

Abstract
Infantile hypertrophic pyloric stenosis (IHPS) has an incidence of 1–8 per 1000 live births and is inherited as a complex sex-modified multifactorial trait with a striking male preponderance. Syndromic and monogenic forms exist, and two loci have been identified. Infants present with vomiting due to gastric-outlet obstruction caused by hypertrophy of the smooth muscle of the pylorus. A genome-wide SNP-based high-density linkage scan was carried out on 81 IHPS pedigrees. Nonparametric and parametric linkage analysis identified loci on chromosomes 11q14-q22 (Zmax = 3.9, p < 0.0001; HLODmax = 3.4, α = 0.34) and Xq23 (Zmax = 4.3, p < 0.00001; HLODmax = 4.8, α = 0.56). The two linked chromosomal regions each harbor functional candidate genes that are members of the canonical transient receptor potential (TRPC) family of ion channels and have a potential role in smooth-muscle control and hypertrophy.
Main Text
Infantile hypertrophic pyloric stenosis (IHPS [MIM #179010]) is the most common cause of gastrointestinal obstruction in the first few months of life, with an incidence of 1–8 per 1000 live births in the white population.1 It typically presents in infants 2–6 weeks after birth with projectile vomiting, weight loss, and dehydration. Surgical treatment by pyloromyotomy was introduced a century ago.2
A genetic predisposition to IHPS is well established. IHPS shows familial aggregation, and the classic studies of Carter defined the disease as a paradigm for the multifactorial sex-modified threshold model of inheritance, with affected males outnumbering females in a 4:1 ratio.3,4 This male preponderance was confirmed by later studies in various populations.5–10 There is a striking variation in incidence between population groups, with the cumulative incidence in American infants being 1.9 per 1000 live births in whites, 1.8 in Hispanics, 0.7 in blacks, and 0.6 in Asians.1
In a reanalysis of data from several studies, λs (the ratio of the disease risk among patient siblings to that among members of the general population) was estimated to be 18.11 The same study concluded that IHPS is determined by two or three loci of moderate effect conferring individual genotype relative risks (GRRs) of up to 5.
IHPS has also been associated with several genetic syndromes, such as Cornelia de Lange12 and Smith-Lemli-Opitz13 syndromes, and chromosomal abnormalities, including translocation of chromosome 8 and 17 and a partial trisomy of chromosome 9.14,15 Autosomal-dominant monogenic forms of IHPS have also been reported in several extended pedigrees.16–18
A region on chromosome 16p13-p12 has been identified as the first locus for monogenic IHPS (IHPS2 [MIM #610260]).18 A second locus for monogenic IHPS on chromosome 16q24 has since been identified (K.V.E., F.C., C.G., B.A.C., A.R., A.P., P.P., R.M.G., and E.M.K.C., unpublished data). The gene encoding neuronal nitric oxide synthase, NOS1 (MIM #163731), on chromosome 12q is also considered to be an IHPS susceptibility locus (IHPS1 [MIM #179010]).19,20
Environmental factors are clearly implicated in the development of IHPS. A period of postnatal enteral feeding is required for the disease to develop. In addition, a recent decline in the incidence of IHPS that parallels that of sudden infant death has been observed, coinciding with the implementation of the “back to sleep campaign”21. This has led to the suggestion that posture and feeding are two important potentially modifiable environmental factors in IHPS. A further environmental factor in some cases is erythromycin, a motilin agonist. A 7-fold increase in the incidence of IHPS has been reported among newborn infants who had received erythromycin for postexposure pertussis prophylaxis.22–25 Erythromycin binds to the motilin receptor, which is expressed in the enteric neurons of the duodenum and colon, and induces intestinal contractions.26 It is administered in low doses to improve gastric emptying, but when used in higher doses as an antibiotic, it can lead to prolonged contraction and hypertrophy.27,28
The aim of this work was the identification of disease-predisposing loci with a genome-wide SNP-based linkage scan in a resource of IHPS pedigrees. A SNP-based approach provides increased genomic coverage and a greater overall information content in comparison to microsatellites.29,30 A model-free nonparametric analysis was initially performed, followed by specific parametric analyses.
