

Abstract
Most genetic disruptions underlying human disease are microlesions, whereas gross lesions are rare with gross deletions being most frequently found (6%). Similar observations have been made in primary immunodeficiency genes, such as BTK, but for unknown reasons the IGHM and DCLRE1C (Artemis) gene defects frequently represent gross deletions (∼60%). We characterized the gross deletion breakpoints in IGHM-, BTK-, and Artemis-deficient patients. The IGHM deletion breakpoints did not show involvement of recombination signal sequences or immunoglobulin switch regions. Instead, five IGHM, eight BTK, and five unique Artemis breakpoints were located in or near sequences derived from transposable elements (TE). The breakpoints of four out of five disrupted Artemis alleles were located in highly homologous regions, similar to Ig subclass deficiencies and Vh deletion polymorphisms. Nevertheless, these observations suggest a role for TEs in mediating gross deletions. The identified gross deletion breakpoints were mostly located in TE subclasses that were specifically overrepresented in the involved gene as compared to the average in the human genome. This concerned both long (LINE1) and short (Alu, MIR) interspersed elements, as well as LTR retrotransposons (ERV). Furthermore, a high total TE content (>40%) was associated with an increased frequency of gross deletions. Both findings were further investigated and confirmed in a total set of 20 genes disrupted in human disease. Thus, to our knowledge for the first time, we provide evidence that a high TE content, irrespective of the type of element, results in the increased incidence of gross deletions as gene disruption underlying human disease.
Introduction
The majority of gene disruptions underlying human inherited diseases are microlesions, i.e., single basepair substitutions (68%) and small (<20 nt) deletions or insertions (25%).1 Gross lesions are less common, with gross deletions being most frequently found (6%).1 For gross deletions to occur, three conditions have to be met: (1) double-stranded DNA breaks in two distinct genomic locations that are (2) physically located in close proximity and joined by (3) incorrect repair. Based on limited sequence data of the junctions of gross deletion breakpoints in disease-causing alleles, two mechanisms have been described: (1) mispairing of homologous sequences and unequal crossing over; and (2) nonhomologous deletions.2 Recent analysis of several gross deletion breakpoints indicates a role for single-strand annealing of repeated fragments in gross lesions.3 Although this mechanism could explain incorrect repair of complex DNA structures, it remains unclear how two genomically distinct sites become located in close proximity.
The human genome contains four classes of frequently recurring sequences, called interspersed repeats, which are derived from transposable elements (TE) and comprise about 45% of the total genome sequence.4 The four classes are short interspersed elements (SINE), long interspersed elements (LINE), LTR retrotransposons, and DNA transposons. Nonhomologous gross deletion breakpoints are frequently located in or near these TEs,5,6 but the role of these elements in mediating gross deletions is currently unknown.
Although inherited disorders of the immune system are rare, multiple gene defects have been identified during the last 15 years, mainly concerning microlesions.7 However, gross deletions have also been found, for example involving the DCLRE1C (Artemis [MIM 605988]), BTK (MIM 300300), and immunoglobulin Cμ heavy chain (IGHM [MIM 147020]) genes.8–13 The gross deletion frequency of BTK disruptions is 6%, whereas more than half of the reported disease-causing alleles of IGHM and Artemis are disrupted by gross deletions.1,8,13 A mechanism underlying this striking difference has never been reported.
The immunoglobulin heavy chain (IgH) is part of the antibody molecule and is produced by B cells after V(D)J recombination of the IGH locus. The IGH locus contains multiple Vh, Dh, and Jh gene segments and constant (Ch) regions. The Vh, Dh, and Jh gene segments are flanked by recombination signal sequences (RSS) that are recognized by the recombinase machinery,14 which mediates rearrangement of the Vh, Dh, and Jh gene segments with deletion of the intervening sequences during precursor-B cell differentiation to generate a functional VDJ exon.15 The VDJ exon is initially spliced to the Cμ exons. During an immune response, genomic Ig class switch recombination can take place to replace the Cμ exons for one of the other Ch regions. This process is mediated by the Ig switch regions.16 Similar to V(D)J recombination, Ig class switch recombination involves the genomic deletion of a large intervening DNA sequence.
Gross deletions involving IGHM can be up to 300 kb and can include upstream Dh and Vh gene segments and downstream constant gene regions.11–13 We hypothesized that the complexity of the IGH locus contributes to the increased incidence of IGHM-disrupting gross deletions. Possible mediators are the RSS and Ig switch regions, which are target sequences for B cell-specific recombination events. Furthermore, germline duplications and deletions have been described in either the Vh region or involving the Ch regions.17–21 Whereas Vh deletions are commonly regarded as polymorphisms,22 constant gene deletions can result in Ig subclass deficiencies.23–25 The Ch regions contain high degrees of sequence homology, and the altered haplotypes are thought to be the result of mispairing between highly homologous regions and subsequent unequal crossing-over during meiosis.18–21,26,27
In this study, we aimed to identify mechanisms that underlie the high frequency of gross deletions disrupting IGHM and Artemis as compared to BTK. Breakpoint analysis suggested either the involvement of TEs or large homologous regions rather than recombination motifs. We considered that the colocalization of gross deletion breakpoints with TEs was not coincidental, and we hypothesized that the TE content of a gene is related to the gross deletion frequency. This hypothesis was tested on 20 genes disrupted in human disease.
Material and Methods
Patient DNA Isolation and PCR Amplification
Genomic DNA was isolated from post-Ficoll PB granulocytes with the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, St. Louis, MO). The sizes of the gross deletions in IGHM-, BTK-, and Artemis-deficient patients were determined with scanning PCRs for gene segments, exons, and subsequently for intronic regions. Seven unrelated IGHM-deficient patients with a homozygous gross deletion were studied. Of these patients, three were previously published as having a gross deletion (F2, F7, and SIOC),11–13 and the other four were newly identified (BN, ID392, ID393, and ID394). Furthermore, we studied three unrelated patients with a hemizygous deletion of BTK (ID113, ID434, ID440) and four unrelated Artemis-deficient patients, three of which are from consanguineous parents carrying a homozygous deletion (ID020, ID024, ID389) and one patient with a compound heterozygous Artemis deletion (ID124). PCR reactions were essentially performed as described before with primers that were designed to specifically amplify 200–600 bp of DNA (primer sequences available upon request).28
Long-Range, Ligation-Mediated PCR and DNA Sequencing for Analysis of the Breakpoint Junction
Long-range (LR-)PCR was performed as described before29 with the appropriate forward and reverse primers that were used to map the gross deletion boundaries.
Ligation-mediated (LM-)PCR was essentially performed as described before30 with newly designed primers. Aliquots of 1 μg high-molecular-weight DNA were digested with blunt-end restriction enzymes (DraI, HincII, PvuII, and StuI), and 50 mM of an adaptor (Clontech, Palo Alto, CA) was ligated to both ends of the restriction fragments. The ligation products were subjected to two rounds of PCR with nested adaptor-specific primers AP1 and AP2 (Clontech) and sets of primers designed upstream of the 5′ end of the breakpoint. Atypical bands that appeared from patient's DNA, but not from control DNA, were excised from the gel, purified with the QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA), and sequenced on an ABI Prism 3100 sequence detection system (Applied Biosystems, Foster City, CA).
Human Disease-Causing Mutation Data
Mutation data of genes disrupted in human disease were extracted from the February 2007 release of the human gene mutation database (HGMD).1 To calculate the gross deletion frequency, we included the newly described gross deletion alleles from IGHM- and Artemis-deficient patients from this study and newly identified BLNK mutations (unpublished results from M.v.d.B. and from M.E.C.).
