient enrichment of the unmethylated DNA fraction. For the germline samples, good-quality-DNA extraction, enrichment, and microarray hybridization was successful for 19 CTRL samples and 20 BD samples. One CTRL sample was excluded on the basis of poor DNA quantity and quality following nucleic-acid extraction.
Microarray Data Preprocessing
Initial array-image processing and quality control was performed with GenePix Pro 6.0 (Molecular Devices). The array signals were background-corrected with NormExp and normalized with weighted block-by-block LOWESS normalization. Spots with ambiguous genome locations, including spots with no sequence or annotation, repetitive spots, and translocation hotspots were removed, leaving a total of 7834 spots.
Normality Testing
Several analyses assumed data to be drawn from a normal distribution; hence the need for normality testing. Log-intensity ratios for each spot were subjected to the Lilliefors test for normality. The resultant p values for all spots were adjusted for multiple testing by use of Benjamini and Hochberg's false-discovery rate (FDR) method.15
Microarray-Data Analysis
For analyses comparing affected individuals to unaffected individuals, affected samples were either grouped by diagnosis (i.e., SZ versus CTRL and BD versus CTRL) or, given increasing evidence for an etiological overlap between SZ and BD,1,16 grouped together into a “major psychosis” group (i.e., psychosis versus CTRL). Limma was used to analyze each array spot for differential methylation between affected and unaffected samples. Each spot was assigned a raw p value based on a moderated t statistic. To correct for multiple testing, the set of raw p values were converted to false-discovery rates (FDR) according to Benjamini and Hochberg.15
Gene-Ontology Analysis
A novel gene-ontological investigation approach was designed to determine if any common functional trends are associated with the genes exhibiting differences between groups. For each group interrogated, only those loci exhibiting a significance value of less than p = 0.01 from a spotwise t test were selected, in order to include only those loci likely to have a true DNA-methylation difference between groups. Gene IDs within 1 kb of these array loci were obtained from the microarray annotation data and cross-referenced with the April 2007 build of the Gene Ontology Database to obtain gene-ontology (GO) categories associated with each microarray locus. All loci and corresponding mean fold change values were sorted into categories on the basis of their GO classifications, and the distribution of each GO category was compared with a paired t test and the more conservative Wilcoxon Signed Rank test. In both cases, p values were adjusted with FDR to correct for multiple testing. Data were then sorted by FDR p value, revealing the most significantly different GO categories.
Network Analysis of Microarray Data
In order to investigate whether DNA methylation is coordinated across different loci, we utilized a novel network-based approach.17 For brain samples, this analysis was performed on twenty male SZ samples and on an equal number of male CTRL samples—the other diagnostic groups were not included in this analysis because of their small sample sizes. We identified the top 700 methylation-variable spots across the samples in each group. The union of these two sets, consisting of 1041 spots, was chosen for network reconstruction. To find connections between methylation at specific genomic regions (nodes), their methylation log intensities were modeled by a linear combination of the methylation log intensities at the remaining spots. After regression, the correlation between the minimized residuals was calculated for measuring the direct association between the two spots. Estimation of correlation and p value was accomplished by a regularized covariance estimator that addresses the issue of small size and a larger number of variables (20 is much smaller than 1042).18 As a control for the network analysis, in each of the 20 CTRL microarrays, we randomized the IDs of the 1041 spots and proceeded with the same estimator. A raw p value of 10−7 was then chosen to cut off the insignificant pairwise correlations. A connection was drawn between a pair of spots whose correlation p value survived the cut. The structure of each network was explored by calculation of the transitivity (quantification of the connectivity between a spot's neighbors) and assortativity (quantification of the tendency of attachment between high-connection spots). The modular structure of a network was detected by a partitioning algorithm17 that maximizes the within-module connection densities at the expense of between-module connection densities. The analysis was repeated on the germline BD and CTRL samples.
Correlation with Lifetime Antipsychotic Use
Linear regression was performed on psychosis patients, with log-intensity ratios for each spot applied as dependent variables and lifetime dosages of antipsychotics applied as independent variables. Base-2 logarithms of the dosages were taken for the regression due to their wide spread. After the regression, p values based on F-statistics were gathered for all spots and converted to FDR to control for multiple testing.
Bisulfite Treatment of Genomic DNA
Bisulfite treatment was performed by use of a standard protocol. In brief, ∼500 ng of genomic DNA was denatured in 0.3 M NaOH for 15 min at 37°C. After the addition of freshly prepared 3.5 M sodium metabisulfite (Sigma) and 1 mM Hydroquinone (Sigma) solution, samples were subjected to a 5 hr incubation at 55°C under exclusion of light. The samples were then purified with QIAGEN DNA-purification columns. Recovered samples were desulfonated in 0.3 M NaOH for 15 min at 37°C and neutralized. DNA was precipitated overnight in ethanol at −20°C and resuspended in 50 μl buffer EB (QIAGEN). Bisulfite-treated DNA was stored at −80°C until needed.
Bisulfite Primer Design and PCR Amplification
Primers were designed with either MethPrimer (available online) or Pyrosequencing Assay Design Software v1.0.6 (Biotage, Uppsala, Sweden). For loci nominated from microarray analyses, primers were designed, where possible, to span a region containing potentially informative HpaII sites in the vicinity of the significant clone on the CpG-island microarray. Where necessary, larger regions were covered by use of several overlapping amplicons. For selected candidate genes, the primary focus of analysis was promoter CpG islands. In some cases (e.g., COMT and BDNF), additional exonic regions in the vicinity of known genetic polymorphisms were also investigated. Where candidate genes had been previously investigated by other groups (RELN and COMT), we aimed to ensure that the same regions were adequately covered by our analyses. A full list of primer sequences and annealing temperatures for each PCR reaction can be found in Table S1. PCR amplifications were performed with a standard hot-start PCR protocol in 25 μl volume reactions containing 3 μl of sodium-bisulfite-treated DNA, 1 μM primers, and a master mix containing hot-start Taq polymerase (Sigma). All PCR reactions were checked on a 1.0% agarose gel to ensure successful amplification and specificity before proceeding with pyrosequencing or MS-SNuPe.
Site-Specific DNA-Methylation Analysis with Pyrosequencing and MS-SNuPe
For pyrosequencing analysis, bisulfite-PCR products were processed according to the manufacturer's standard protocol (Biotage). In brief, 4 μl of streptavidin-sepharose beads (Amersham Biosciences, Piscataway, NJ, USA) and 40 μl of binding buffer (10 mM Tris-HCl, 1 mM EDTA, 2 M NaCl) were mixed with 40 μl of PCR product for 10 min at room temperature. The reaction mixture was placed onto a MultiScreen-HV, Clear Plate (Millipore, Billerica, MA, USA). After application of the vacuum, the beads were treated with a denaturation solution (0.2 N NaOH) for 1 min and washed twice with washing buffer (10 mM Tris-acetate at pH 7.6). The beads were then suspended with 24 μl of annealing buffer (20 mM Tris-acetate, 2 mM Mg-acetate at pH 7.6) containing 8 pmol of sequencing primer. The template-sequencing primer mixture was transferred onto a PSQ 96 Plate (Biotage), heated to 90°C for 2 min followed by 60°C for 10 min, and finally cooled to room temperature. Sequencing reactions were performed with a PSQ 96 SNP Reagent Kit (Biotage) according to the manufacturer's instructions. The percentage methylation at each CpG site was calculated from the raw data by use of Pyro-Q-CpG Software (Biotage). MS-SNuPe analysis was performed with ABI SNaPshot reagents (Applied Biosystems) by use of a method developed in our laboratory. Extension products were separated on an ABI3100 Genetic Analyzer (Applied Biosystems). Methylation data from pyrosequencing and MS-SNuPe analysis were analyzed by use of SPSS v14 (Lead Technologies) with standard t tests and ANOVA.