Eighty-one pedigrees, all of European descent, were ascertained from the UK and Ireland via pediatric units in four major centers: St George's Healthcare National Health Service (NHS) Trust, London; Great Ormond Street Hospital for Children NHS Trust, London; Barts and the London NHS Trust; and Our Lady's Children's Hospital, Dublin. There were a total of 302 individuals, with 206 affected. This resource has sufficient power to detect loci of moderate effect in a subset of these pedigrees. Thirty-nine families had two affecteds; 25 had three affecteds; seven had four affecteds; eight had five affecteds; and two had seven affecteds. Full details are contained in the Supplemental Data available online. The ratio of male to female affecteds was 3 to 1. The diagnosis in all cases was confirmed at pyloromyotomy. The study was approved by the Ethics Committee of University College London Hospital and by the appropriate local insitutional review boards at all other participating hospitals. Informed consent was obtained from all participating families.
Genomic DNA was extracted from cheek swab, saliva, or blood samples according to standard kits and protocols (Oragene and Simhelix DNA Isolation kits). Samples were whole-genome ampliflied as required with the REPLI-g kit. Of the total 302 individuals included in the study, whole-genome-amplified DNA was used for 182 samples.
A genome-wide scan was carried out with the Illumina Linkage IV Panel of 6008 SNPs; 5942 SNPs (98.9%) were successfully typed. These SNPs are designed to be evenly distributed throughout the genome with an average genetic distance between SNPs of 0.64 cM and an average physical distance of 482 kb. Fifteen whole-genome amplification (WGA) samples (8.2%) could not be genotyped at all; none of the genomic DNA samples failed completely. This genotyping was performed by Illumina, San Diego CA.
Multipoint nonparametric linkage (NPL) analysis was performed with MERLIN 1.0.1. The Minx version of this software was used for the analysis of the X chromosome data.31 Thirteen of the 81 families in the resource were excluded by the program from X chromosome analysis because no part of the pedigree was consistent with X-linked inheritance (i.e., only male-to-male disease transmission occurred). MERLIN 1.0.1 checks for and removes erroneous genotypes prior to analysis. Z scores were derived at each SNP position along with a corresponding p value. The Z score is compared to a normal distribution to test for deviation. The assumption of a normal distribution is conservative, as are the associated p values.32 Thresholds for suggestive evidence for linkage and significant evidence for linkage were based on those proposed by Lander and Kruglyak33 but are slightly less stringent. Suggestive linkage in this study is defined by a p value less than 10−3, and the threshold for significant linkage is a p value less than 10−4. These values are equivalent to one-tailed Z scores of 3.1 and 3.7, respectively, which are themselves equivalent to maximized logarithm of odds (LOD) scores of 2.4 and 3.3.
The results of the multipoint NPL analysis for each chromosome are shown in Figure 1. The X chromosome and chromosome 11 demonstrated significant evidence for linkage with an NPL Zmax of 4.3 (p < 0.00001) for the X chromosome and an NPL Zmax of 3.9 (p < 0.00005) for chromosome 11. A comparison of Zmax scores for each family for the “critical” regions on chromosome 11 and the X chromosome is given in the Supplemental Data. The critical region is arbitrarily defined as extending to where the Z score falls below 3. This is a conservative approach to defining the critical region so that no possible candidate genes are overlooked. Most families were consistent with linkage to either chromosome 11 or the X chromosome (or both); 12 families were not consistent with linkage to either locus. An expanded view of each linked region is shown in Figure 2 and detailed in Tables 1 and 2. Haplotypes were derived with the MERLIN 1.0.1 best-estimate approach, which is based on the most likely pattern of gene flow. The pedigree structure and haplotypes for the linked regions for the most informative families is given in the Supplemental Data. Suggestive evidence for linkage with a Zmax of 3.1 (p < 0.001) was found at chromosome 22q11.1.
Figure 1.