Sequence Analysis of Genes and Breakpoint Regions
Sequences of the IGH locus (NCBI: NG_001019.5) and the BTK, Artemis (DCLRE1C), BLNK, APC, ATM, BRCA1, BRCA2, CFTR, DMD, Factor VIII (F8), FANCA, HPRT1, LDLR, MECP2, MLH1, MSH2, NF1, RB1, and VWF genes including 10 kb upstream and downstream sequences, extracted from Ensembl v42 (Dec 2006),31 were annotated with TE-derived interspersed repeats by the CENSOR software tool of the Repbase database.32 To obtain representative TE content frequencies, only genes spanning more than 50 kb were included.
The identified breakpoint regions were aligned with the gene sequences extracted from public databases. The Genewindow website was used to identify whether mismatches with standard sequences were previously described polymorphisms.33 Complexity analysis of ±25 bp flanking the breakpoint regions was performed to examine the potential contribution of local sequence structure to the mechanism of gross deletion in the IGH locus and the BTK and Artemis genes.3,34 In addition, the same region was scanned for the presence of 24 sequence motifs known to be associated with site-specific recombination, mutation, cleavage, and gene rearrangement.2 The sequences ±1000 bp flanking the breakpoint regions were annotated with TEs by the CENSOR software tool of the Repbase database.32 The nucleic acid dot plot tool was used to study the homology between the Artemis gene and the genomic region 82 kb 5′ of Artemis. A 19 nucleotide window size was chosen in which the mismatch limit was set at 0.
Statistical Analysis
Differences in gross deletion frequencies were analyzed with the nonparametric Mann-Whitney U test (exact test; one-tailed; p < 0.05) in both the SPSS and the MatLab programs, yielding exactly similar results.
Results
Identification of Five Unique Gross Deletion Breakpoint Junctions in Seven Homozygous IGHM-Deficient Patients
DNA material was collected from four previously described and three newly identified unrelated patients with a full block in precursor-B cell differentiation in bone marrow resulting from a homozygous deletion of IGHM. For each of these seven patients, the extent of the deletions was studied, which resulted in the characterization of five unique breakpoint junctions within the IGH loci of the seven IGHM-deficient patients (Figure 1A).
Figure 1.

Gross Deletion Breakpoints of Seven Unrelated Homozygous IGHM-Deficient Patients
(A) IGHM gross deletions ranging in size from 71 to 732 kb were identified with a PCR approach. An additional 33 kb deletion 1.6 kb upstream of the IGHM deletion disrupts three Vh gene segments in patient BN.
(B) Sequences of the five unique IGHM-deleting breakpoint junctions and one novel Vh deletion polymorphism. The IGHM-deletion breakpoint junction of patient BN shows a 9-nucleotide insertion (caps). The two mismatches with the 5′ breakpoint region are previously described SNPs. The 33 kb Vh-deletion polymorphism in patient BN shows a dinucleotide microhomology (boxed). The IGHM deletion in patients ID393 and ID394 shows a trinucleotide microhomology. The IGHM deletion in patients ID392 and F7 shows one nucleotide microhomology. The small deletion and the mismatch with the 5′ breakpoint region are not previously described polymorphisms. The IGHM deletion in patient F2 shows a 51 nucleotide insertion that corresponds to the intronic region between Dh4-23 and Dh5-24. The 5′ junctions showed one nucleotide microhomology, whereas the 3′ junction showed three nucleotides of microhomology. The mismatch within the 51 nt insertion is not a previously described SNP. Complexity analysis of ±25 bp flanking the breakpoints showed that the deletions in patients ID393, ID394, and SIOC are potentially mediated by a hairpin loop (inverted repeats). The deletion in patients ID392 and F7 could have been mediated by a knot structure. Furthermore, the deletion in patient F2 is potentially mediated by combination of a knot structure for the 5′ joint and a Möbius loop for the 3′ joint.
Patient BN was found to carry a homozygous IGHM deletion of 732 kb starting 5′ of Vh3-37 and ending 3′ of IGHGP (Figure 1A). The junction contained 9 bp of unique sequence that could not be assigned to the 5′ or 3′ breakpoint region sequences (Figure 1B). The two mismatches with the 5′ breakpoint region corresponded to previously described single nucleotide polymorphisms (SNP).
In addition to the IGHM deletion, the homozygous IGH locus of patient BN had a 33 kb deletion involving the Vh3-39, Vh7-40, and Vh3-41 gene segments. About 1.3 kb sequence between the two gross deletions, including the Vh3-38 gene segment, was maintained at the locus. The junction of the 33 kb deletion contains 2 bp microhomology and shows high degrees of sequence homology between the regions downstream of the breakpoints (Figure 1B). This deletion appeared similar to a deletion (Del III) that was mapped before but not cloned.22 None of the other six IGHM patients carried this deletion, and it was found in 15 of 66 controls, who did not have the 732 kb IGHM deletion; 7 were heterozygous and 8 homozygous (23 alleles). Consequently, the 33 kb deletion of Vh3-39, Vh7-40, and Vh3-41 is a common polymorphism present in 17% of the IGH alleles in controls.
Identical breakpoint junctions were found in two unrelated Caucasian patients: ID393 and ID394. Both patients lack 578 kb of genomic DNA starting 5′ of the Vh3-19 gene segment and ending 3′ of the IGHG4 constant region. The two breakpoints were fused with a three-nucleotide microhomology region (Figure 1B). Analysis of 21 SNPs in Vh3-20, IGHE, and IGHA2 revealed no differences between the patients, indicating that they carry the same allele.
The two unrelated Turkish patients ID392 and F7 also carried deleted alleles with identical breakpoint junctions. The deletions span 126 kb of genomic DNA starting 5′ of the KIAA0125 element between the Vh6-1 and Dh1-1 gene segments and ending 3′ of IGHD (Figure 1B). One nucleotide microhomology was found at the breakpoint fusion (Figure 1B). Interestingly, 5′ of the breakpoint junction of both patients, a small deletion of 19 bp with 6 bp microhomology in the fusion was found. This small deletion was not seen in 82 controls. We suggest that both the small and the gross deletion occurred in the same event. SNP analysis of the breakpoint fusion region, Vh3-20, VhIV-20.1, and IGHE PCR amplicons showed no polymorphic differences between the alleles of patients ID392 and F7, indicating that both patients inherited the allele from a common ancestor.
The gross deletion in patient F2 spanned 71 kb from Dh1-1 to IGHM. Interestingly, the junction contained a 51 nt insertion, which aligned almost perfectly with the intronic region between Dh4-23 and Dh5-24, 37 kb downstream of the 5′ breakpoint (Figure 1A). Consequently, two deletions were found; the two junctions contained 1 and 3 nucleotides of microhomology, respectively (Figure 1B). The 5′ deletion that does not involve IGHM was not found in 66 controls. Therefore, it does not appear to be a common polymorphism.
Patient SIOC had a gross deletion of 146 kb extending from Dh6-13 to IGHG3 (Figure 1A). The junction contains four nucleotides of microhomology (Figure 1B).
We identified five unique breakpoint junctions in seven unrelated homozygous IGHM-deficient patients. Two previously identified IGHM-deficient patients, who were heterozygous for a gross deletion,13 did not carry one of the identified breakpoint junctions and each is likely to carry a unique deletion.
IGH Breakpoints Are Associated with TEs
The five identified IGH breakpoints were located in intronic sequences without any apparent homology between the 5′ and 3′ breakpoint flanking regions. In order to study the involvement of TEs, 2 kb DNA sequences flanking the breakpoint regions were screened against a reference collection of repeats. As shown in Figure 2, we found that 2 of the 10 IGHM-deleting breakpoints were located in or near a type 1 LINE and 6 were located in or near endogenous retroviral (ERV) sequences, which belong to the class of LTR retrotransposons. The remaining two breakpoints were located in unique sequences. Consequently, in almost all gross deletions involving IGHM, the breakpoints of fusion partners are located in or near TEs of the same class. TEs are present throughout the genome and they have been implicated in genomic rearrangements,5,6 so the localization of gross deletions in or near TEs could be a general phenomenon.
Figure 2.