Genotyping of COMT and BDNF SNPs
Nonsynonymous SNPs in COMT (rs4680–val108/158met) and BDNF (rs6265–val66met) were genotyped with the pyrosequencing assays designed to interrogate the density of methylated cytosines in these regions. In addition, genotypes were double-checked by use of the ABI TaqMan Allelic discrimination method utilizing Assay-on-Demand reagents provided by the manufacturer (Applied Biosystems) and the ABI 7900HT Sequence Detection System. DNA methylation at surrounding CpG sites was compared between samples grouped by genotype with standard t tests.
Results
Overview of Experimental Strategy
This study utilized two complimentary approaches for detection of DNA-methylation differences associated with major psychosis. Our first strategy involved enrichment of the unmethylated fraction of genomic DNA and subsequent hybridization on CpG-island microarrays. SZ and BD samples were compared to unaffected CTRL samples both separately and, given increasing evidence for an etiological overlap between the two disorders,1,16 as a combined major-psychosis group. Of the genomic regions showing a significant difference between affected and unaffected groups, a number of loci were of particular interest given our prior knowledge about the etiology of the disorder, and a subset of these were selected for subsequent bisulfite-based fine-mapping of methylated cytosines for verification of our microarray approach. We also employed gene-ontology analysis to uncover functional pathways epigenetically altered in major psychosis, and we utilized novel network-based analyses to investigate epigenetic modularity in affected individuals and controls. Using demographic data available from the brain samples, we investigated correlations between DNA methylation and variables including lifetime antipsychotic intake. Our second major strategy involved bisulfite-based fine-mapping across ten candidate genes previously implicated in major psychosis and principally nominated from genetic association and expression studies of both SZ and BD.
Methylomic Profiling of Brain DNA
Figure 1 illustrates raw p values for microarray signal intensity versus fold change observed for comparisons between brain DNA from major-psychosis patients and unaffected controls, matched for sex. Significant (FDR < 0.05) mean differences were found for spots associated with a number of genes (Table S2 and Figure 2). Many of these loci are consistent with our knowledge about the neurobiological and genetic systems involved in major psychosis, including several glutamatergic and GABAergic genes, loci involved in neuronal development, and loci in regions highlighted in genetic linkage studies (Table S3).
Figure 1.
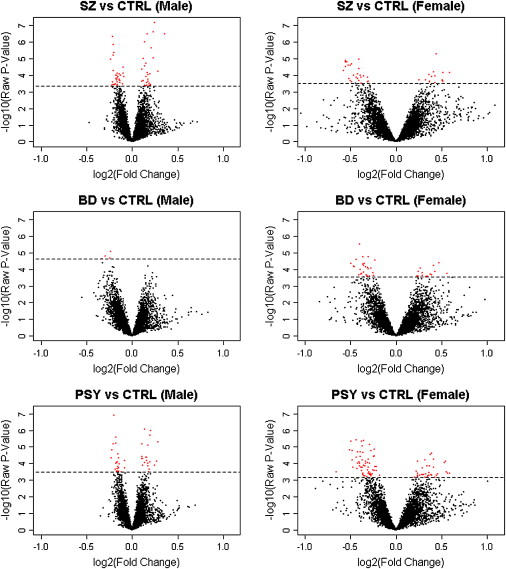
Frontal-Cortex DNA-Methylation Differences
Volcano plots showing raw p values for differential DNA methylation versus fold change observed when the hypomethylated fraction of brain DNA from psychosis patients was compared to that from unaffected individuals by use of 12K CpG-island microarrays. Each spot (red and black) represents an array probe averaged across all individuals in a group, with red spots denoting probes with FDR < 5%. SZ indicates schizophrenia, BD indicates bipolar disorder, and PSY indicates combined psychosis group (SZ and BD).
Figure 2.
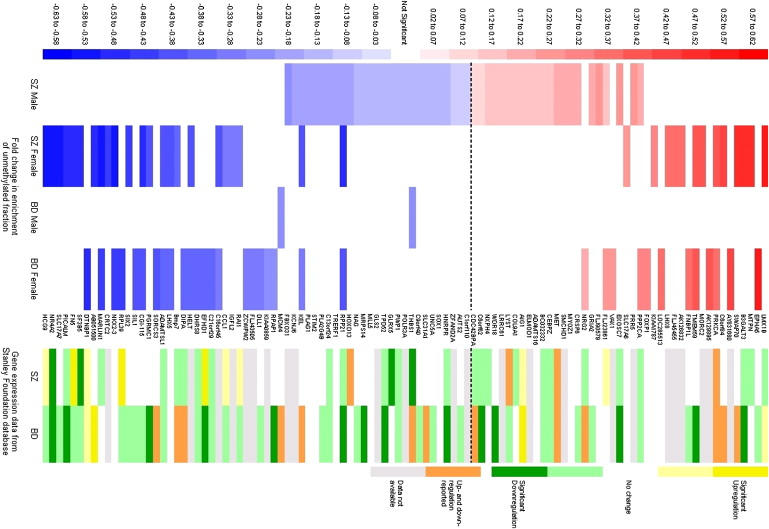
FDR-Significant DNA-Methylation Differences Associated with Major Psychosis
A positive fold change (red) corresponds to lower DNA methylation in the affected group, and a negative fold change (blue) corresponds to higher DNA methylation in the affected group. Also shown are gene-expression data from the same group of samples obtained from the Stanley Research Foundation database. Dark yellow/green corresponds to significant transcript up-/downregulation in a meta-analysis of all expression studies performed on these samples, and light yellow/green corresponds to evidence of significant up-/downregulation from at least one study. Orange indicates that expression has been reported as altered in both directions in different studies. Grey denotes missing data.
Although our initial analyses separated samples by sex, we were also interested in the overlap between males and females. DNA methylation in SZ males and SZ females was significantly correlated (r2 = 0.13, p = 8.1e-26), suggesting that there are SZ-associated epigenetic changes common to both sexes. Of particular interest was evidence for FDR-significant hypermethylation in both male and female samples in the vicinity of two genes. The first is RPP21 (male SZ FDR = 0.025; female SZ FDR = 0.021), which encodes a component of ribonuclease P, a protein complex that generates mature tRNA molecules by cleaving their 5′-ends19. The second is KEL (male SZ FDR = 0.04; female SZ FDR = 0.044), encoding the Kell blood-group glycoprotein.20 Interestingly, both regions are also FDR-significantly hypermethylated in female BD samples (RPP21 FDR = 0.045; KEL FDR = 0.045). For BD, however, no significant correlation was found between males and females, suggesting that sex-specific etiological factors may play a stronger role.
Gene-expression data for the samples used in this study are available from the Stanley Medical Research Institute Online Genomics Database.21 Figure 3 illustrates two examples of the available expression data obtained from this database. Figure 3A highlights downregulation of NR4A2, a gene found to be hypermethylated in female SZ samples (FDR = 0.021). For NR4A2, nine out of 24 studies on SZ samples show significantly reduced expression, with the overall analysis across all mRNA studies in the Stanley Array Collection being highly significant (p < 0.00001). Figure 3B highlights significant downregulation of GLRX5, which we found to be hypermethylated in male SZ samples (FDR = 0.04), across expression studies on SZ brains in the Stanley Array Collection (p = 0.003). Figure 2 summarizes the available expression data for all FDR-significant DNA-methylation changes. Interestingly, 82% of the loci found to be hypermethylated in major-psychosis samples for which expression data are available (40 out of 49) are significantly downregulated in at least one gene-expression study, with 24% (12 out of 49) showing significant downregulation of expression averaged across all mRNA studies performed on these samples in either SZ, BD, or both. The story for hypomethylated samples is more complex, with only 34% of loci for which gene expression data are available (11 out of 32) showing significant upregulation in at least one gene-expression study.
Figure 3.
