

Abstract
Single-gene disorders offer unique opportunities to shed light upon fundamental physiological processes in humans. We investigated an autosomal-recessive phenotype characterized by alopecia, progressive neurological defects, and endocrinopathy (ANE syndrome). By using homozygosity mapping and candidate-gene analysis, we identified a loss-of-function mutation in RBM28, encoding a nucleolar protein. RBM28 yeast ortholog, Nop4p, was previously found to regulate ribosome biogenesis. Accordingly, electron microscopy revealed marked ribosome depletion and structural abnormalities of the rough endoplasmic reticulum in patient cells, ascribing ANE syndrome to the restricted group of inherited disorders associated with ribosomal dysfunction.
Introduction
The nucleolus is associated with the regulation of a number of major physiological cellular processes including ribosome assembly, cell mitosis, stress response, and generation of ribonucleoprotein complexes.1 More than 700 nucleolar proteins have been identified in humans,2,3 most of which seem to be involved in the processing and maturation of ribosomal RNAs (rRNAs) and in the generation of mature ribosomes.1 Despite major advances in our understanding of the nucleolus role, much uncertainty remains concerning the precise physiological importance of individual constituents of the various nucleolar complexes, possibly as a result of the fact that in vitro systems used to probe nucleolar-protein function often lack direct relevance to in vivo physiological conditions. More specifically, very little is currently known about the pathophysiological consequences resulting from nucleolar-protein defective function in humans. With the recognition of the major limitations inherent to genetically engineered mouse models, the study of rare inherited single-gene disorders represents a powerful investigative tool for the delineation of biological functions in humans.4,5 In the present report, we characterize through the study of a rare syndrome the clinical manifestations resulting from decreased expression of RBM28, a recently described nucleolar component of the spliceosomal small nuclear ribonucleoproteins (snRNPs).
Material and Methods
Patients
All affected and healthy control participants or their legal guardian provided written and informed consent according to a protocol approved by the institutional review board and by Israel National Committee for Human Genetic Studies in adherence to the Helsinki guidelines.
SNP Genotyping
Genome-wide homozygosity mapping was performed with the Mapping NspI 250K Array Set from Affymetrix (Affymetrix). Genomic DNA was digested with NspI, ligated to an adaptor, and subjected to polymerase chain reaction (PCR) amplification with adaptor-specific primers. The PCR products were digested with DNaseI and labeled with a biotinylated nucleotide analog. The labeled DNA fragments were hybridized to the microarray, stained by streptavidin-phycoerythrin conjugates, washed with the Affymetrix Fluidics Station 450, and scanned with GeneChip scanner 3000.
Microsatellite Analysis
Polymorphic microsatellite markers spanning the ANE syndrome critical interval were selected from the GDB database; genotypes were established by PCR amplification of genomic DNA with Taq polymerase, Q solution (QIAGEN) and fluorescently labeled oligonucleotide pairs (Syntezza Bioscience) according to the manufacturer's recommendations. PCR conditions were 5 min at 95°C; this was followed by 35 cycles for 30 s at 94°C, 45 s at 57°C, and 1 min 30 s at 72°C and a final extension step at 72°C for 7 min. PCR products were separated by PAGE on an ABI 310 sequencer system, and allele sizes were determined with Genescan 3.1 and Genotyper 2.0 software. Parsimonious haplotypes were subsequently established for each individual.
Mutation Analysis
RBM28 was sequenced with oligonucleotide primer pairs spanning the entire coding sequence of the gene (Table S1 available online), Taq polymerase, and Q solution (QIAGEN) with the following cycling conditions: 94°C for 5 min and then by 35 cycles at 95°C for 30 s, 57°C for 45 s, and 72°C for 1 min 30 s. Gel-purified (QIAquick gel extraction kit, QIAGEN) amplicons were subjected to bidirectional DNA sequencing with the BigDye terminator system on an ABI Prism 3100 sequencer (PE Applied Biosystems).
To screen for p.L351P mutation, we PCR amplified a 324bp fragment, with forward primer 5′-CAAGGGAGAGTTGATATCTAAC-3′ and reverse primer 5′-GCTGGATGGACTGTGAAC-3′. The mutation creates a recognition site for endonuclease BSAJI (New England Biolabs). After incubation at 37°C for 4 hr, digested PCR products were electrophoresed in a 2% agarose gel.
Cell Cultures and Reagents
Fibroblast cell cultures were established from punch biopsies obtained from two patients and two healthy controls after written informed consent was obtained, and were maintained in DMEM supplemented with 20% fetal calf serum, 1% L-Glutamine and 1% penicillin/streptomycin (Beit-Ha-Emek).