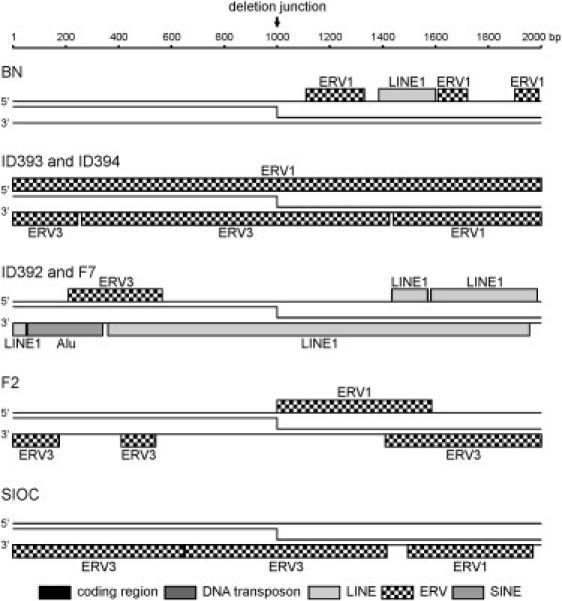
IGHM Deletion Breakpoints Are Located in or near Transposable Elements
±1000 bp flanking the 10 unique breakpoints involved in IGHM deletions were analyzed for the presence of transposon-derived repetitive elements. No Vh, Dh, Jh gene segments or constant gene exons were present in these regions.
Similar to IGHM, Gross Deletion Breakpoints in BTK and Artemis Frequently Occur in TEs
In addition to IGH, gross deletions disrupting BTK or Artemis have been found in primary immunodeficiencies (PID). Although only 5 of 37 identified breakpoints have been analyzed at sequence level, similar to IGHM gross deletions, most of the BTK breakpoints were located in TEs.35,36
In addition to the published sequences, we identified three additional gross deletions in BTK-deficient patients. The eight deletions are spread throughout the gene and range in size from 2.6 to 38.2 kb deleting one or more exons (Figure 3A). The results of the breakpoint sequence analyses of our three patients were in line with previous observations (Figures 3B and 3C). In six of the eight patients, both the 5′ and 3′ breakpoints were located in Alu elements (SINE) and showed high degrees of sequence homology (Figure 3B).35,36 Interestingly, two Alu motifs were each involved in two independent recombination events; the 3′ breakpoints of patients ID434 and P4 and the 3′ breakpoint of patient 0850 and the 5′ breakpoint of patient P4 were located in the same Alu element. In both cases, the breaks occurred at distinct sites within the Alu motifs, although the 3′ breakpoint of patient P4 is located closely to the 3′ breakpoint of patient ID434 (Figure 3B).
Figure 3.

Gross Deletions in BTK Are Mainly Associated with Alu-Alu Mispairing and Crossing Over
(A) Mapping of the BTK gross deletions in three unrelated patients by a PCR approach. The gross deletions in patients 0703, 0850, 2430, 2433,35 and P436 have been described before.
(B) Sequences of the three newly identified breakpoint junctions compared to control sequences. The breakpoint junction of patient ID440 shows a TT insertion, which could be palindromic nucleotides. Microhomology regions at the junctions of patients ID113 and ID434 are boxed. Complexity analysis of ±25 bp flanking the breakpoints did not result in a likely mediator for patient ID440, whereas the breakpoint in patients ID113, 0850, ID434, P4, 2430, and 2433 appear to be mediated by homologous recombination and in patient 0703 by a knot structure (inversion of inverted repeats). The 3′ breakpoint of patient P4 was located close to the 3′ breakpoint of patient ID434, and the six nucleotides of microhomology are indicated by bold font in the 3′ breakpoint region of ID434.
(C) ±1000 bp flanking the 16 unique BTK gross deletion breakpoints were analyzed for transposon-derived repetitive elements. In addition, BTK exons are indicated. The 3′ breakpoints of patients ID434 and P4 were located in the same Alu element, and the 3′ breakpoint of patient 0850 and the 5′ breakpoint of patient P4 were located in the same Alu element.
The gross deletion breakpoints in patients ID440 and 0703 were not located in Alu elements and showed no sequence homology within 50 bp. Two nucleotides were inserted in the junction of patient ID440, whereas patient 0703 had one nucleotide of microhomology at the junction.35 In patient ID440, the breakpoints were located closely to SINE elements: the 5′ breakpoint downstream of a MIRb and the 3′ breakpoint downstream of an Alu motif. The 5′ breakpoint of patient 0703 was located near a MIRb (SINE2) sequence, whereas the 3′ breakpoint was located in a LINE1 element and near an Alu motif (Figure 3C).
In general, the gross deletions in BTK were much smaller than those characterized in the IGH locus. This was especially noted for the Alu-Alu recombination events. Detailed TE annotation of breakpoint flanking regions (Figure 3C) showed that all eight gross deletion breakpoints in BTK were located in or near TEs.
In contrast to BTK, Artemis gross deletion breakpoints have not been sequenced to date. We identified two unique breakpoint regions in four Artemis-deficient patients (Figure 4A). Although the breakpoint regions appeared similar, four unique breakpoint junctions were identified in patients ID020, ID024, and ID124, all resulting in the loss of 82 kb including exons 1–3 (Figure 4B). All eight 5′ and 3′ breakpoint regions contained a high density of TEs (Figure 4C), and the breakpoints were located either in (ID020 and ID024) or near (ID124, both alleles) a TE. Strikingly, the regions that contained the 5′ breakpoints were highly homologous to the regions that contained the 3′ breakpoints. Sequence alignment between the Artemis gene and −80 to −55 kb upstream region revealed a highly homologous region of ∼12 kb (Figure 4D). Thus on top of TE involvement, these breakpoints show high degrees of sequence homology.
Figure 4.
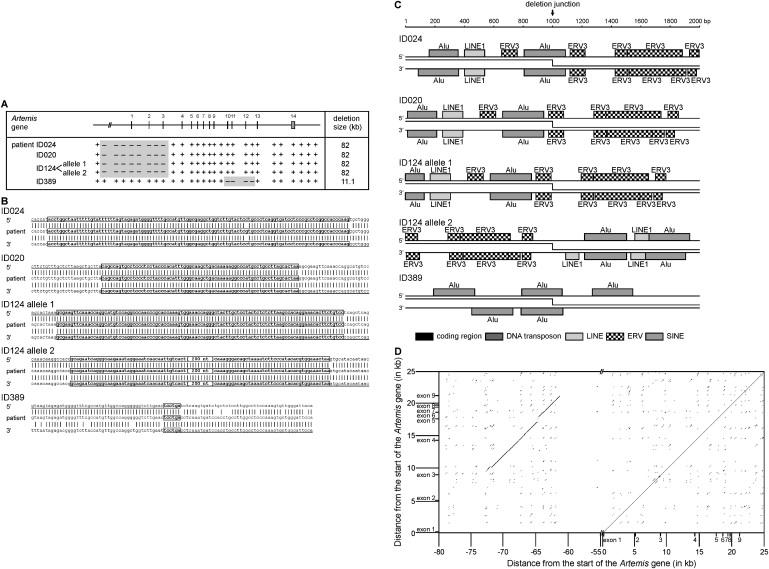
Gross Deletions in the Artemis Gene Suggest Alu-Alu Mispairing and Crossing Over
(A) Mapping of the homozygous Artemis gross deletions in four unrelated patients with a PCR approach.
(B) The 5′ and 3′ unique breakpoint regions in patients ID020, ID024, and ID124 are located within 1 kb in two highly homologous 12 kb regions. The mismatch in the breakpoint region of patient ID024 with both the 5′ and 3′ control sequences is not a previously identified SNP. Complexity analysis of local sequences indicates that the deletions in these patients and in patient ID389 are potentially mediated by homologous recombination.
(C) The breakpoint regions identified in patients ID020, ID024, and ID124 show a high density of TEs. The deletion breakpoints of patients ID024 and ID389 are located in Alu elements, and of patient ID020 in an LTR retrotransposon.