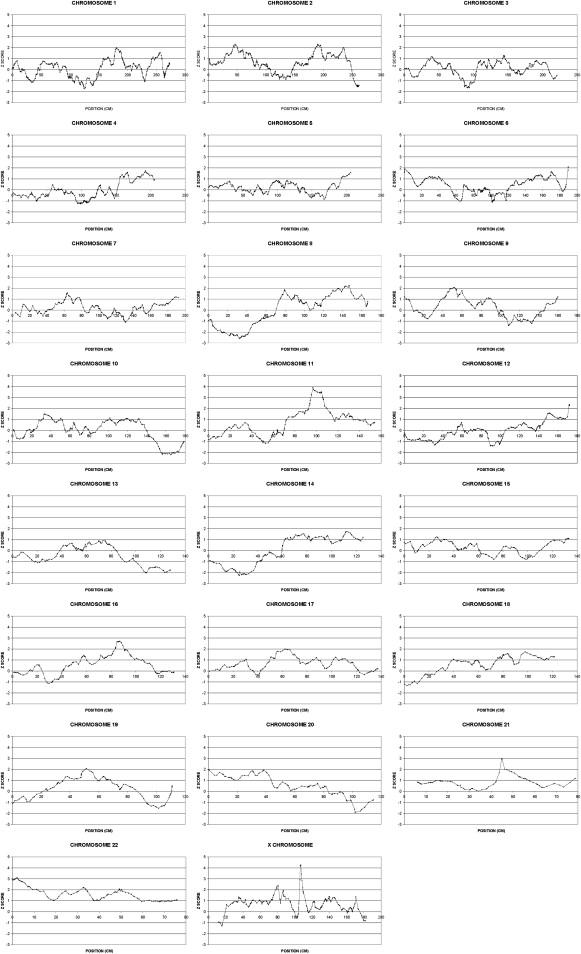
Multipoint NPL Z Scores for All Chromosomes
Position refers to genetic distance from the telomere of the short arm of the chromosome.
Figure 2.
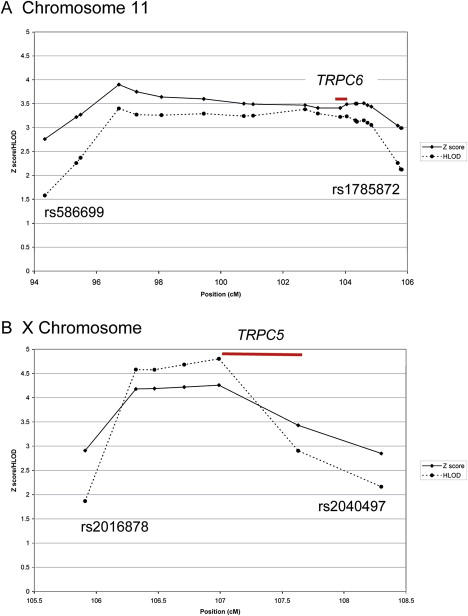
NPL Z Score and Parametric HLOD Plots for the Linked Regions on Chromosomes 11 and X
NPL Z score and parametric HLOD plots for chromosome 11 (A) and the X chromosome (B) for the regions showing evidence for linkage; position refers to genetic distance from the telomere of the short arm of the chromosome. Each marked point corresponds to a genotyped SNP. The SNPs flanking the critical region are labeled. Chromosome 11 was analyzed under the assumption of autosomal-dominant inheritance. Parametric analysis of the X chromosome assumed X-linked recessive inheritance. Both analyses assumed a disease allele frequency of 0.001 and a reduced penetrance of 0.6. Also shown, with red lines, are the approximate positions of two candidate genes, TRPC6 and TRPC5.
Table 1.
Linkage-Analysis Data for the Critical Region on Chromosome 11
| SNP | Physical Position (Mb) | Genetic Position (cM) | Z Score | p Value |
|---|---|---|---|---|
| rs586699 | 91929382 | 94.34 | 2.76 | 0.003 |
| rs1944108 | 93103823 | 95.35 | 3.22 | 0.0006 |
| rs584109 | 93652402 | 95.49 | 3.27 | 0.0005 |
| rs541821 | 94801223 | 96.72 | 3.9 | 0.00005 |
| rs1255182 | 95112039 | 97.29 | 3.75 | 0.00009 |
| rs483884 | 95551040 | 98.09 | 3.64 | 0.00014 |
| rs2919032 | 96132850 | 99.45 | 3.6 | 0.0002 |
| rs921562 | 96806939 | 100.74 | 3.5 | 0.0002 |
| rs723513 | 97588753 | 101.04 | 3.49 | 0.0002 |
| rs1002122 | 99566587 | 102.71 | 3.47 | 0.0003 |
| rs622675 | 100165933 | 103.12 | 3.41 | 0.0003 |
| rs618291 | 100186421 | 103.121 | 3.41 | 0.0003 |
| rs1943752 | 100601234 | 103.84 | 3.41 | 0.0003 |
| rs948734 | 101434119 | 104.05 | 3.49 | 0.0002 |
| rs1711399 | 101965987 | 104.33 | 3.5 | 0.0002 |
| rs1784405 | 101975664 | 104.331 | 3.5 | 0.0002 |
| rs1276278 | 102028851 | 104.37 | 3.5 | 0.0002 |
| rs1298740 | 102057989 | 104.38 | 3.5 | 0.0002 |
| rs536107 | 102404003 | 104.6 | 3.51 | 0.0002 |
| rs601535 | 102605727 | 104.72 | 3.47 | 0.0003 |
| rs559345 | 102802726 | 104.84 | 3.44 | 0.0003 |
| rs540819 | 104371671 | 105.69 | 3.04 | 0.0012 |
| rs1785872 | 104474890 | 105.79 | 2.99 | 0.0014 |
Table 2.