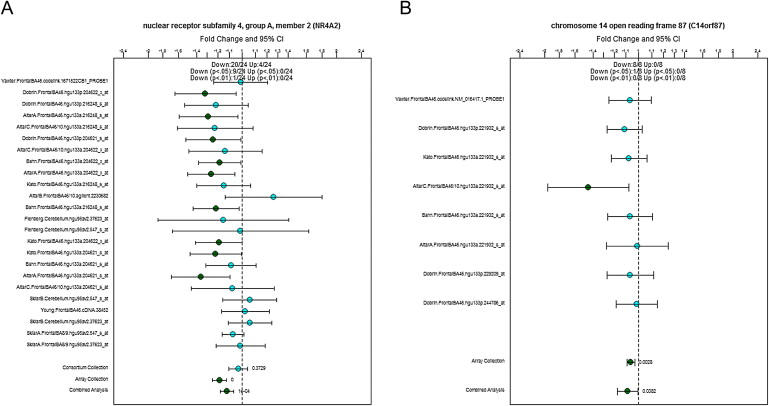
Examples of Data Obtained from the Stanley Research Foundation Expression Database
Shown is down-regulation of (A) NR4A2 and (B) GLRX5 (alternative name C14orf87) in SZ samples obtained from the Stanley Array Consortium. Data points refer to the relative expression fold change in the affected group relative to control samples. Scale bars denote 95% confidence intervals.
Analysis of Demographic Data, Brain-Tissue Parameters, and Lifetime Antipsychotic Use
No FDR-significant correlations were found between any of the available demographic variables (PMI, brain weight, brain pH, lifetime alcohol use, and lifetime illicit-drug use) and DNA methylation. Methylation of a CpG island located ∼30kb upstream of the gene-encoding mitogen-activated protein kinase kinase I (MEK1) was found to be significantly correlated with lifetime antipsychotic use in male SZ samples (n = 25), with higher lifetime antipsychotic use associated with lower DNA methylation (Figure 4A) (r2 = 0.6, p = 6.8E-06, FDR = 0.04). Interestingly, a similar correlation in the same direction is also observed in female SZ samples (Figure 4B), although this does not reach FDR significance due to the small number (n = 9) of samples with available medication data (r2 = 0.5, p = 0.04). No such correlation is observed in the BD samples.
Figure 4.
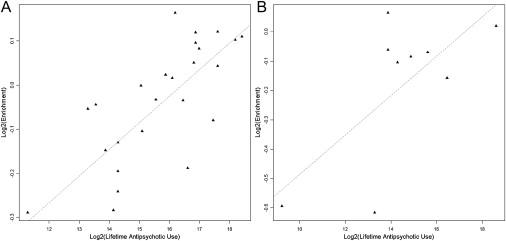
DNA Methylation and Lifetime Antipsychotic Use
Correlation between DNA methylation in the promoter of the mitogen-activated protein kinase kinase I gene (MEK1) and lifetime antipsychotic use in (A) male schizophrenia samples (r2 = 0.6, p = 6.76E-06) and (B) female schizophrenia samples (r2 = 0.5, p = 0.04).
Methylomic Profiling of Germline DNA
Figure 5 illustrates raw p values for array signal intensity versus fold change observed in our comparison of germline DNA from BD patients and unaffected CTRLs. No FDR-significant differences were observed between the two groups. A comparison of the largest psychosis-associated DNA-methylation differences in the germline analysis with those in the brain DNA analysis, taking loci with a raw p < 0.001, found no overlap between datasets.
Figure 5.
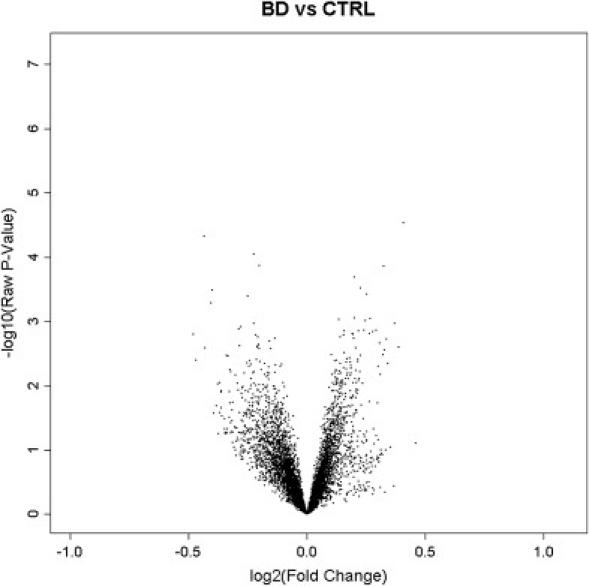
Germline DNA-Methylation Differences
Volcano plot showing raw p values for differential DNA methylation versus fold change observed when the hypomethylated fraction of germline DNA from BD patients was compared to that from unaffected individuals by use of 12K CpG-island microarrays. No FDR-significant DNA-methylation differences were observed.
Site-Specific CpG-Methylation Analysis in Selected Genes
After microarray analysis, we tested a number of loci to further verify the microarray approach. From the genes listed in Table S3, we quantitatively measured site-specific CpG methylation upstream of DTNBP1 (n = 30), GRIA2 (n = 39), HCG9 (n = 31), HELT (n = 26), KCNJ6 (n = 26), LHX5 (n = 24), MARLIN-1 (n = 28), NR4A2 (n = 24), RPL39 (n = 25), SLC17A7 (n = 24), TMEM59 (n = 30), and WDR18 (n = 29). Given that our enrichment strategy was based on differential cleavage of HpaII sites, we focused primarily on these and surrounding CpG positions located in or near genomic regions corresponding to specific microarray probes.
Our site-specific CpG analyses show good agreement with data obtained from microarray analysis, although the absolute differences observed are generally small. Two examples are shown in Figure 6 for regions upstream of the genes WDR18 and RPL39. Microarray analysis (Figure 6A) predicted these regions to be hypomethylated in SZ male samples and hypermethylated in BD female samples, respectively. Figure 6B shows pyrosequencing data confirming WDR18 hypomethylation in male SZ samples compared to controls (n = 29, average methylation 17% versus 25%, p < 0.001). This region contains a putative binding site for the brain-expressed transcription factor c-myb, known to be blocked by CpG methylation22(Figure 6C). Pyrosequencing also verified RPL39 hypermethylation in BD female samples (n = 25, 28% versus 22%, p = 0.009), especially at a CpG located within putative binding sites for several brain-expressed transcription factors, PAX-523 and NF-kB,24 which are known to be affected by DNA methylation (Figure 6C). Example pyrograms across both regions showing representative affected and unaffected individuals are shown in Figure 6D.
Figure 6.

Bisulfite Modification and Pyrosequencing Verification of CpG-Methylation Differences in Two Genes Nominated from Microarray Analysis
(A) Microarray signal intensity for probes located in CpG islands in the promoter region of WDR18 and RPL39. Scale bars denote 95% confidence intervals. For WDR18, male SZ samples had significantly higher intensities than did unaffected control samples (raw p = 4.5E-05, FDR corrected = 0.05), indicating hypomethylation in affected individuals. For RPL39, female BD samples had significantly lower intensities than unaffected control samples (raw p = 4.0E-05, FDR corrected = 0.02), indicating hypermethylation in affected individuals.
(B) Bisulfite mapping across amplicons spanning regions interrogated by CpG-island microarrays confirms DNA-methylation changes predicted by microarrays: shown are WDR18 hypomethylation in male SZ samples (p < 0.001) and RPL39 hypermethylation in female BD samples (p = 0.009).
(C) Predicted transcription-factor binding sites in the regions of WDR18 and RPL39 analyzed by pyrosequencing. Boxes indicate CpG sites with the largest DNA-methylation differences in affected individuals.
(D) Example pyrograms demonstrating relative WDR18 hypomethylation in a SZ male sample and RPL39 hypermethylation in a BD female sample.
In addition to WDR18 and RPL39, we were able to confirm significant DNA-methylation differences in MARLIN-1, postulated to be hypermethylated in affected female samples from microarray analysis. Average methylation of a HpaII site located in the genomic region spanning the microarray clone and falling in a putative binding site for a Pbx1/Meis1 heterodimer was 84% in unaffected controls compared to 93% in SZ females (p = 0.04) and 91% in the combined major-psychosis female group (p = 0.03). Interestingly, whereas only 13% of unaffected control samples were fully methylated, 71% of SZ females and 47% of the combined female major-psychosis group were fully methylated. Other significant DNA-methylation differences were observed for CpG sites upstream of DTNBP1 and HCG9, confirming hypermethylation in affected female samples relative to controls, as predicted by microarray data for both genes (DTNBP1: 62% versus 58%, p = 0.04; HCG9: 20% versus 15%, p = 0.04).