(D) A DNA dot plot generated from the alignment between exon 1 to intron 9 of the Artemis gene with a region 82 kb 5′ of Artemis shows a highly homologous region of ∼12 kb. The 5′ and 3′ breakpoints of patients ID020, ID024, and ID124 are located in the homologous regions.
Patient ID389 had an 11.1 kb genomic deletion including Artemis exons 10, 11, and 12. The 5′ and 3′ breakpoints were located in Alu sequences and the joint showed high degrees of sequence homology (Figures 4B and 4D).
High Total TE Content Associated with Increased Incidence of Gene-Disrupting Gross Deletions
Although the incidence of gross deletions in IGHM, BTK, and Artemis differs greatly (58%, 6%, and 56% of disease-causing mutations, respectively), the majority of gross deletions in all three genes show involvement of TEs. To address this difference, we determined the total TE content in these genes and compared this TE content to that of the total human genome (Figure 5A). We found that the TE content of IGH (41%) and Artemis (45%) was higher than the average of the human genome (37%). In contrast, only 29% of the BTK gene consists of TE-derived repeats. Similar analysis was performed for BLNK (MIM 604515), which is also a PID gene of >50 kb in size. Only four disease-causing mutations in BLNK have been identified, which were all microlesions (unpublished observations from M.v.d.B. and M.E.C.).37 Similar to BTK, a reduced fraction of the BLNK gene consisted of TEs (30%; Figure 5A). These observations suggest that a high total TE content of a gene is associated with an increased gross deletion frequency found in gene disruptions underlying human disease.
Figure 5.
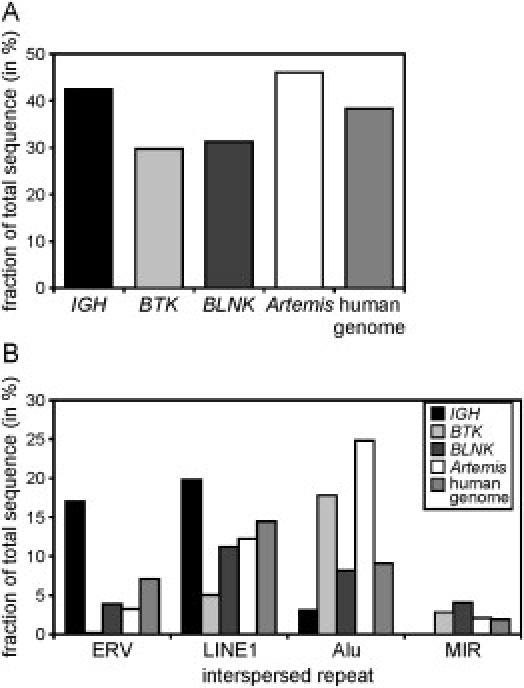
Highly Variable TE Content in Four Genes
(A) The IGH and Artemis loci contain a high TE content compared to the average of the human genome, whereas the BTK and BLNK genes have decreased TE content.
(B) The LINE, ERV, Alu, and MIR elements are the most common TEs found in the human genome. IGH consists of increased ERV and LINE repeats, whereas BTK and Artemis mainly contain Alu elements. BLNK does not display increased presence of any of the four types of elements as compared to the average of the human genome.
Of the four types of TEs that are most frequently found in the human genome (LINE1, ERV, Alu, MIR), the ERV and LINE1 content is over-represented in IGH compared to the average in the human genome (Figure 5B). In contrast, BTK and Artemis have very few of these TE elements, whereas twice as much of their genomic sequence is derived from Alu motifs as compared to the average in the human genome. None of the four dominant types of TEs was overrepresented in BLNK. Whereas the total TE content seemed to correlate to the gross deletion frequency, gross deletions involved mainly TEs belonging to a class that is overrepresented in a gene, irrespective of the total TE content. The involvement of ERV elements in 4 out of 10 IGH gross deletion breakpoints and the involvement of Alu elements in 12 out of 16 BTK breakpoints is relatively high when considering that they constitute only 16% and 18% of the IGH and BTK gene sequences, respectively. However, ERV elements constitute 39% of TEs in IGH and Alu elements constitute 62% of TEs in BTK. Therefore, if one assumes that gross deletions mainly occur in TEs, the involvement of relatively overrepresented TE subclasses appears to be as expected by chance.
TEs and Gross Deletions in Human Disease Genes
We identified a trend between the TE content and the gross deletion frequency of four genes implicated in primary immunodeficiencies. This is a small set of genes and with the exception of BTK, only a limited number of disease-causing mutations have been identified in these genes: 12 IGHM mutations, 13 Artemis mutations, and 4 BLNK mutations (Table 1). Therefore, we decided to test our model in a larger set of more extensively studied human disease genes. A literature search yielded a set of 16 additional genes for which >200 unique mutations were recorded in the HGMD and that spanned >50 kb of genomic DNA. The gross deletion frequency of these genes in human disease was extracted from the HGMD,1 and the TE content was determined with CENSOR.32 Similar to what was observed for the initial four genes, the genes with a higher TE content tended to show an increased frequency of gross deletions (Table 1). A strong increase in the gross deletion frequency was noted in genes that showed >40% TE content (Table 1). The median gross deletion frequency was 6% for genes with ≤40% repetitive elements, compared with 14% for genes with >40% repetitive elements (Figure 6A). This difference was found to be highly significant between both groups of genes (p = 0.01).
Table 1.
Correlation of Gross Deletions Incidence and TE Content
| Gene | Gene Size (kb) Incl. ±10 kb Sequence | Gross Deletion Frequencya (# Gross Deletions/# Total Mutations) | Motif Fractionb (% Total Sequence) | ERVc (% Total Sequence) | LINE1c (% Total Sequence) | Aluc (% Total Sequence) | MIRc (% Total Sequence) | Breakpoint Mediators | References |
|---|---|---|---|---|---|---|---|---|---|
| IGHMd (MIM 147020) | 1280 | 58% (7/12) | 41% | 16.1 | 19.1 | 2.8 | 0.2 | ERV, LINE1 | |
| BTK (MIM 300300) | 57 | 7% (37/523) | 29% | 0.1 | 4.6 | 18.0 | 2.9 | Alu, MIR | 35,36 |
| BLNKe (MIM 604515) | 100 | 0% (0/4) | 30% | 3.8 | 10.9 | 7.9 | 4.2 | – | |
| Artemisd (MIM 605988) | 66 | 56% (9/16) | 45% | 3.1 | 11.3 | 23.3 | 4.0 | Alu | |
| CFTR (MIM 602421) | 209 | 3% (35/1208) | 27% | 5.2 | 9.8 | 4.1 | 3.0 | ? | |
| DMD (MIM 300377) | 2112 | 35% (255/735) | 29% | 5.3 | 11.8 | 5.3 | 1.9 | Alu, LINE1 | 5 |
| MECP2 (MIM 300005) | 96 | 34% (108/319) | 31% | 1.7 | 8.8 | 19.4 | 1.0 | Alu | 43,44 |
| APC (MIM 175100) | 128 | 5% (34/696) | 32% | 3.5 | 7.9 | 13.3 | 2.2 | Alu | 45,46 |
| VWF (MIM 193400) | 196 | 6% (12/206) | 33% | 3.3 | 12.1 | 10.9 | 2.7 | Alu | 47 |
| MLH1 (MIM 120436) | 77 | 11% (46/415) | 36% | 2.4 | 8.2 | 16.5 | 2.3 | Alu, LINE1 | 48 |
| NF1 (MIM 162200) | 299 | 6% (45/718) | 37% | 4.6 | 13.3 | 13.8 | 1.5 | Alu | 49 |
| BRCA2 (MIM 600185) | 104 | 1% (7/504) | 39% | 4.2 | 8.3 | 13.8 | 2.0 | Alu | 50 |
| BRCA1 (MIM 113705) | 101 | 6% (45/750) | 40% | 2.1 | 1.7 | 31.5 | 2.9 | Alu | 51 |
| ATM (MIM 607585) | 166 | 8% (35/421) | 40% | 1.3 | 20.3 | 11.7 | 1.3 | Alu, LINE1 | 52 |
| FANCA (MIM 607139) | 99 | 28% (66/234) | 42% | 4.3 | 3.7 | 30.4 | 2.6 | Alu | 53,54 |
| HPRT1 (MIM 308000) | 60 | 12% (28/232) | 43% | 3.8 | 6.4 | 21.9 | 4.2 | Alu, LINE1 | 55 |
| MSH2 (MIM 609309) | 100 | 16% (56/346) | 45% | 4.1 | 5.9 | 26.2 | 5.7 | Alu | 48,56 |
| RB1 (MIM 180200) | 198 | 8% (35/418) | 47% | 4.1 | 26.4 | 6.9 | 1.7 | ? | |
| LDLR (MIM 606945) | 64 | 11% (94/866) | 51% | 2.0 | 1.9 | 41.0 | 4.5 | Alu | 57–61 |
| F8 (MIM 306700) | 207 | 11% (95/885) | 58% | 10.7 | 32.3 | 7.0 | 1.0 | Alu, LINE1 | 62–66 |
| Total | 3.2 × 106f | 6% (2962/53200) | 37%f | 6.8f | 14.6f | 9.3f | 1.8f | – | |
Bold text indicates values clearly higher than the total values in the bottom row.