Linkage-Analysis Data for the Critical Region on the X Chromosome
| SNP | Physical Position (Mb) | Genetic Position (cM) | Z Score | p Value |
|---|---|---|---|---|
| rs2016878 | 109820582 | 105.91 | 2.91 | 0.002 |
| rs1890141 | 110232670 | 106.32 | 4.18 | 0.00001 |
| rs926412 | 110380673 | 106.47 | 4.19 | 0.00001 |
| rs1016231 | 110619665 | 106.71 | 4.22 | 0.00001 |
| rs3027802 | 110894171 | 106.99 | 4.26 | 0.00001 |
| rs7049660 | 111445482 | 107.63 | 3.43 | 0.0003 |
| rs2040497 | 111642086 | 108.3 | 2.85 | 0.002 |
Parametric analysis, also with MERLIN 1.0.1, was performed on those genomic regions that showed significant evidence for linkage with NPL analysis (chromosomes 11 and X). A disease allele frequency of 0.001 and penetrance of 0.6 was assumed. A heterogeneity LOD score (HLOD) was derived at each SNP position with a corresponding alpha, where α equals the proportion of families demonstrating linkage to that locus.
Analysis of chromosome 11 with the assumption of autosomal-dominant inheritance demonstrated significant evidence for linkage with a maximum HLOD (HLODmax) of 3.4 (α = 0.34). Analysis of the X chromosome with the assumption of X-linked recessive inheritance also demonstrated significant evidence for linkage with an HLODmax of 4.8 (α = 0.56). Figure 2 shows the results of the NPL and parametric analyses for these regions.
The genetic architecture of common familial traits that do not segregate in a Mendelian fashion is beginning to emerge with the increased power provided by completion of the human-genome and HapMap projects. As anticipated, the genetic basis of such complex traits covers a wide spectrum. The number of loci involved (oligo- to polygenicity), their effect size and mode of interaction, and the frequency and heterogeneity of causal alleles is clearly variable.34 Linkage-analysis strategies remain a valid approach for those traits for which there is reasonable expectation that loci of moderate effect exist, provided that family resources adequate to offset the erosion of power engendered by locus heterogeneity can be assembled.
Syndromic and monogenic forms of IHPS clearly exist, but the most common form segregates as a complex multifactorial trait.11 The male preponderance could not be explained solely by X-linked recessive inheritance because male-to-male transmission is frequently observed. Two loci for monogenic IHPS have been identified by SNP-based genome-wide scans in two different multigeneration pedigrees consistent with autosomal dominant inheritance: chromosome 16p13-p12 (IHPS2 [MIM #610260])18 and chromosome 16q24 (K.V.E., F.C., C.G., B.A.C., A.R., A.P., P.P., R.M.G., and E.M.K.C., unpublished data).
Suggestive evidence of linkage between IHPS and an intragenic microsatellite polymorphism in NOS1 (MIM #163731) on chromosome 12q24.2-q24.31 was shown with 27 IHPS families from our resource,19 although this finding has not been replicated.35 More recently, an association between IHPS and a NOS1 promoter 1c regulatory polymorphism has been reported20,36. The NOS1 gene encodes neuronal nitric oxide synthase (nNOS), an enzyme that catalyzes the formation of nitric oxide (NO) in neurons of the peripheral nervous system, where it plays a role in smooth-muscle relaxation. Transient pyloric relaxation is essential for the passage of stomach contents into the small bowel, and this is mainly mediated by the action of NO. A defect in NO production, leading to a failure of pyloric relaxation and subsequent alteration of luminal transit, was therefore proposed to underlie at least some cases of IHPS. NOS1-deficient mice are also reported to have features of IHPS.36 Consequently, NOS1 is considered to be a predisposing locus for IHPS (IHPS1 [MIM #179010]).
The statistical significance of linkage data generated by analysis of markers across the entire genome remains controversial, but thresholds adopted merely determine the extent to which investigators wish to tolerate false-positive results and waste resources in following up signals that have arisen by chance.37,38 Nonparametric analysis was adopted initially so that potential problems in mis-specification of inheritance parameters could be avoided. A parametric analysis was then carried out in those regions showing significant linkage. It can be argued that the similar results obtained suggest that the parameters chosen might be close to the true pattern of inheritance at these loci.