Overall significant DNA-methylation differences were observed in five of the 12 specific regions tested. However, even in the regions where no overall significant CpG-methylation differences were detected, changes were consistently in the direction predicted by our microarray analysis. The regions tested with bisulfite sequencing are somewhat arbitrary, and it is likely that the specific critical CpG sites causing differential enrichment (and thus microarray signal intensity) were not included in all the amplicons tested. Five confirmed loci out of 12 tested (∼40%) is thus a good rate of verification, especially given that no DNA-methylation differences were observed across any of the 12 arbitrarily chosen negative-control array regions tested (ACTL6A, BCL11A, DDX1, DUSP10, GNL1, GPR160, LGI2, REV3L, PTPNS1, PHGDHL1, STIL, and ZNF218) or across any of the a priori psychosis-candidate genes, which were not selected on the basis of microarray analysis (see below). These data suggest that a substantial proportion of epigenetic differences detected in our microarray experiments are real and that the conclusions drawn from the full array dataset are likely to be based on genuine DNA-methylation changes.
Gene-Ontology Analysis of Brain Methylomic Data
The top 60 GO categories for each diagnostic group can be seen in Figure 7, and Table S4 lists all significant GO categories with a p < 0.01. GO categories detected by this analysis included genes involved in the epigenetic regulation of transcription and development. Of particular interest to the etiology of psychosis were the FDR-significant associations for “response to stress” in male BD samples and for “brain development” in both female BD samples and female SZ samples. In addition, consistent with the postulated link between mitochondrial function and psychosis,25 several “mitochondrial function” GO categories showed significantly different distributions in the affected individuals compared to controls.
Figure 7.

Gene-Ontology Analysis
The top 60 GO categories for each diagnostic group (SM = SZ male, SF = SZ female, BM = BD male, BF = BD female). Colors denote raw p values. See Table S4 for additional data.
Modularity in DNA-Methylation Microarray Data
In the brain, the average number of connections between nodes (representing correlated methylation observed between different genomic loci) is higher in the SZ group compared to the CTRL group (2.7 versus 1.7) (Figure 8A). The large clustering coefficient (CTRL = 0.17, SZ = 0.22), and its decrease with increasing connections in both sample groups, suggests that both are hierarchically modular. The lack of clustering in a series of simulated “random” datasets suggests that this modularity is likely to be a real biological phenomenon (Figure 8B). Assortativity is higher in SZ (knn = 9) compared to CTRL (knn = 6), probably reflecting the higher number of connections between nodes in SZ (Figure 8C). Although the number of modules (CTRL = 42, SZ = 43) and median size of modules (CTRL = 10, SZ = 11) is approximately the same in both groups, the degree of modularity is higher in CTRL (0.56) than in SZ (0.44), suggesting some epigenetic dysfunction in affected individuals (Figure 9). A similar pattern is seen in the germline, with higher connectivity (3.9 versus 1.5) and assortativity (15 versus 7) in affected individuals compared to controls, a high degree of clustering in both groups (CTRL = 0.14, BD = 0.17), but higher modularity in unaffected individuals (0.42 versus 0.33).
Figure 8.
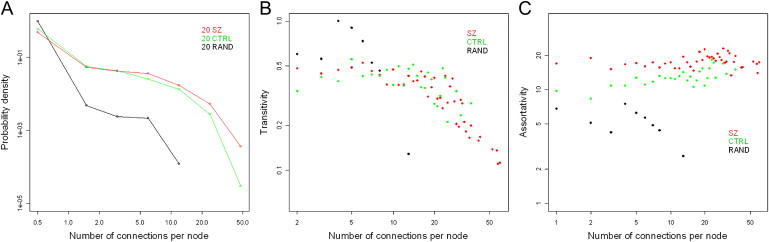
Network Analysis of DNA-Methylation Microarray Data
(A) The average number of connections between nodes is higher in the SZ sample group (2.7) compared to the CTRL sample group (1.7).
(B) The clustering coefficient is high in both groups (CTRL = 0.17, SZ = 0.22) and decreases with increasing connections, suggesting that both groups are hierarchical to the same degree.
(C) Assortativity was found to be higher in SZ (knn = 9) compared to CTRL (knn = 6), reflecting the higher number of connections between nodes in the SZ data. A simulated dataset generated by random shuffling of microarray data produced a network with a low number of connections and a low clustering coefficient.
Figure 9.
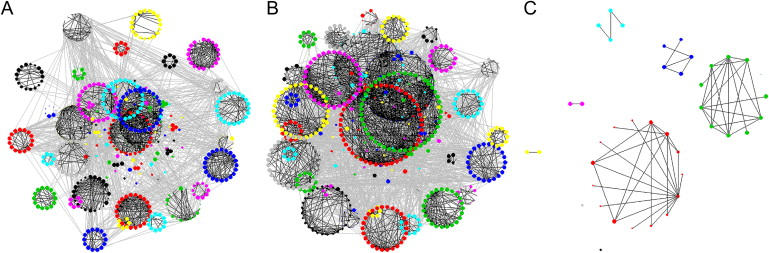
Reduced Epigenetic Modularity in Major Psychosis
Partial-correlation network analysis of microarray data illustrating significant connections between nodes (p < 1e-7) demonstrating strong hierarchical modularity for (A) male CTRL samples (n = 20) and (B) male SZ samples (n = 20) but not for (C) a randomly shuffled dataset. Although modularity is apparent in both sample groups, it is lower in the SZ group (0.44) than in the CTRL group (0.56). A similar pattern of modularity is seen in the comparison of methylation between germline male BD samples (0.33) and unaffected CTRLs (0.47).
DNA-Methylation Analysis of Psychosis-Candidate Genes in Brain DNA
The second approach utilized in this study assessed DNA methylation across specific candidate-gene regions with sodium bisulfite treatment and subsequent pyrosequencing analysis to quantitatively measure the density of methylated cytosines. We found little evidence of any psychosis-associated DNA-methylation differences in any of the ten genes tested (Table S5), including the promoter regions of COMT and RELN found to be differentially methylated in previous studies. Nonsynonymous SNPs in both COMT (rs4680–val108/158met) and BDNF (rs6265–val66met) create or abolish exonic CpG sites. In COMT, surrounding CpG sites were highly methylated (>95%) in all samples tested, with no correlation between genotype and DNA methylation. In BDNF there was modest evidence for an association between genotype and DNA methylation. Of the samples tested, 74% were CC (val homozygotes) and 26% were CT or TT (met carriers). Val homozygotes had significantly higher DNA methylation across the exonic region profiled with pyrosequencing (average methylation = 83% versus 78%, p = 0.02), with CpG1 (86% versus 77%, p = 0.01) and CpG3 (79% versus 71%, p = 0.03) showing the largest differences (see Figure 10).
Figure 10.
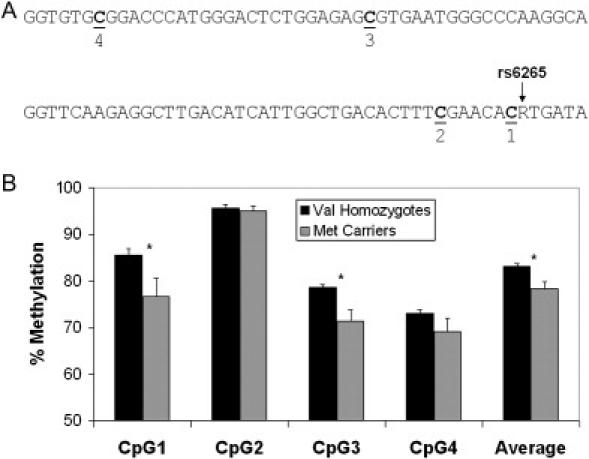
BDNF genotype and DNA Methylation
Association of BDNF genotype with DNA methylation at nearby exonic CpG sites in DNA obtained from frontal-cortex brain tissue.