HGMD February 2007.
TE content was determined with CENSOR.
The four most abundant TE subclasses; ERVs are LTR retrotransposons, LINE1s are the most common LINEs, and Alu and MIR are the most common SINEs.
Statistics include alleles from patients that are described here.
Statistics include alleles from unpublished patients (M.v.d.B., M.E.C.).
Data obtained on the total human genome sequence.
Figure 6.
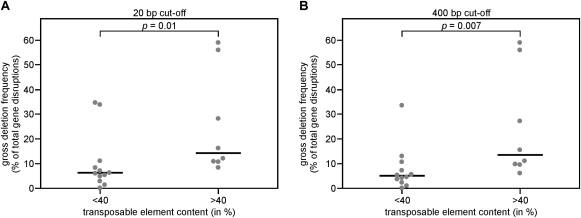
Genes Disrupted in Human Inherited Diseases with a Transposon-Derived Repetitive Element Content of >40% Are Significantly More Frequently Disrupted by Gross Deletions than Genes with ≤40% TE Content
The frequency of gross deletions was determined from 20 genes disrupted in human inherited disease. A surprising increase in gross deletion frequency was found in genes with >40% TE content.
(A) The gross deletion frequency of 20 genes is shown for genes with ≤40% and >40% TE content. Every dot represents an individual gene. The gross deletion frequency was significantly increased in genes with >40% TE content as determined with a nonparametric test.
(B) The significance increases slightly when the cut-off set for gross deletions is increased from >20 bp to >400 bp.
Gross deletions are currently defined as >20 bp, which is a crude cut-off value. TEs in the human genome range in size from about 300 bp to several kb. This would imply that only gross deletions spanning more than a few hundred bp can show involvement of two independent TEs in a gross deletion. To test whether this holds true, additional analyses were performed with cut-off values for gross deletions of 200 and 400 bp. These analyses mainly showed a remarkable reduction in gross deletion frequency of MECP2 (300005) from 34% to 13% with a cut-off of 400 bp. No difference was found for the median values and the significance for the 200 bp cut-off, whereas the medians were 5% and 13% for genes with ≤40% and >40% sequence derived from TEs, respectively, with p = 0.007 (Figure 6B). In conclusion, genes with a TE content of >40% have a significantly increased gross deletion frequency in mutations underlying human disease. The significance increased slightly when the cut-off for gross deletions is increased to 400 bp.
In addition to the total TE content, the content of the four most abundant TE subclasses (ERV, LINE1, Alu, and MIR) was determined for the 20 genes to study whether the overrepresented elements were specifically involved in gross deletions (Table 1). Although the breakpoint fusion sequence of only a small fraction of the reported gross deletions has been determined, mainly the overrepresented TE subclasses appeared to be involved in gross deletion breakpoints (Table 1).
Discussion
We characterized the gross deletion breakpoints in IGHM-, BTK-, and Artemis-deficient patients to explain the high frequency of gross deletions in IGHM and Artemis compared to BTK. IGHM gross deletion breakpoints were not located in or near RSS or Ig switch regions, but similar to most gross deletions involving BTK or Artemis, they were associated with TE-derived repeats. We analyzed potential TE involvement in gross deletion frequencies on a total set of 20 genes disrupted in human disease. We found that genes with a high TE content (>40%) showed an increased frequency of gross deletions when disrupted in human disease. We conclude that, similar to suggestions based on limited sequence data from other genes disrupted in human disease, TEs are likely mediators of gross deletions in IGHM, BTK, and Artemis. Consequently, we propose a role for TEs in the high frequency of gross deletions involving IGHM, whereas the high gross deletion frequency of Artemis gene disruptions is potentially mediated by the high TE content and facilitated by mispairing of the ∼12 kb region that encompasses exons 3–9 with a region >60 kb upstream of the Artemis gene.
In this study, five unique breakpoint regions were characterized in seven unrelated IGHM-deficient patients. The breakpoints were scattered throughout the locus in introns within the Vh, Dh, and constant regions, thereby indicating that there was no preferential targeting of a specific region. Interestingly, no colocalization with RSS or Ig switch regions was found. Whereas such sequences are the potent recombination sequences in specific stages during B cell development, they do not appear to be prone to recombination in germline cells. It is therefore highly unlikely that the B cell-specific recombination characteristics of the IGH locus underlie the high frequency of gross deletions involving IGHM.
The five IGHM deletions did not show high sequence homology between the 5′ and the 3′ breakpoint regions. Therefore, it is unlikely that the gross deletions were the result of mispairing of homologous sequences and unequal crossing-over as has been observed in recurrent Ig subclass deletions.27 The five breakpoint junctions did show the characteristics of NHEJ, with microhomology in four joints and an insertion of six nucleotides in the fifth. No common sequence motifs were found in the breakpoint regions, whereas complexity analysis revealed that short repetitive sequences are likely mediators of the unequal repair.
The majority of IGHM-deleting gross deletion breakpoints were located in or near TE-derived repeats. Interestingly, the involved elements (LINEs and LTR retrotransposons) were overrepresented in the IGH locus as compared to the average in the human genome. On the other hand, the gross deletions identified in BTK- and Artemis-deficient patients mainly involved SINEs. In contrast to IGH, these SINEs are specifically overrepresented in BTK and Artemis. Furthermore, the high frequency of gross deletions in IGH and Artemis as compared to BTK and BLNK was associated with a high total TE content. These observations suggest a role for TEs in mediating gross deletions. Involvement of TEs has been shown before.5,6 However, by analysis of 20 genes disrupted in human disease, we are the first (to our knowledge) to show that the total TE content is associated with a significantly increased frequency of gross deletions as cause of gene disruption.
The gene disruptions included in this study were originally identified because they resulted in a genetic disorder. We used the relative number of disruptions caused by gross deletions, thereby assuming that the chance that a mutation or a deletion results in disease is similar for every gene. Furthermore, the detection of gross lesions is more difficult than microlesions. Because in our study we included multiple relatively large genes that have been extensively studied with respect a genetic disorder, we assumed that this evened out in the statistical analysis.
The role of TEs in mediating gross deletions is difficult to examine. In general, the breakpoints analyzed in this study involved at least two double-stranded DNA breaks in distant genomic locations (2.6–732 kb) that were placed in physical proximity and incorrectly repaired. Complexity analysis showed that small repeat fragments are likely mediators of the incorrect repair. This mechanism is quite similar to what is seen in microlesions, which are thought to result from slipped mispairing of single-stranded DNA in the replication fork. Because the stretches of single-stranded DNA in the replication fork do not extend beyond 1000–2000 bp, additional factors are required to mediate colocalization of two distant genomic regions and double-stranded DNA break induction.