Adopting significance thresholds corresponding to those proposed by Lander and Kruglyak,33 we found evidence for significant linkage at chromosome 11q14-q22 and Xq23, and suggestive evidence of linkage was found at chromosome 22q. No evidence of linkage was obtained at the NOS1 locus (IHPS1 [MIM #179010]) on chromosome 12q24 or at the loci for monogenic IHPS on chromosome 16. This does not of course exclude that small-effect alleles at any of these loci might contribute to the trait in some or even all of these pedigrees. It is consistent with the infrequency with which loci for Mendelian forms of complex traits emerge as susceptibility loci for common cases. The results in any event confirm that locus heterogeneity exists for IHPS.
Linkage data from multiple small pedigrees segregating in a non-Mendelian fashion cannot reveal unequivocal recombinant breakpoints that allow a clear definition of the critical linked region. On the pragmatic assumption that the regions extend to where Z scores fall below 3.0, a region of approximately 12 Mb on chromosome 11q14-q22 and approximately 2 Mb on chromosome Xq23 can be defined. The former encompasses about 100 known or predicted genes and the latter about 12 genes.
Knowledge of the underlying pathophysiology of IHPS is sufficiently advanced to allow evaluation of positional candidate genes within the critical regions according to their expression and putative function. Information was collected from a number of human-genome resources including the Ensembl, National Center for Biotechnology Information (NCBI), and University of California-Santa Cruz (UCSC) Browsers (genome build NCBI36.1), in addition to the PubMed and Unigene databases.
Smooth-muscle hypertrophy in the pyloric canal underlies the gastric-outlet obstruction in IHPS. This develops after birth and requires a period of enteral feeding.2 IHPS appears to arise from a defect in gastrointestinal motility that shows both development- and location-specific expression, rather than a defect in the developmental anatomy of the gastrointestinal tract. It is noteworthy that each of these two chromosomal regions harbors a gene from the same family of canonical transient receptor potential (TRPC) cation channels: TRPC6 on chromosome 11q21-22 and TRPC5 on chromosome Xq23. These genes represent the strongest functional candidate in each region.
Mammalian TRP proteins form a superfamily of 28 genes encoding six-transmembrane (6-TM) permeable cation-channel subunits grouped into six subfamilies by amino acid sequence homology. TRP channels are calcium-permeable cation channels and appear to act as signal integrators in a wide variety of biological contexts.39
The canonical TRP channel subfamily has seven members. TRPC channel expression tends to be ubiquitous and abundant in brain and neural tissues, but TRPC5 and TRPC6 have been detected in tissues of the gut and smooth muscle cells. TRPC6 is known to be highly expressed in pulmonary and vascular smooth muscle, and a specific role in regulating smooth-muscle function is emerging for this channel.40,41 Overexpression of TRPC6 is implicated in increased pulmonary smooth-muscle proliferation,42 and it is possible that there might be a similar effect on the smooth muscle cells of the pylorus. Furthermore, TRPC6 also acts as a sensor of membrane stretch,43 which could be important in the gastrointestinal tract for correct maintenance of peristalsis. TRPC6-deficient mice display increased vascular smooth muscle contractility, indicating a role for TRPC6 in control of smooth-muscle tone.44 Gain-of-function mutations in TRPC6 that enhance channel activity cause a proteinuric kidney disease, focal and segmental glomerulosclerosis (FSGS),45 but this does not exclude quite different pathology arising from allelic variants with different functional effects.
TRPC5 is predominantly expressed in the brain, but it is also detected in smooth muscle cells and in the stomach, where a role in gastrointestinal-tract muscle contraction has been proposed.46,47 There is evidence that TRPC5 might be involved in cardiac-muscle hypertrophy,48 and it has been shown to be activated by nitric oxide,49 which is important in smooth-muscle relaxation in the gastrointestinal tract.
An X-linked recessive locus accounting for a subset of IHPS families could obviously contribute to the male preponderance in IHPS. In our resource of familial and sporadic cases, the ratio of males to females is more skewed among the sporadic population (3:1 and 5:1, respectively). It is difficult to quantify the potential contribution of X-linked recessive inheritance to male preponderance in our limited resource. However, within the pedigrees, the ratio of males to females is approximately 3.5:1 in families consistent with linkage to Xq23 and 2.5:1 in families consistent with autosomal linkage.