Quantitative RT-PCR
For quantitative real-time PCR, cDNA was synthesized from 1 μg of total RNA with the Reverse-iT first-strand synthesis kit (ABgene) and random hexamers. cDNA PCR amplification was carried out with the SYBR Green JumpStart Taq ReadyMix (Sigma) on a Mx3000p/5p multi-filter system (Stratagene) with gene-specific intron-crossing oligonucleotide pairs (Table S2). To ensure the specificity of the reaction conditions, at the end of the individual runs, we measured the melting temperature (Tm) of the amplified products to confirm its homogeneity. Cycling conditions were as follows: 95°C for 10 min and 95°C for 10 s, 62°C for 15 s, and 72°C for 25 s for a total of 40 cycles. Each sample was analyzed in triplicate. For quantification, standard curves were obtained with serially diluted cDNA amplified in the same real-time PCR run. Results were normalized to ACTB and GAPDH mRNA levels.
Immunofluorescence Microscopy
Fibroblast cells were grown on glass coverslips, fixed with 4% paraformaldehyde, and incubated with a rabbit polyclonal antibody against RBM286 for 60 min at 20°C. Staining was detected after incubation with Cy2-conjugated goat anti-rabbit IgG (Jackson Immunoresearch Laboratories). The slides were observed under a confocal microscope (Axiovert 200M LSM 510 Meta, Carl Zeiss MicroImaging GmbH) with Image-Pro Plus LSM image examiner software (Media Cybernetics).
Immunohistochemistry
Formaldehyde-fixed 5 μm paraffin-embedded sections were treated with 3% H2O2 in methanol for 15 min at room temperature, warmed in a microwave oven in citrate buffer for 15 min at 90°C, and stained with polyclonal anti-RBM28, anti-β-catenin antibodies (Abnova Corporation) or preimmune rabbit antiserum for 1 hr at room temperature. After extensive washings in phosphate buffered saline, the antibodies were revealed with the ABC technique (Zymed), and the slides were counterstained with hematoxylin.
Western Blotting
Cells were homogenized in lysis buffer (25 mM HEPES, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and protease inhibitors mix including 1 mM PMSF, 1 mg/ml aprotinin, and leupeptin; Sigma, St Louis, MO, USA) or directly in loading buffer (100 mM Tris [pH 6.8], 4% SDS, 20% glycerol, and 0.2% Bromophenol blue). After centrifugation at 10,000 g for 10 min at 4°C, proteins were electrophoresed through a 10% SDS-PAGE and transferred onto a nitrocellulose membrane (Trans-Blot Bio-Rad). After 1 hr blocking with 1× TBS (20 mM Tris and 150 mM NaCl) with 5% skim milk and 0.01% Tween 20, blots were incubated with primary rabbit anti-RBM28 or mouse anti-β-actin antibodies (Aviva System Biology and ABcam). The blots were washed three times with TBS-Tween (20 mM Tris HCl, 4 mM Tris base, 140 mM NaCl, 1 mM EDTA, and 0.1% Tween-20). After incubation with secondary HRP-conjugated anti-rabbit or anti-mouse antibody (Sigma-Aldrich) and subsequent washings, proteins were detected with the EZ-ECL chemiluminescence detection kit (Biological Industries).
Electron Microscopy
Cell pellets were fixed in half-strength Karnowski's fixative and 1% osmium tetroxide. The pellets were dehydrated with ethanol and embedded in EPON812 (Taab). Ultrathin sections were stained with uranyl acetate and lead citrate.
Bioinformatics and Computational Modeling
Linkage analysis was performed with the Superlink platform.7 Sequence alignment was performed with MAFFT, a multiple-sequence-alignment program. The sequence of the N-terminal part of RBM28 was modeled with the Phyre homology recognition engine server.8
Results
ANE Syndrome: A Pleiotropic and Clinically Heterogeneous Disorder
We assessed a consanguineous kindred of Arab Moslem descent comprising five male individuals affected with a complex syndrome consisting of alopecia, neurological defects, and endocrinopathy (ANE syndrome) (Table 1). Patients were found to display hair loss of widely varying severity (Figures 1A–1D). A skin biopsy obtained from scalp skin revealed absence of mature hair follicles; instead, only rudimentary infundibula and epithelial cysts were observed in the dermis (Figure 1I). Neurological impairment consisted of moderate to severe mental retardation and progressive motor deterioration, which started in all affected siblings during their second decade of life. Progressive motor decline was found to result from combined upper and lower motor dysfunction. Extensive endocrinological evaluation revealed central hypogonadotropic hypogonadism manifesting with delayed or absent puberty and central adrenal insufficiency. Brain magnetic resonance imaging (MRI) revealed a hypoplastic pituitary gland with preserved hypothalamus (Figure 1H) but no evidence for basal ganglia or white-matter disease. Additional features included short stature, microcephaly, gynecomastia (Figure 1E), flexural reticulate hyperpigmentation (Figure 1F), hypodontia (Figure 1G), kyphoscoliosis, multiple facial pigmented nevi, ulnar deviation of hands, and loss of subcutaneous fat.
Table 1.