As recently discussed by Hedges and Deininger, the disruptive potential of TEs might result from two mechanisms.6 First, the sequence homology between the involved elements facilitates incorrect homologous repair. Second, because the endonuclease products of TEs are designed to preferentially target TEs, they might contribute to genomic instability at these sites. The first mechanism potentially contributes to the gross deletions in BTK and Artemis that showed high levels of homology between the 5′ and 3′ breakpoint regions. The endonuclease product of LINE1, on the other hand, might contribute to gross deletions in IGHM.
The accumulation of transposon-derived repetitive elements in heterochromatin might be a third mechanism that contributes to gross deletion formation (reviewed by Dimitri et al.38). This localization of TEs could result from preferential insertion of these elements in DNA that is packed in heterochromatin.39 However, it is well possible that it is a defense mechanism of the host cell to repress expression of TE products by actively packing these elements in heterochromatin.40,41 Irrespective of this, heterochromatic regions are compact clusters of genomic DNA. It seems therefore likely that genomic regions with high TE content are tightly packed in heterochromatin, and this is a mechanism by which TEs mediate colocalization of distant genomic regions.
Microinsertions and microdeletions are assumed to result from slipped mispairing, but this mechanism has been proposed to underlie some of the identified deletions that were >20 bp.42 This is not remarkable because it is suggested to result from mispairing of ssDNA in the replication fork and the length of ssDNA is 1000–2000 bp. However, the size of TEs ranges from few hundred to a few thousand bp. Therefore, the involvement of TEs in the generation of deletions smaller than a few hundred bp is unlikely. We suggest defining gross deletions to be larger than 400 or even 1000 bp because this better reflects the mechanism underlying these lesions. Ultimately, additional sequence data and analysis of deletion breakpoints should allow for a more accurate discrimination of micro- and gross deletions based on the underlying mechanism.
Not all deletions in Artemis were solely associated with TEs. A recurrent deletion of exons 1–3 was associated with ∼12 kb homology between the 5′ and 3′ breakpoint regions. Thus, similar to the Vh deletion polymorphism in patient BN and previously reported Ch deletions,27 the recurrent Artemis exon 1–3 deletion likely results from mispairing between highly homologous sequences.
In this study, we identified and characterized IGHM, BTK, and Artemis deletions in patients with primary immunodeficiency. Whereas the human IGH locus contains multiple functionally proven recombination sequences, these were not observed near IGHM gross deletion breakpoints. Consequently, we conclude that RSS and Ig switch regions are unlikely to be mediators of germline deletions. Analysis of the IGHM, BTK, and Artemis deletion junctions did show involvement of TEs and formed the basis of our hypothesis that a high total TE content is correlated to an increased frequency of gene disruption by gross deletions, which was confirmed by analysis of 20 genes disrupted in human disease. Although more sequence data are needed to draw firm conclusions on the underlying mechanisms, this study shows a novel statistical correlation between TE density and the frequency of gene disruption by gross deletions in human disease.
Web Resources
The URLs for data presented herein are as follows:
Genewindow, http://genewindow.nci.nih.gov:8080/home.jsp
Human Gene Mutation Database (HGMD), http://www.hgmd.cf.ac.uk/ac/index.php
Molecular Toolkit (nucleic acid dot plot tool), http://www.vivo.colostate.edu/molkit/dnadot/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
The authors acknowledge W.M. Comans-Bitter for assistance with preparing the figures. This work was supported by grant 349 from the foundation “Sophia Kinderziekenhuis Fonds” (SKF) to M.C.v.Z., R. de Groot, and J.J.M.v.D. and Veni grant 916.56.107 from ZonMw to M.v.d.B. The authors state that they have no competing financial interests.
References
- 1.Stenson P.D., Ball E.V., Mort M., Phillips A.D., Shiel J.A., Thomas N.S., Abeysinghe S., Krawczak M., Cooper D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 2.Abeysinghe S.S., Chuzhanova N., Krawczak M., Ball E.V., Cooper D.N. Translocation and gross deletion breakpoints in human inherited disease and cancer I: nucleotide composition and recombination-associated motifs. Hum. Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- 3.Chuzhanova N., Abeysinghe S.S., Krawczak M., Cooper D.N. Translocation and gross deletion breakpoints in human inherited disease and cancer II: potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum. Mutat. 2003;22:245–251. doi: 10.1002/humu.10253. [DOI] [PubMed] [Google Scholar]
- 4.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 5.McNaughton J.C., Cockburn D.J., Hughes G., Jones W.A., Laing N.G., Ray P.N., Stockwell P.A., Petersen G.B. Is gene deletion in eukaryotes sequence-dependent? A study of nine deletion junctions and nineteen other deletion breakpoints in intron 7 of the human dystrophin gene. Gene. 1998;222:41–51. doi: 10.1016/s0378-1119(98)00466-1. [DOI] [PubMed] [Google Scholar]
- 6.Hedges D.J., Deininger P.L. Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 2007;616:46–59. doi: 10.1016/j.mrfmmm.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Notarangelo L., Casanova J.L., Conley M.E., Chapel H., Fischer A., Puck J., Roifman C., Seger R., Geha R.S. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee Meeting in Budapest, 2005. J. Allergy Clin. Immunol. 2006;117:883–896. doi: 10.1016/j.jaci.2005.12.1347. [DOI] [PubMed] [Google Scholar]
- 8.Moshous D., Callebaut I., de Chasseval R., Corneo B., Cavazzana-Calvo M., Le Deist F., Tezcan I., Sanal O., Bertrand Y., Philippe N. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 9.Noordzij J.G., Verkaik N.S., van der Burg M., van Veelen L.R., de Bruin-Versteeg S., Wiegant W., Vossen J.M., Weemaes C.M., de Groot R., Zdzienicka M.Z. Radiosensitive SCID patients with Artemis gene mutations show a complete B-cell differentiation arrest at the pre-B-cell receptor checkpoint in bone marrow. Blood. 2003;101:1446–1452. doi: 10.1182/blood-2002-01-0187. [DOI] [PubMed] [Google Scholar]
- 10.Conley M.E., Mathias D., Treadaway J., Minegishi Y., Rohrer J. Mutations in btk in patients with presumed X-linked agammaglobulinemia. Am. J. Hum. Genet. 1998;62:1034–1043. doi: 10.1086/301828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yel L., Minegishi Y., Coustan-Smith E., Buckley R.H., Trubel H., Pachman L.M., Kitchingman G.R., Campana D., Rohrer J., Conley M.E. Mutations in the mu heavy-chain gene in patients with agammaglobulinemia. N. Engl. J. Med. 1996;335:1486–1493. doi: 10.1056/NEJM199611143352003. [DOI] [PubMed] [Google Scholar]