In conclusion, our results confirm locus heterogeneity for IHPS and identify loci on chromosomes 11q and Xq. Strong functional candidate genes, TRPC5 and TRPC6, exist at these loci, and further work is required for the evaluation of their putative role by resequencing and SNP-based association studies in our resource of IHPS trios.
Acknowledgments
This work was supported by Birth Defects Foundation Newlife and Action Medical Research. We are very grateful to all the families that participated in this study. We would like to thank K. Rogers, N. Johnson, A. Massoud, and J. Mulligan for their contribution to the family-ascertainment effort.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl genome browser, http://www.ensembl.org/Homo_sapiens/index.html
Illumina, http://www.illumina.com/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/OMIM/
UniGene, http://www.ncbi.nlm.nih.gov/UniGene/
References
- 1.Schechter R., Torfs C.P., Bateson T.F. The epidemiology of infantile hypertrophic pyloric stenosis. Paediatr. Perinat. Epidemiol. 1997;11:407–427. doi: 10.1046/j.1365-3016.1997.d01-32.x. [DOI] [PubMed] [Google Scholar]
- 2.Murtagh K., Perry P., Corlett M., Fraser I. Infantile hypertrophic pyloric stenosis. Dig. Dis. 1992;10:190–198. doi: 10.1159/000171357. [DOI] [PubMed] [Google Scholar]
- 3.Carter C.O., Powell B.W. Two-generation pyloric stenosis. Lancet. 1954;266:746–748. doi: 10.1016/s0140-6736(54)92713-0. [DOI] [PubMed] [Google Scholar]
- 4.Carter C.O. The inheritance of congenital pyloric stenosis. Br. Med. Bull. 1961;17:251–254. doi: 10.1093/oxfordjournals.bmb.a069918. [DOI] [PubMed] [Google Scholar]
- 5.Applegate M.S., Druschel C.M. The epidemiology of infantile hypertrophic pyloric stenosis in New York State, 1983 to 1990. Arch. Pediatr. Adolesc. Med. 1995;149:1123–1129. doi: 10.1001/archpedi.1995.02170230077011. [DOI] [PubMed] [Google Scholar]
- 6.Cui W., Ma C.X., Tang Y., Chang V., Rao P.V., Ariet M., Resnick M.B., Roth J. Sex differences in birth defects: A study of opposite-sex twins. Birth Defects Res. A. Clin. Mol. Teratol. 2005;73:876–880. doi: 10.1002/bdra.20196. [DOI] [PubMed] [Google Scholar]
- 7.Habbick B.F., To T. Incidence of infantile hypertrophic pyloric stenosis in Saskatchewan, 1970–85. CMAJ. 1989;140:395–398. [PMC free article] [PubMed] [Google Scholar]
- 8.Lammer E.J., Edmonds L.D. Trends in pyloric stenosis incidence, Atlanta, 1968 to 1982. J. Med. Genet. 1987;24:482–487. doi: 10.1136/jmg.24.8.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen L., Green A., Hansen L.P. The epidemiology of infantile hypertrophic pyloric stenosis in a Danish population, 1950–84. Int. J. Epidemiol. 1989;18:413–417. doi: 10.1093/ije/18.2.413. [DOI] [PubMed] [Google Scholar]
- 10.Walpole I.R. Some epidemiological aspects of pyloric stenosis in British Columbia. Am. J. Med. Genet. 1981;10:237–244. doi: 10.1002/ajmg.1320100307. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell L.E., Risch N. The genetics of infantile hypertrophic pyloric stenosis. A reanalysis. Am. J. Dis. Child. 1993;147:1203–1211. doi: 10.1001/archpedi.1993.02160350077012. [DOI] [PubMed] [Google Scholar]
- 12.Jackson L., Kline A.D., Barr M.A., Koch S. de Lange syndrome: A clinical review of 310 individuals. Am. J. Med. Genet. 1993;47:940–946. doi: 10.1002/ajmg.1320470703. [DOI] [PubMed] [Google Scholar]
- 13.Smith D.W., Lemli L., Opitz J.M. A newly recognized syndrome of multiple contenital anomalies. J. Pediatr. 1964;64:210–217. doi: 10.1016/s0022-3476(64)80264-x. [DOI] [PubMed] [Google Scholar]
- 14.Heller A., Seidel J., Hubler A., Starke H., Beensen V., Senger G., Rocchi M., Wirth J., Chudoba I., Claussen U., Liehr T. Molecular cytogenetic characterisation of partial trisomy 9q in a case with pyloric stenosis and a review. J. Med. Genet. 2000;37:529–532. doi: 10.1136/jmg.37.7.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodgson S.V., Berry A.C., Dunbar H.M. Two brothers with an unbalanced 8;17 translocation and infantile pyloric stenosis. Clin. Genet. 1995;48:328–330. doi: 10.1111/j.1399-0004.1995.tb04120.x. [DOI] [PubMed] [Google Scholar]