(A) the exonic region of BDNF covered by our bisulfite mapping indicating the four CpG sites tested.
(B) DNA methylation at the four CpG sites in the vicinity of rs6265. Asterisk denotes t test of p < 0.05, bars denote mean standard error.
Discussion
In this study we performed a microarray-based epigenomic scan using CpG-island microarrays and found psychosis-associated brain-DNA-methylation differences in numerous loci, including many genes that have been functionally linked to disease etiology. Consistent with increasing evidence for altered glutamatergic and GABAergic neurotransmission in the pathogenesis of major psychosis,26,27 we identified epigenetic changes in loci associated with both of these neurotransmitter pathways.
Glutamate is the most abundant fast excitatory neurotransmitter in the mammalian nervous system, with a critical role in synaptic plasticity. Several lines of evidence link the glutamate system to psychosis, in particular the observation that glutamate-receptor agonists can cause psychotic symptoms in unaffected individuals. Probes associated with two glutamate-receptor genes—one near WDR18, located ∼10 kb upstream of the NMDA-receptor-subunit gene NR3B (also known as GRIN3B) and another in the promoter of the AMPA-receptor-subunit gene GRIA2—were found to be hypomethylated in SZ and major-psychosis males. Dysregulation of both NMDA and AMPA glutamate receptors is important in the etiology of major psychosis,28 and GRIA2 expression is altered in the brains of SZ patients.29
Various types of glutamate transporters are present in the plasma membranes of glial cells and neurons. Our data suggest that two vesicular glutamate transporters (VGLUTs), which pack glutamate into synaptic vesicles, are epigenetically altered in major psychosis. Given the link between DNA methylation and gene transcription, our data concur with data from gene expression studies and the observation that VGLUT1 and VGLUT2 are expressed in a complementary manner in cortical neurons.30 VGLUT1, which was hypermethylated in SZ female samples, is downregulated in the brains of SZ patients.31 In addition, VGLUT2, which is upregulated in SZ patients,32 is hypomethylated in SZ females.
Several other glutamatergic genes showed evidence of epigenetic dysregulation in major psychosis. GLS2, which encodes a glutaminase enzyme that catalyzes the hydrolysis of glutamine to glutamate, was hypermethylated in SZ male samples. Previous studies report that glutaminase expression is altered in the pathology of SZ.33 The gene encoding Secretogranin II (SCG2), a secretory protein located in neuronal vesicles that is known to stimulate the release of glutamate, was hypomethylated in major-psychosis females relative to unaffected controls. SCG2 expression is known to be modulated by both chronic PCP exposure, which mimics symptoms of major psychosis,34 and lithium treatment.35
Unlike glutamate, which is a strong excitatory neurotransmitter, GABA acts as a potent inhibitory neurotransmitter. Hypofunctioning GABAergic interneurons appear to be important in the etiology of major psychosis.27 Our data suggest that MARLIN-1, a RNA-binding protein that is widely expressed in the brain and regulates the production of functional GABA(B) receptors,36 is hypermethylated in SZ, BD, and major-psychosis female samples. In addition, KCNJ6, a G protein-coupled inwardly rectifying potassium channel that has been linked to the regulation of GABA neurotransmission,37 was found to be hypermethylated in SZ and major-psychosis males. Increasing evidence suggests that both the glutamate and GABA systems are synergistically involved in major psychosis,26 supporting our observation of increased HELT-promoter methylation in SZ and BD female samples. HELT is known to determine GABAergic over glutamatergic neuronal fate in the developing mesencephalon.38
We observed evidence for epigenetic dysregulation near several genes involved in neuronal development in the brain. WNT1, an integral part of the Wnt signaling pathway that is critical for neurodevelopment, which is differentially expressed in SZ brains,39 was significantly hypermethylated in major-psychosis females relative to controls. The transcriptionally inducible nuclear-receptor NR4A2, downregulated in both SZ and BD (see Figure 3A and40), was found to be hypermethylated in SZ females. FOSB, which encodes a protein controlling cell proliferation in the brain known to be expressed following chronic antipsychotic treatment,41 was hypomethylated in major-psychosis females relative to controls. Finally, the LIM homeobox transcription factors LMX1B and LHX5, linked to normal learning and motor functions,42 also showed significant methylation changes in female psychosis samples, with LMX1B demonstrating putative hypomethylation and LHX5 demonstrating putative hypermethylation.
Several other genes with links to major psychosis were found to be epigenetically altered. Given that phospholipid metabolism is disturbed in SZ,43 it is noteworthy that the phospholipase gene PLA2G4B was hypermethylated in SZ male, major-psychosis male, and major-psychosis female samples. RAI1, hypermethylated in SZ female samples, is located in an unstable genomic region encoding a polymorphic polyglutamine tract associated with SZ and response to antipsychotic medication.44 AUTS2, hypermethylated in SZ male samples, spans a translocation breakpoint associated with mental retardation and autism.45 Finally, a probe located ∼90 kb upstream of one of our prenominated “psychosis-candidate genes,” DTNBP1, was hypermethylated in affected females.
There is considerable clinical, epidemiological, genetic, and neurochemical evidence to support a role for sex-specific factors in the etiology of both SZ46 and BD.47 For these reasons, our initial analyses considered males and females separately. It can be hypothesized that sex-specific differences represent underlying differences in etiology that may be mediated by epigenetic processes.48 For example, although sex hormones cannot change DNA sequence, it is known they can be potent modifiers of epigenetic status and gene expression. There are several reports of the female sex hormone estrogen, for example, altering the chromatin-configuration and DNA-methylation profile of specific loci in the genome,49,50 potentially controlling gene expression in a sex-specific manner. We were also interested in the overlap between data from male and female psychosis samples. Methylomic array data from SZ males and SZ females were significantly correlated, signifying an overlap in the genomic regions epigenetically altered in both sexes. Of particular interest was evidence for FDR-significant hypermethylation in both male and female samples in the vicinity of two genes. The first is RPP21, which encodes a component of ribonuclease P, a protein complex that generates mature tRNA molecules by cleaving their 5′-ends19. The second is KEL, encoding the Kell blood-group glycoprotein.20 Interestingly, abnormal expression of Kell antigens is one cause of McLeod Syndrome [MIM 314850], which is known to manifest itself in symptoms of schizophrenia.51 Of note, both RPP21 and KEL are also FDR-significantly hypermethylated in female BD samples. For BD, however, no significant overall correlation was found between males and females, suggesting that sex-specific etiological factors could play a stronger role in BD than SZ. Taken together, these data reinforce the benefits of performing epigenetic studies separated by sex, but they also indicate that there is significant overlap between males and females for DNA-methylation profiles associated with SZ.