- 12.Milili M., Antunes H., Blanco-Betancourt C., Nogueiras A., Santos E., Vasconcelos J., Castro e Melo J., Schiff C. A new case of autosomal recessive agammaglobulinaemia with impaired pre-B cell differentiation due to a large deletion of the IGH locus. Eur. J. Pediatr. 2002;161:479–484. doi: 10.1007/s00431-002-0994-9. [DOI] [PubMed] [Google Scholar]
- 13.Lopez Granados E., Porpiglia A.S., Hogan M.B., Matamoros N., Krasovec S., Pignata C., Smith C.I., Hammarstrom L., Bjorkander J., Belohradsky B.H. Clinical and molecular analysis of patients with defects in micro heavy chain gene. J. Clin. Invest. 2002;110:1029–1035. doi: 10.1172/JCI15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schatz D.G. V(D)J recombination. Immunol. Rev. 2004;200:5–11. doi: 10.1111/j.0105-2896.2004.00173.x. [DOI] [PubMed] [Google Scholar]
- 15.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 16.Odegard V.H., Schatz D.G. Targeting of somatic hypermutation. Nat. Rev. Immunol. 2006;6:573–583. doi: 10.1038/nri1896. [DOI] [PubMed] [Google Scholar]
- 17.Lefranc M.P. Nomenclature of the human immunoglobulin heavy (IGH) genes. Exp. Clin. Immunogenet. 2001;18:100–116. doi: 10.1159/000049189. [DOI] [PubMed] [Google Scholar]
- 18.Bech-Hansen N.T., Cox D.W. Duplication of the human immunoglobulin heavy chain gamma 2 gene. Am. J. Hum. Genet. 1986;38:67–74. [PMC free article] [PubMed] [Google Scholar]
- 19.Bottaro A., Cariota U., DeMarchi M., Carbonara A.O. Pulsed-field electrophoresis screening for immunoglobulin heavy-chain constant-region (IGHC) multigene deletions and duplications. Am. J. Hum. Genet. 1991;48:745–756. [PMC free article] [PubMed] [Google Scholar]
- 20.Brusco A., Cariota U., Bottaro A., Boccazzi C., Plebani A., Ugazio A.G., Galanello R., van Leeuwen A.M., DeLange G.G., Depelchin S. Structural and immunologic analysis of gene triplications in the Ig heavy chain constant region locus. J. Immunol. 1994;152:129–135. [PubMed] [Google Scholar]
- 21.Brusco A., Cariota U., Bottaro A., Boccazzi C., Plebani A., Ugazio A.G., Galanello R., Guerra M.G., Carbonara A.O. Variability of the immunoglobulin heavy chain constant region locus: a population study. Hum. Genet. 1995;95:319–326. doi: 10.1007/BF00225201. [DOI] [PubMed] [Google Scholar]
- 22.Chimge N.O., Pramanik S., Hu G., Lin Y., Gao R., Shen L., Li H. Determination of gene organization in the human IGHV region on single chromosomes. Genes Immun. 2005;6:186–193. doi: 10.1038/sj.gene.6364176. [DOI] [PubMed] [Google Scholar]
- 23.Plebani A., Carbonara A.O., Bottaro A., Gallina R., Boccazzi C., Crispino P., Ruggeri L., Salvioni F., Duina M., Negrini A. Gene deletion as a cause of associated deficiency of IgA1, IgG2, IgG4 and IgE. Immunodeficiency. 1993;4:245–248. [PubMed] [Google Scholar]
- 24.Terada T., Kaneko H., Li A.L., Kasahara K., Ibe M., Yokota S., Kondo N. Analysis of Ig subclass deficiency: first reported case of IgG2, IgG4, and IgA deficiency caused by deletion of C alpha 1, psi C gamma, C gamma 2, C gamma 4, and C epsilon in a Mongoloid patient. J. Allergy Clin. Immunol. 2001;108:602–606. doi: 10.1067/mai.2001.118293. [DOI] [PubMed] [Google Scholar]
- 25.Pan Q., Hammarstrom L. Molecular basis of IgG subclass deficiency. Immunol. Rev. 2000;178:99–110. doi: 10.1034/j.1600-065x.2000.17815.x. [DOI] [PubMed] [Google Scholar]
- 26.Rabbani H., Pan Q., Kondo N., Smith C.I., Hammarstrom L. Duplications and deletions of the human IGHC locus: evolutionary implications. Immunogenetics. 1996;45:136–141. doi: 10.1007/s002510050181. [DOI] [PubMed] [Google Scholar]
- 27.Brusco A., Saviozzi S., Cinque F., Bottaro A., DeMarchi M. A recurrent breakpoint in the most common deletion of the Ig heavy chain locus (del A1-GP-G2–G4-E) J. Immunol. 1999;163:4392–4398. [PubMed] [Google Scholar]
- 28.Van Dongen J.J., Langerak A.W., Bruggemann M., Evans P.A., Hummel M., Lavender F.L., Delabesse E., Davi F., Schuuring E., Garcia-Sanz R. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 29.Noordzij J.G., de Bruin-Versteeg S., Comans-Bitter W.M., Hartwig N.G., Hendriks R.W., de Groot R., van Dongen J.J. Composition of precursor B-cell compartment in bone marrow from patients with X-linked agammaglobulinemia compared with healthy children. Pediatr. Res. 2002;51:159–168. doi: 10.1203/00006450-200202000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Przybylski G.K., Dik W.A., Wanzeck J., Grabarczyk P., Majunke S., Martin-Subero J.I., Siebert R., Dolken G., Ludwig W.D., Verhaaf B. Disruption of the BCL11B gene through inv(14)(q11.2q32.31) results in the expression of BCL11B-TRDC fusion transcripts and is associated with the absence of wild-type BCL11B transcripts in T-ALL. Leukemia. 2005;19:201–208. doi: 10.1038/sj.leu.2403619. [DOI] [PubMed] [Google Scholar]
- 31.Hubbard T.J., Aken B.L., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohany O., Gentles A.J., Hankus L., Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staats B., Qi L., Beerman M., Sicotte H., Burdett L.A., Packer B., Chanock S.J., Yeager M. Genewindow: an interactive tool for visualization of genomic variation. Nat. Genet. 2005;37:109–110. doi: 10.1038/ng0205-109. [DOI] [PubMed] [Google Scholar]
- 34.Gusev V.D., Nemytikova L.A., Chuzhanova N.A. On the complexity measures of genetic sequences. Bioinformatics. 1999;15:994–999. doi: 10.1093/bioinformatics/15.12.994. [DOI] [PubMed] [Google Scholar]
- 35.Rohrer J., Minegishi Y., Richter D., Eguiguren J., Conley M.E. Unusual mutations in Btk: an insertion, a duplication, an inversion, and four large deletions. Clin. Immunol. 1999;90:28–37. doi: 10.1006/clim.1998.4629. [DOI] [PubMed] [Google Scholar]
- 36.Jo E.K., Wang Y., Kanegane H., Futatani T., Song C.H., Park J.K., Kim J.S., Kim D.S., Ahn K.M., Lee S.I. Identification of mutations in the Bruton's tyrosine kinase gene, including a novel genomic rearrangements resulting in large deletion, in Korean X-linked agammaglobulinemia patients. J. Hum. Genet. 2003;48:322–326. doi: 10.1007/s10038-003-0032-4. [DOI] [PubMed] [Google Scholar]
- 37.Minegishi Y., Rohrer J., Coustan-Smith E., Lederman H.M., Pappu R., Campana D., Chan A.C., Conley M.E. An essential role for BLNK in human B cell development. Science. 1999;286:1954–1957. doi: 10.1126/science.286.5446.1954. [DOI] [PubMed] [Google Scholar]
- 38.Dimitri P., Corradini N., Rossi F., Mei E., Zhimulev I.F., Verni F. Transposable elements as artisans of the heterochromatic genome in Drosophila melanogaster. Cytogenet. Genome Res. 2005;110:165–172. doi: 10.1159/000084949. [DOI] [PubMed] [Google Scholar]
- 39.Dimitri P., Junakovic N. Revising the selfish DNA hypothesis: new evidence on accumulation of transposable elements in heterochromatin. Trends Genet. 1999;15:123–124. doi: 10.1016/s0168-9525(99)01711-4. [DOI] [PubMed] [Google Scholar]