- 16.Finsen V.R. Infantile hypertrophic pyloric stenosis–unusual familial incidence. Arch. Dis. Child. 1979;54:720–721. doi: 10.1136/adc.54.9.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fried K., Aviv S., Nisenbaum C. Probable autosomal dominant infantile pyloric stenosis in a large kindred. Clin. Genet. 1981;20:328–330. doi: 10.1111/j.1399-0004.1981.tb01043.x. [DOI] [PubMed] [Google Scholar]
- 18.Capon F., Reece A., Ravindrarajah R., Chung E. Linkage of monogenic infantile hypertrophic pyloric stenosis to chromosome 16p12-p13 and evidence for genetic heterogeneity. Am. J. Hum. Genet. 2006;79:378–382. doi: 10.1086/505952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung E., Curtis D., Chen G., Marsden P.A., Twells R., Xu W., Gardiner M. Genetic evidence for the neuronal nitric oxide synthase gene (NOS1) as a susceptibility locus for infantile pyloric stenosis. Am. J. Hum. Genet. 1996;58:363–370. [PMC free article] [PubMed] [Google Scholar]
- 20.Saur D., Vanderwinden J.M., Seidler B., Schmid R.M., De Laet M.H., Allescher H.D. Single-nucleotide promoter polymorphism alters transcription of neuronal nitric oxide synthase exon 1c in infantile hypertrophic pyloric stenosis. Proc. Natl. Acad. Sci. USA. 2004;101:1662–1667. doi: 10.1073/pnas.0305473101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Persson S., Ekbom A., Granath F., Nordenskjold A. Parallel incidences of sudden infant death syndrome and infantile hypertrophic pyloric stenosis: A common cause? Pediatrics. 2001;108:E70. doi: 10.1542/peds.108.4.e70. [DOI] [PubMed] [Google Scholar]
- 22.Cooper W.O., Griffin M.R., Arbogast P., Hickson G.B., Gautam S., Ray W.A. Very early exposure to erythromycin and infantile hypertrophic pyloric stenosis. Arch. Pediatr. Adolesc. Med. 2002;156:647–650. doi: 10.1001/archpedi.156.7.647. [DOI] [PubMed] [Google Scholar]
- 23.Honein M.A., Paulozzi L.J., Himelright I.M., Lee B., Cragan J.D., Patterson L., Correa A., Hall S., Erickson J.D. Infantile hypertrophic pyloric stenosis after pertussis prophylaxis with erythromcyin: A case review and cohort study. Lancet. 1999;354:2101–2105. doi: 10.1016/s0140-6736(99)10073-4. [DOI] [PubMed] [Google Scholar]
- 24.Mahon B.E., Rosenman M.B., Kleiman M.B. Maternal and infant use of erythromycin and other macrolide antibiotics as risk factors for infantile hypertrophic pyloric stenosis. J. Pediatr. 2001;139:380–384. doi: 10.1067/mpd.2001.117577. [DOI] [PubMed] [Google Scholar]
- 25.SanFilippo A. Infantile hypertrophic pyloric stenosis related to ingestion of erythromycine estolate: A report of five cases. J. Pediatr. Surg. 1976;11:177–180. doi: 10.1016/0022-3468(76)90283-9. [DOI] [PubMed] [Google Scholar]
- 26.Feighner S.D., Tan C.P., McKee K.K., Palyha O.C., Hreniuk D.L., Pong S.S., Austin C.P., Figueroa D., MacNeil D., Cascieri M.A. Receptor for motilin identified in the human gastrointestinal system. Science. 1999;284:2184–2188. doi: 10.1126/science.284.5423.2184. [DOI] [PubMed] [Google Scholar]
- 27.Coulie B., Tack J., Peeters T., Janssens J. Involvement of two different pathways in the motor effects of erythromycin on the gastric antrum in humans. Gut. 1998;43:395–400. doi: 10.1136/gut.43.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Lorenzo C., Flores A.F., Tomomasa T., Hyman P.E. Effect of erythromycin on antroduodenal motility in children with chronic functional gastrointestinal symptoms. Dig. Dis. Sci. 1994;39:1399–1404. doi: 10.1007/BF02088040. [DOI] [PubMed] [Google Scholar]
- 29.John S., Shephard N., Liu G., Zeggini E., Cao M., Chen W., Vasavda N., Mills T., Barton A., Hinks A. Whole-genome scan, in a complex disease, using 11,245 single-nucleotide polymorphisms: Comparison with microsatellites. Am. J. Hum. Genet. 2004;75:54–64. doi: 10.1086/422195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans D.M., Cardon L.R. Guidelines for genotyping in genomewide linkage studies: Single-nucleotide-polymorphism maps versus microsatellite maps. Am. J. Hum. Genet. 2004;75:687–692. doi: 10.1086/424696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 32.Whittemore A.S., Halpern J. A class of tests for linkage using affected pedigree members. Biometrics. 1994;50:118–127. [PubMed] [Google Scholar]