No correlation was found between any demographic variables or postmortem brain parameters and DNA methylation. Given the dynamic nature of the epigenome, however, and evidence linking drug exposure to DNA methylation, we also examined the epigenetic effect of antipsychotic treatment. Methylation of a CpG island located upstream of MEK1 was found to be strongly correlated with antipsychotic use in SZ. This correlation was particularly strong in male SZ samples, but it was also present in female SZ samples. No correlation was seen in BD samples. The link between MEK1 and antipsychotic exposure in SZ is striking given the involvement of mitogen-activated protein-kinase (MAPK) signaling pathways in mediation of intraneuronal signaling and the observation that clozapine, a widely used medication in the treatment of SZ, selectively activates this pathway via an interaction with MEK1.52
GO analysis allows the investigation of functionally linked biological pathways in microarray datasets.53 Several interesting GO categories are highlighted by our analysis, including several involved in various epigenetic processes, transcription, and development. In addition, we find an association with genes involved in brain development in both female BD and SZ samples and in response to stress in male BD samples, consistent with the popular diathesis-stress hypothesis of psychosis susceptibility. In addition, given the postulated link between mitochondrial dysfunction, oxidative stress, and psychosis,25 it is interesting that a number of mitochondrial GO categories show significantly different distributions in affected individuals. Our methylome results are in close agreement with a parallel microarray-based transcriptomics, proteomics, and metabolomics study, also performed on brain tissue obtained from the Stanley Foundation, in which genes and proteins associated with mitochondrial function and oxidative stress responses were the most altered group.25
Traditional etiological studies of complex disease, both genetic and epigenetic, have tended to investigate discrete regions of DNA in isolation. It is plausible, however, that the epigenome, like many other biological systems, comprises a complex network of interacting processes and that DNA methylation in different genomic regions is interdependent. Understanding the system-level features of biological organization across the epigenome is an important aspect of elucidating the epigenetic changes associated with disease. In order to investigate whether DNA methylation is coordinated across different loci, we utilized a novel network-based approach to test the modularity of our methylome data. In this way, a network comprises distinct clusters of elements, termed “modules,” which are highly connected within themselves but have fewer connections with the rest of the network.17 The study of interaction networks has proven fruitful in many areas of biological research, highlighting distinct modularity in metabolic networks,54 cellular networks,55 and protein-interaction networks.56 Although such an approach has not been previously applied to the epigenome, recent evidence suggests the involvement of coordinated epigenetic silencing across large genomic regions in cancer.57
The goal of our network analysis was twofold: first, to see whether there is modularity in the methylome; second, if such epigenetic modularity exists, to see whether there are any differences between affected and unaffected groups. For both brain and germline DNA, we found evidence for significant epigenetic modularity in both groups analyzed. No modules were observed in a series of simulated “random” datasets, suggesting that the modular structure of the methylome is a real biological phenomenon and that the epigenome can be split into distinct groups of correlated loci, potentially corresponding to distinct functional pathways and/or physical regions. Although DNA methylation in both affected and unaffected groups is clearly modular, the number of interconnections between specific genomic regions is higher in the affected group compared to the CTRL group, resulting in more between-module interference, in both brain and germline DNA. Given that modules within such biological networks are likely to have specific functional tasks separate to those of other modules,17 the lower degree of DNA-methylation modularity observed in the major-psychosis samples points to some degree of systemic epigenetic dysfunction associated with major psychosis.
In addition to the microarray-based screening for epigenetic changes, our second approach utilized in this study focused on DNA methylation in the vicinity of genes with a priori evidence for an etiological role in major psychosis. These regions were profiled directly with bisulfite modification and pyrosequencing, with assays designed to span CpG-rich promoter regions, along with some exonic and intronic regions for several genes. Little evidence was found to suggest that DNA methylation in these genes is associated with either SZ or BD. Our analyses included the promoter regions of both COMT and RELN that have been previously shown to be epigenetically altered in psychosis in previous studies.9,10,58 Unlike these studies that report COMT hypomethylation and RELN hypermethylation in SZ samples, we found no evidence for DNA-methylation changes in these genes associated with either SZ or BD. Our data are in agreement with a previous study on COMT reporting no association between promoter methylation and major psychosis12 and a recent study reporting very low levels of methylation across the RELN region and no association with major psychosis.11 It should be noted that some of the methods used in previous studies of these genes, for example methylation-specific PCR, can lead to biased assessment of methylated cytosines and are not able to assess epigenetic changes in a truly quantitative manner as is possible with the pyrosequencing methodology utilized in this study.
The observation of an association between genotype at a nonsynonymous SNP (rs6265) in BDNF and DNA methylation at surrounding CpG sites in DNA from frontal-cortex brain tissue adds to the increasing evidence that DNA sequences can influence epigenetic profiles (e.g., 59,60). Although DNA alleles and haplotypes can be subject to differential epigenetic modification, it appears that epigenetic status cannot be unequivocally deduced from DNA-sequence data alone. The notion that epigenetic changes might be associated with DNA-sequence variation is relevant to the inconsistent genetic-association studies in complex diseases and suggests that a comprehensive epigenetic analysis of candidate SNPs and haplotypes is warranted.
Our tandem use of two complementary approaches allowed us to test both a priori hypotheses and identify novel regions of the genome that may be epigenetically dysfunctional in major psychosis. The unbiased microarray approach was far more productive in identification of differentially methylated loci than was the focused candidate-gene approach; this has implications for the design of future epigenetic studies of complex disease. Of note is the observation that a high proportion of the microarray-nominated loci can be considered good functional and/or positional candidates. Given the relatively large number of differences observed between affected and unaffected individuals in our microarray screen and the laborious nature of current bisulfite-based mapping approaches, it was unfeasible to further investigate each nominated gene at the level of specific CpG nucleotides in the course of this study. Our analyses were stringently controlled for multiple testing by use of the FDR statistic, but as with all microarray-based experiments, it is possible that some of the genes uncovered are false-positives; more in-depth screening of specific gene regions will be needed to verify the specific DNA-methylation changes involved. In general, the actual DNA-methylation differences observed between major-psychosis and CTRL groups appear quite subtle but consistent across affected individuals. This pattern of findings reflects that seen for other etiological approaches in major psychosis (e.g., gene-association studies, gene-expression studies, and brain-imaging studies) and is as expected given the highly complex and heterogeneous nature of the phenotypes being studied.
It is plausible that differences in the cellular composition of our brain-tissue samples can actually account for some of the differences we observed. Conversely, however, we cannot exclude the possibility that actual epigenetic defects are more substantial than reported here but only present in a specific cell type in the brain. This point is highlighted by a recent study identifying epigenetic changes occurring specifically in cortical-interneuron cells, suggesting that different cell populations within a single tissue type can have quite distinct epigenetic profiles.61 Of course, access to postmortem brain tissue from affected individuals is difficult, and at present it would probably be very difficult, logistically and financially, to perform a microarray-based study focused on various types of cell populations. However, with the rapid technological advances currently taking place, a future line of research could be the search for cell-type-specific epimutations by use of laser-capture microdissection technology, which enables the isolation of single cells from whole tissue, thus avoiding the confounding effects of cell-type variation.
Several strategies exist for enriching DNA prior to microarray hybridization, dependent upon DNA methylation status. While each method has distinct advantages and disadvantages, we utilized a method based on cleavage with methylation-sensitive restriction enzymes.14 We have previously verified this method, and shown it to be both sensitive and repoducible.14,59 Because the majority of CpG sites in the genome are methylated, it has been shown that enrichment of the unmethylated DNA fraction is more effective at detecting small DNA methylation changes than alternative methods based on enrichment of the methylated DNA fraction, such as methylated DNA immunoprecipitation (MeDIP).62 Given the small DNA methylation changes likely associated with complex diseases such as psychosis, in part verified by the absolute changes we detected in our study, such sensitivity is likely to be a major advantage of our approach. One potential limitation to all enrichment strategies is that they can be affected by the presence of SNPs and copy number variation (CNV). Until the full extent (and location) of CNV in the genome is ascertained, it will be hard to fully address this issue. Our bisulfite-mapping results suggest that neither SNPs nor CNVs are systemically influencing our microarray data, but both should be taken into consideration when investigating further the epigenetic differences reported here.
Proving a direct causal link between epigenetic factors and disease is not straightforward. It is possible that the epigenetic differences observed actually result from psychosis-induced changes in the brain. Tissues that are not the disease site or directly affected by antipsychotic medications, could thus actually be very useful in elucidating epigenetic changes associated directly with major psychosis. It has been proposed that germline epimutations may be important in disease etiology,63 and potentially transmitted between generations. A recent study from our group reported considerable intra- and inter-individual epigenetic variation in male germ cells.59 Our microarray analysis of a BD germline DNA sample, however, did not detect any FDR-significant DNA methylation differences associated with BD (Figure 3). The reason for discrepant brain and the germline results could be numerous. For example, epigenetic changes predisposing one to or causing major psychosis might be tissue-specific and restricted to the main sites of disease manifestation (e.g., the brain). In addition, previous studies of germline epimutations have found that aberrantly methylated cytosines may be present at very low frequencies (<1%),63 and such small changes cannot be accurately detected with current microarray technology. Recent technological advances in DNA methylation analysis, such as the deep-sequencing of bisulfite-treated DNA, may make the detection of such minute DNA methylation differences more easily detectable.