- 40.Lippman Z., Gendrel A.V., Black M., Vaughn M.W., Dedhia N., McCombie W.R., Lavine K., Mittal V., May B., Kasschau K.D. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430:471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 41.Slotkin R.K., Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 42.Chen J.M., Chuzhanova N., Stenson P.D., Ferec C., Cooper D.N. Meta-analysis of gross insertions causing human genetic disease: novel mutational mechanisms and the role of replication slippage. Hum. Mutat. 2005;25:207–221. doi: 10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- 43.Laccone F., Junemann I., Whatley S., Morgan R., Butler R., Huppke P., Ravine D. Large deletions of the MECP2 gene detected by gene dosage analysis in patients with Rett syndrome. Hum. Mutat. 2004;23:234–244. doi: 10.1002/humu.20004. [DOI] [PubMed] [Google Scholar]
- 44.Schollen E., Smeets E., Deflem E., Fryns J.P., Matthijs G. Gross rearrangements in the MECP2 gene in three patients with Rett syndrome: implications for routine diagnosis of Rett syndrome. Hum. Mutat. 2003;22:116–120. doi: 10.1002/humu.10242. [DOI] [PubMed] [Google Scholar]
- 45.Su L.K., Steinbach G., Sawyer J.C., Hindi M., Ward P.A., Lynch P.M. Genomic rearrangements of the APC tumor-suppressor gene in familial adenomatous polyposis. Hum. Genet. 2000;106:101–107. doi: 10.1007/s004399900195. [DOI] [PubMed] [Google Scholar]
- 46.Cao X., Eu K.W., Seow-Choen F., Zhao Y., Cheah P.Y. Topoisomerase-I- and Alu-mediated genomic deletions of the APC gene in familial adenomatous polyposis. Hum. Genet. 2001;108:436–442. doi: 10.1007/s004390100492. [DOI] [PubMed] [Google Scholar]
- 47.Xie F., Wang X., Cooper D.N., Chuzhanova N., Fang Y., Cai X., Wang Z., Wang H. A novel Alu-mediated 61-kb deletion of the von Willebrand factor (VWF) gene whose breakpoints co-locate with putative matrix attachment regions. Blood Cells Mol. Dis. 2006;36:385–391. doi: 10.1016/j.bcmd.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 48.van der Klift H., Wijnen J., Wagner A., Verkuilen P., Tops C., Otway R., Kohonen-Corish M., Vasen H., Oliani C., Barana D. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC) Genes Chromosomes Cancer. 2005;44:123–138. doi: 10.1002/gcc.20219. [DOI] [PubMed] [Google Scholar]
- 49.Gervasini C., Venturin M., Orzan F., Friso A., Clementi M., Tenconi R., Larizza L., Riva P. Uncommon Alu-mediated NF1 microdeletion with a breakpoint inside the NF1 gene. Genomics. 2005;85:273–279. doi: 10.1016/j.ygeno.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 50.Casilli F., Tournier I., Sinilnikova O.M., Coulet F., Soubrier F., Houdayer C., Hardouin A., Berthet P., Sobol H., Bourdon V. The contribution of germline rearrangements to the spectrum of BRCA2 mutations. J. Med. Genet. 2006;43:e49. doi: 10.1136/jmg.2005.040212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armaou S., Konstantopoulou I., Anagnostopoulos T., Razis E., Boukovinas I., Xenidis N., Fountzilas G., Yannoukakos D. Novel genomic rearrangements in the BRCA1 gene detected in greek breast/ovarian cancer patients. Eur. J. Cancer. 2007;43:443–453. doi: 10.1016/j.ejca.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 52.Cavalieri S., Funaro A., Porcedda P., Turinetto V., Migone N., Gatti R.A., Brusco A. ATM mutations in Italian families with ataxia telangiectasia include two distinct large genomic deletions. Hum. Mutat. 2006;27:1061. doi: 10.1002/humu.9454. [DOI] [PubMed] [Google Scholar]
- 53.Levran O., Doggett N.A., Auerbach A.D. Identification of Alu-mediated deletions in the Fanconi anemia gene FAA. Hum. Mutat. 1998;12:145–152. doi: 10.1002/(SICI)1098-1004(1998)12:3<145::AID-HUMU2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 54.Centra M., Memeo E., d'Apolito M., Savino M., Ianzano L., Notarangelo A., Liu J., Doggett N.A., Zelante L., Savoia A. Fine exon-intron structure of the Fanconi anemia group A (FAA) gene and characterization of two genomic deletions. Genomics. 1998;51:463–467. doi: 10.1006/geno.1998.5353. [DOI] [PubMed] [Google Scholar]
- 55.Williams M., Rainville I.R., Nicklas J.A. Use of inverse PCR to amplify and sequence breakpoints of HPRT deletion and translocation mutations. Environ. Mol. Mutagen. 2002;39:22–32. doi: 10.1002/em.10040. [DOI] [PubMed] [Google Scholar]
- 56.Li L., McVety S., Younan R., Liang P., Du Sart D., Gordon P.H., Hutter P., Hogervorst F.B., Chong G., Foulkes W.D. Distinct patterns of germ-line deletions in MLH1 and MSH2: the implication of Alu repetitive element in the genetic etiology of Lynch syndrome (HNPCC) Hum. Mutat. 2006;27:388. doi: 10.1002/humu.9417. [DOI] [PubMed] [Google Scholar]
- 57.Hobbs H.H., Brown M.S., Goldstein J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1992;1:445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 58.Kim S.H., Bae J.H., Chae J.J., Kim U.K., Choe S.J., Namkoong Y., Kim H.S., Park Y.B., Lee C.C. Long-distance PCR-based screening for large rearrangements of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Clin. Chem. 1999;45:1424–1430. [PubMed] [Google Scholar]
- 59.Chae J.J., Park Y.B., Kim S.H., Hong S.S., Song G.J., Han K.H., Namkoong Y., Kim H.S., Lee C.C. Two partial deletion mutations involving the same Alu sequence within intron 8 of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Hum. Genet. 1997;99:155–163. doi: 10.1007/s004390050331. [DOI] [PubMed] [Google Scholar]
- 60.Peeters A.V., Van Gaal L.F., du Plessis L., Lombardi M.P., Havekes L.M., Kotze M.J. Mutational and genetic origin of LDL receptor gene mutations detected in both Belgian and Dutch familial hypercholesterolemics. Hum. Genet. 1997;100:266–270. doi: 10.1007/s004390050503. [DOI] [PubMed] [Google Scholar]
- 61.Simard L.R., Viel J., Lambert M., Paradis G., Levy E., Delvin E.E., Mitchell G.A. The Delta>15 Kb deletion French Canadian founder mutation in familial hypercholesterolemia: rapid polymerase chain reaction-based diagnostic assay and prevalence in Quebec. Clin. Genet. 2004;65:202–208. doi: 10.1111/j.0009-9163.2004.00223.x. [DOI] [PubMed] [Google Scholar]
- 62.Woods-Samuels P., Kazazian H.H., Antonarakis S.E. Nonhomologous recombination in the human genome: deletions in the human factor VIII gene. Genomics. 1991;10:94–101. doi: 10.1016/0888-7543(91)90489-2. [DOI] [PubMed] [Google Scholar]
- 63.Vidal F., Farssac E., Tusell J., Puig L., Gallardo D. First molecular characterization of an unequal homologous alu-mediated recombination event responsible for hemophilia. Thromb. Haemost. 2002;88:12–16. [PubMed] [Google Scholar]
- 64.Van de Water N., Williams R., Ockelford P., Browett P. A 20.7 kb deletion within the factor VIII gene associated with LINE-1 element insertion. Thromb. Haemost. 1998;79:938–942. [PubMed] [Google Scholar]
- 65.Nakaya S.M., Hsu T.C., Geraghty S.J., Manco-Johnson M.J., Thompson A.R. Severe hemophilia A due to a 1.3 kb factor VIII gene deletion including exon 24: homologous recombination between 41 bp within an Alu repeat sequence in introns 23 and 24. J. Thromb. Haemost. 2004;2:1941–1945. doi: 10.1111/j.1538-7836.2004.00963.x. [DOI] [PubMed] [Google Scholar]
- 66.Rossetti L.C., Goodeve A., Larripa I.B., De Brasi C.D. Homeologous recombination between AluSx-sequences as a cause of hemophilia. Hum. Mutat. 2004;24:440. doi: 10.1002/humu.9288. [DOI] [PubMed] [Google Scholar]