- 33.Lander E., Kruglyak L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 34.Glazier A.M., Nadeau J.H., Aitman T.J. Finding genes that underlie complex traits. Science. 2002;298:2345–2349. doi: 10.1126/science.1076641. [DOI] [PubMed] [Google Scholar]
- 35.Soderhall C., Nordenskjold A. Neuronal nitric oxide synthase, nNOS, is not linked to infantile hypertrophic pyloric stenosis in three families. Clin. Genet. 1998;53:421–422. doi: 10.1111/j.1399-0004.1998.tb02758.x. [DOI] [PubMed] [Google Scholar]
- 36.Huang P.L., Dawson T.M., Bredt D.S., Snyder S.H., Fishman M.C. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 37.Sawcer S., Jones H.B., Judge D., Visser F., Compston A., Goodfellow P.N., Clayton D. Empirical genomewide significance levels established by whole genome simulations. Genet. Epidemiol. 1997;14:223–229. doi: 10.1002/(SICI)1098-2272(1997)14:3<223::AID-GEPI1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Zhao L.P., Prentice R., Shen F., Hsu L. On the assessment of statistical significance in disease-gene discovery. Am. J. Hum. Genet. 1999;64:1739–1753. doi: 10.1086/512072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang C.L. The transient receptor potential superfamily of ion channels. J. Am. Soc. Nephrol. 2004;15:1690–1699. doi: 10.1097/01.asn.0000129115.69395.65. [DOI] [PubMed] [Google Scholar]
- 40.Inoue R., Okada T., Onoue H., Hara Y., Shimizu S., Naitoh S., Ito Y., Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca(2+)-permeable cation channel. Circ. Res. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- 41.Jung S., Strotmann R., Schultz G., Plant T.D. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am. J. Physiol. Cell Physiol. 2002;282:C347–C359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- 42.Yu Y., Fantozzi I., Remillard C.V., Landsberg J.W., Kunichika N., Platoshyn O., Tigno D.D., Thistlethwaite P.A., Rubin L.J., Yuan J.X. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spassova M.A., Hewavitharana T., Xu W., Soboloff J., Gill D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA. 2006;103:16586–16591. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dietrich A., Mederos Y., Schnitzler M., Gollasch M., Gross V., Storch U., Dubrovska G., Obst M., Yildirim E., Salanova B. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol. Cell. Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winn M.P., Conlon P.J., Lynn K.L., Farrington M.K., Creazzo T., Hawkins A.F., Daskalakis N., Kwan S.Y., Ebersviller S., Burchette J.L. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 46.Dietrich A., Chubanov V., Kalwa H., Rost B.R., Gudermann T. Cation channels of the transient receptor potential superfamily: their role in physiological and pathophysiological processes of smooth muscle cells. Pharmacol. Ther. 2006;112:744–760. doi: 10.1016/j.pharmthera.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 47.Lee Y.M., Kim B.J., Kim H.J., Yang D.K., Zhu M.H., Lee K.P., So I., Kim K.W. TRPC5 as a candidate for the nonselective cation channel activated by muscarinic stimulation in murine stomach. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;284:G604–G616. doi: 10.1152/ajpgi.00069.2002. [DOI] [PubMed] [Google Scholar]
- 48.Bush E.W., Hood D.B., Papst P.J., Chapo J.A., Minobe W., Bristow M.R., Olson E.N., McKinsey T.A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 2006;281:33487–33496. doi: 10.1074/jbc.M605536200. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida T., Inoue R., Morii T., Takahashi N., Yamamoto S., Hara Y., Tominaga M., Shimizu S., Sato Y., Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.