To conclude, consistent with the epigenetic theory of major psychosis, a number of loci were found to be epigenetically altered in the brain of SZ and BD patients relative to unaffected controls. This study clearly demonstrates that epigenomic studies can be done cost-effectively with current technologies. Microarray technology is advancing at a tremendous pace, and it will soon be economically feasible to perform similar experiments on very high-density tiling microarrays covering the entire genome. Future studies can also be broadened to include other epigenetic processes such as histone modifications and small non-coding RNA molecules.
Supplemental Data
Five Supplemental tables are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Raw microarray data, www.epigenomics.ca
Stanley Foundation Brain Collection, http://www.stanleyresearch.org/programs/brain_collection.asp
Gene-expression data for the Stanley Brain samples, https://www.stanleygenomics.org/
MethPrimer, http://www.urogene.org/methprimer/index1.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Gene Ontology Database, www.geneontology.org
Acknowledgments
Postmortem brains were donated by The Stanley Medical Research Institute's Brain Collection courtesy of Michael B. Knable, E. Fuller Torrey, Maree J. Webster, and Robert H. Yolken. This project was supported by the Ontario Mental Health Foundation (OHMF), Canadian Institutes for Health and Research (CIHR), the National Alliance for Research on Schizophrenia and Depression, and the Stanley Foundation. Development of some analytical tools used in this study was funded by the National Institute of Mental Health (R01 MH074127-01). A.P. is an OMHF Senior Fellow, and J.M. was supported by a CIHR postdoctoral fellowship.
References
- 1.Craddock N., O'Donovan M.C., Owen M.J. Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr. Bull. 2006;32:9–16. doi: 10.1093/schbul/sbj033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Craddock N., O'Donovan M.C., Owen M.J. The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J. Med. Genet. 2005;42:193–204. doi: 10.1136/jmg.2005.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petronis A. The origin of schizophrenia: genetic thesis, epigenetic antithesis, and resolving synthesis. Biol Psychiatry. 2004;55:965–970. doi: 10.1016/j.biopsych.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Craddock N., Jones I. Genetics of bipolar disorder. J. Med. Genet. 1999;36:585–594. doi: 10.1136/jmg.36.8.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGuffin P., Asherson P., Owen M., Farmer A. The strength of the genetic effect. Is there room for an environmental influence in the aetiology of schizophrenia? Br. J. Psychiatry. 1994;164:593–599. doi: 10.1192/bjp.164.5.593. [DOI] [PubMed] [Google Scholar]
- 6.Henikoff S., Matzke M.A. Exploring and explaining epigenetic effects. Trends Genet. 1997;13:293–295. doi: 10.1016/s0168-9525(97)01219-5. [DOI] [PubMed] [Google Scholar]
- 7.Riggs A.D., Xiong Z., Wang L., LeBon J.M. Methylation dynamics, epigenetic fidelity and X chromosome structure. Novartis Found. Symp. 1998;214:214–225. doi: 10.1002/9780470515501.ch13. [DOI] [PubMed] [Google Scholar]
- 8.Richards E.J. Inherited epigenetic variation–revisiting soft inheritance. Nat. Rev. Genet. 2006;7:395–401. doi: 10.1038/nrg1834. [DOI] [PubMed] [Google Scholar]
- 9.Abdolmaleky H.M., Cheng K.H., Faraone S.V., Wilcox M., Glatt S.J., Gao F., Smith C.L., Shafa R., Aeali B., Carnevale J. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum. Mol. Genet. 2006;15:3132–3145. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grayson D.R., Jia X., Chen Y., Sharma R.P., Mitchell C.P., Guidotti A., Costa E. Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. USA. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tochigi M., Iwamoto K., Bundo M., Komori B., Sasaki T., Kato N., Kato T. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol. Psychiatry. 2008;63:530–533. doi: 10.1016/j.biopsych.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Dempster E.L., Mill J., Craig I.W., Collier D.A. The quantification of COMT mRNA in post mortem cerebellum tissue: diagnosis, genotype, methylation and expression. BMC Med. Genet. 2006;7:10. doi: 10.1186/1471-2350-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnston-Wilson N.L., Sims C.D., Hofmann J.P., Anderson L., Shore A.D., Torrey E.F., Yolken R.H. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol. Psychiatry. 2000;5:142–149. doi: 10.1038/sj.mp.4000696. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher A., Kapranov P., Kaminsky Z., Flanagan J., Assadzadeh A., Yau P., Virtanen C., Winegarden N., Cheng J., Gingeras T. Microarray-based DNA methylation profiling: technology and applications. Nucleic Acids Res. 2006;34:528–542. doi: 10.1093/nar/gkj461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statist. Soc. Ser. B. Methodological. 1995;57:289–300. [Google Scholar]
- 16.Craddock N., Owen M.J. Rethinking psychosis: the disadvantages of a dichotomous classification now outweigh the advantages. World Psychiatry. 2007;6:20–27. [PMC free article] [PubMed] [Google Scholar]
- 17.Newman M.E. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA. 2006;103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schafer J., Strimmer K. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat. Appl. Genet. Mol. Biol. 2005;4 doi: 10.2202/1544-6115.1175. Article32. [DOI] [PubMed] [Google Scholar]
- 19.Mann H., Ben-Asouli Y., Schein A., Moussa S., Jarrous N. Eukaryotic RNase P: role of RNA and protein subunits of a primordial catalytic ribonucleoprotein in RNA-based catalysis. Mol. Cell. 2003;12:925–935. doi: 10.1016/s1097-2765(03)00357-5. [DOI] [PubMed] [Google Scholar]
- 20.Westhoff C.M., Reid M.E. Review: the Kell, Duffy, and Kidd blood group systems. Immunohematol. 2004;20:37–49. [PubMed] [Google Scholar]
- 21.Higgs B.W., Elashoff M., Richman S., Barci B. An online database for brain disease research. BMC Genomics. 2006;7:70. doi: 10.1186/1471-2164-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klempnauer K.H. Methylation-sensitive DNA binding by v-myb and c-myb proteins. Oncogene. 1993;8:111–115. [PubMed] [Google Scholar]
- 23.Maier H., Colbert J., Fitzsimmons D., Clark D.R., Hagman J. Activation of the early B-cell-specific mb-1 (Ig-alpha) gene by Pax-5 is dependent on an unmethylated Ets binding site. Mol. Cell. Biol. 2003;23:1946–1960. doi: 10.1128/MCB.23.6.1946-1960.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cowled P., Kanter I., Leonardos L., Jackson P. Uroplakin Ib gene transcription in urothelial tumor cells is regulated by CpG methylation. Neoplasia. 2005;7:1091–1103. doi: 10.1593/neo.05364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prabakaran S., Swatton J.E., Ryan M.M., Huffaker S.J., Huang J.T., Griffin J.L., Wayland M., Freeman T., Dudbridge F., Lilley K.S. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9:684–697. doi: 10.1038/sj.mp.4001511. 643. [DOI] [PubMed] [Google Scholar]
- 26.Coyle J.T. The GABA-glutamate connection in schizophrenia: which is the proximate cause? Biochem. Pharmacol. 2004;68:1507–1514. doi: 10.1016/j.bcp.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 27.Benes F.M., Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 28.Lau C.G., Zukin R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 29.Gupta D.S., McCullumsmith R.E., Beneyto M., Haroutunian V., Davis K.L., Meador-Woodruff J.H. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse. 2005;57:123–131. doi: 10.1002/syn.20164. [DOI] [PubMed] [Google Scholar]
- 30.Fremeau R.T., Troyer M.D., Pahner I., Nygaard G.O., Tran C.H., Reimer R.J., Bellocchio E.E., Fortin D., Storm-Mathisen J., Edwards R.H. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 31.Eastwood S.L., Harrison P.J. Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons. Schizophr. Res. 2005;73:159–172. doi: 10.1016/j.schres.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Smith R.E., Haroutunian V., Davis K.L., Meador-Woodruff J.H. Vesicular glutamate transporter transcript expression in the thalamus in schizophrenia. Neuroreport. 2001;12:2885–2887. doi: 10.1097/00001756-200109170-00026. [DOI] [PubMed] [Google Scholar]
- 33.Bruneau E.G., McCullumsmith R.E., Haroutunian V., Davis K.L., Meador-Woodruff J.H. Increased expression of glutaminase and glutamine synthetase mRNA in the thalamus in schizophrenia. Schizophr. Res. 2005;75:27–34. doi: 10.1016/j.schres.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Marksteiner J., Weiss U., Weis C., Laslop A., Fischer-Colbrie R., Humpel C., Feldon J., Fleischhacker W.W. Differential regulation of chromogranin A, chromogranin B and secretogranin II in rat brain by phencyclidine treatment. Neuroscience. 2001;104:325–333. doi: 10.1016/s0306-4522(01)00081-1. [DOI] [PubMed] [Google Scholar]
- 35.McQuillin A., Rizig M., Gurling H.M. A microarray gene expression study of the molecular pharmacology of lithium carbonate on mouse brain mRNA to understand the neurobiology of mood stabilization and treatment of bipolar affective disorder. Pharmacogenet. Genomics. 2007;17:605–617. doi: 10.1097/FPC.0b013e328011b5b2. [DOI] [PubMed] [Google Scholar]
- 36.Couve A., Restituito S., Brandon J.M., Charles K.J., Bawagan H., Freeman K.B., Pangalos M.N., Calver A.R., Moss S.J. Marlin-1, a novel RNA-binding protein associates with GABA receptors. J. Biol. Chem. 2004;279:13934–13943. doi: 10.1074/jbc.M311737200. [DOI] [PubMed] [Google Scholar]
- 37.Koyrakh L., Lujan R., Colon J., Karschin C., Kurachi Y., Karschin A., Wickman K. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J. Neurosci. 2005;25:11468–11478. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakatani T., Minaki Y., Kumai M., Ono Y. Helt determines GABAergic over glutamatergic neuronal fate by repressing Ngn genes in the developing mesencephalon. Development. 2007;134:2783–2793. doi: 10.1242/dev.02870. [DOI] [PubMed] [Google Scholar]
- 39.Miyaoka T., Seno H., Ishino H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 1999;38:1–6. doi: 10.1016/s0920-9964(98)00179-0. [DOI] [PubMed] [Google Scholar]
- 40.Xing G., Zhang L., Russell S., Post R. Reduction of dopamine-related transcription factors Nurr1 and NGFI-B in the prefrontal cortex in schizophrenia and bipolar disorders. Schizophr. Res. 2006;84:36–56. doi: 10.1016/j.schres.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 41.Kontkanen O., Lakso M., Wong G., Castren E. Chronic antipsychotic drug treatment induces long-lasting expression of fos and jun family genes and activator protein 1 complex in the rat prefrontal cortex. Neuropsychopharmacology. 2002;27:152–162. doi: 10.1016/S0893-133X(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 42.Paylor R., Zhao Y., Libbey M., Westphal H., Crawley J.N. Learning impairments and motor dysfunctions in adult Lhx5-deficient mice displaying hippocampal disorganization. Physiol. Behav. 2001;73:781–792. doi: 10.1016/s0031-9384(01)00515-7. [DOI] [PubMed] [Google Scholar]
- 43.Horrobin D.F., Glen A.I., Vaddadi K. The membrane hypothesis of schizophrenia. Schizophr. Res. 1994;13:195–207. doi: 10.1016/0920-9964(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 44.Toulouse A., Rochefort D., Roussel J., Joober R., Rouleau G.A. Molecular cloning and characterization of human RAI1, a gene associated with schizophrenia. Genomics. 2003;82:162–171. doi: 10.1016/s0888-7543(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 45.Kalscheuer V.M., FitzPatrick D., Tommerup N., Bugge M., Niebuhr E., Neumann L.M., Tzschach A., Shoichet S.A., Menzel C., Erdogan F. Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet. 2007;121:501–509. doi: 10.1007/s00439-006-0284-0. [DOI] [PubMed] [Google Scholar]
- 46.Hafner H. Gender differences in schizophrenia. Psychoneuroendocrinology. 2003;28(Suppl 2):17–54. doi: 10.1016/s0306-4530(02)00125-7. [DOI] [PubMed] [Google Scholar]
- 47.Arnold L.M. Gender differences in bipolar disorder. Psychiatr. Clin. North Am. 2003;26:595–620. doi: 10.1016/s0193-953x(03)00036-4. [DOI] [PubMed] [Google Scholar]
- 48.Kaminsky Z., Wang S.C., Petronis A. Complex disease, gender and epigenetics. Ann. Med. 2006;38:530–544. doi: 10.1080/07853890600989211. [DOI] [PubMed] [Google Scholar]
- 49.Saluz H.P., Jiricny J., Jost J.P. Genomic sequencing reveals a positive correlation between the kinetics of strand-specific DNA demethylation of the overlapping estradiol/glucocorticoid-receptor binding sites and the rate of avian vitellogenin mRNA synthesis. Proc. Natl. Acad. Sci. USA. 1986;83:7167–7171. doi: 10.1073/pnas.83.19.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yokomori N., Moore R., Negishi M. Sexually dimorphic DNA demethylation in the promoter of the Slp (sex-limited protein) gene in mouse liver. Proc. Natl. Acad. Sci. USA. 1995;92:1302–1306. doi: 10.1073/pnas.92.5.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung H.H., Haker H. Schizophrenia as a manifestation of X-linked Mcleod-Neuroacanthocytosis syndrome. J. Clin. Psychiatry. 2004;65:722–723. doi: 10.4088/jcp.v65n0520c. [DOI] [PubMed] [Google Scholar]
- 52.Browning J.L., Patel T., Brandt P.C., Young K.A., Holcomb L.A., Hicks P.B. Clozapine and the mitogen-activated protein kinase signal transduction pathway: implications for antipsychotic actions. Biol. Psychiatry. 2005;57:617–623. doi: 10.1016/j.biopsych.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ravasz E., Somera A.L., Mongru D.A., Oltvai Z.N., Barabasi A.L. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 55.Rives A.W., Galitski T. Modular organization of cellular networks. Proc. Natl. Acad. Sci. USA. 2003;100:1128–1133. doi: 10.1073/pnas.0237338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spirin V., Mirny L.A. Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. USA. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frigola J., Song J., Stirzaker C., Hinshelwood R.A., Peinado M.A., Clark S.J. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 2006;38:540–549. doi: 10.1038/ng1781. [DOI] [PubMed] [Google Scholar]
- 58.Abdolmaleky H.M., Cheng K.H., Russo A., Smith C.L., Faraone S.V., Wilcox M., Shafa R., Glatt S.J., Nguyen G., Ponte J.F. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2005;134:60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- 59.Flanagan J.M., Popendikyte V., Pozdniakovaite N., Sobolev M., Assadzadeh A., Schumacher A., Zangeneh M., Lau L., Virtanen C., Wang S.C. Intra- and interindividual epigenetic variation in human germ cells. Am. J. Hum. Genet. 2006;79:67–84. doi: 10.1086/504729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murrell A., Heeson S., Cooper W.N., Douglas E., Apostolidou S., Moore G.E., Maher E.R., Reik W. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: interaction between genotype and epigenotype. Hum. Mol. Genet. 2004;13:247–255. doi: 10.1093/hmg/ddh013. [DOI] [PubMed] [Google Scholar]
- 61.Veldic M., Guidotti A., Maloku E., Davis J.M., Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc. Natl. Acad. Sci. USA. 2005;102:2152–2157. doi: 10.1073/pnas.0409665102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weber M., Davies J.J., Wittig D., Oakeley E.J., Haase M., Lam W.L., Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 63.Suter C.M., Martin D.I., Ward R.L. Germline epimutation of MLH1 in individuals with multiple cancers. Nat. Genet. 2004;36:497–501. doi: 10.1038/ng1342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.