ANE Syndrome: Clinical Delineation
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | |
|---|---|---|---|---|---|
| Sex | male | male | male | male | male |
| Age (years) | 39 | 36 | 30 | 27 | 20 |
| MR | severe | moderate | severe | moderate | severe |
| Alopecia | universalis | sparse hair trunk, axilla | universalis | partial | universalis |
| Height in cm (centile) | 150 (−3.5 SD) | 166 (7) | 167 (8) | 169 (10) | 148 (−3.5 SD) |
| Head circumference in cm (centile) | 52.5 (−2.5 SD) | 55 (40) | 52 (−2.5 SD) | 53.5 (5) | 49.8 (−3 SD) |
| Dental carries | +++ | ++ | +++ | +++ | +++ |
| Hypodontia/early teeth loss | yes | no | yes | no | yes |
| Gynecomastia | +++ | +++ | + | ++ | no |
| Kyphoscoliosis | severe | no | moderate | moderate | moderate |
| Limb contractures | + | no | ++ | +++ | ++ |
| Motor deterioration | +++ | +/− | ++ | ++ | ++ |
| UMN dysfunction | +++ | +/− | +++ | ++ | +++ |
| Muscle atrophy | no | no | yes | yes | yes |
| Penile length | 4 cm | 8 cm | 5 cm | 9.5 cm | 4 cm |
| Tanner stage | 1 (prepubertal) | 3–4 | 1 (prepubertal) | 3 | 1 (prepubertal) |
| Hypogonadotropic hypogonadism | yes | yes | yes | yes | yes |
| Central adrenal insufficiency | yes | yes | yes | yes | yes |
| Brain CT | ND | ND | normal | normal | normal |
| Brain MRI | ND | ND | ND | ND | hypoplastic pituitary |
Abbreviations are used as follows: MR, mental retardation; SD, standard deviation; UMN, upper motor neuron; and ND, not done.
Figure 1.
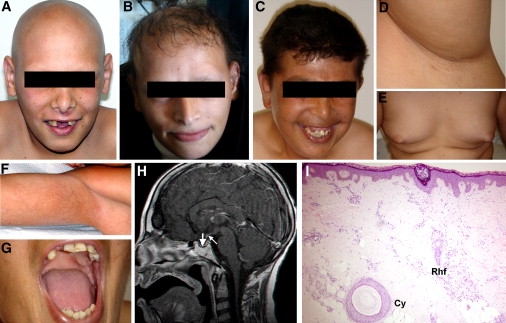
Clinical Features of ANE Syndrome
Patients display varying patterns of alopecia including (A) alopecia universalis, (B) hypotrichosis, (C) near-normal scalp hair associated with absence of body and (D) axillary hair; additional features include (E) gynecomastia, (F) flexural reticulate hyperpigmentation and (G) hypodontia and tooth malplacement; (H) cerebral MRI (T1 weighted after gadolinium injection) shows normal hypothalamus, hypoplastic thin anterior pituitary gland at the bottom of the sella turcica (thick arrow), and ectopic posterior pituitary hyperintense signal located at a proximal level of the pituitary stalk (thin arrow); (I) a skin biopsy obtained from patient scalp reveals absence of mature hair follicles, and instead, presence of dermal cysts (Cy) as well as elongated vertically oriented epithelial structures associated with sebaceous glands, corresponding to rudimentary hair follicles (Rhf).
A Deleterious Homozygous Mutation in RBM28 Underlies ANE Syndrome
To identify the ANE syndrome locus, we initially excluded candidate regions previously linked to alopecia, alopecia with mental retardation, or alopecia associated with dermal cysts on histology as seen in our patients (Figure 1I) on 3q26.2-q26.31, 3q26.33-q27.3, 8p12, 12q13.11, and 18q11.2-q12.29–12 (not shown). We then genotyped all affected individuals for 250K SNP markers distributed across the genome and identified a single 6.45 Mb region of homozygosity on 7q31.32-7q32 shared by all patients. By using microsatellite markers spanning the disease interval, we confirmed a gene locus for ANE syndrome in this region, with a maximum multipoint LOD score of 3.64 (θ = 0.0) at marker D7S530 (Figure 2A). Haplotype analysis (Figure 2B) defined a critical ∼6 Mb interval between rs17147033 and rs205763. This interval was found to contain 72 genes, of which 24 were fully sequenced.
Figure 2.
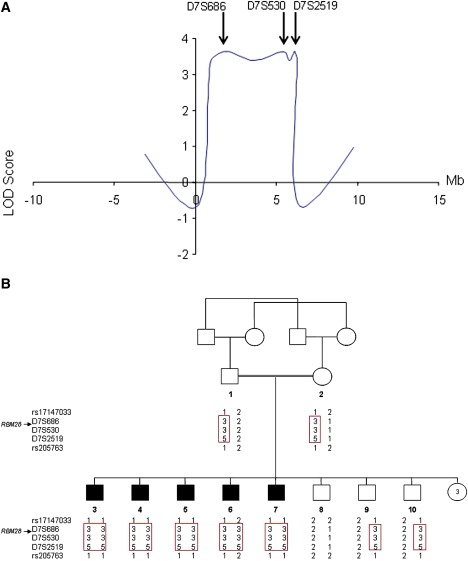
Genetic Mapping of ANE Syndrome
(A) Multipoint LOD score analysis in ANE syndrome. The LOD scores are plotted against the physical distances separating the markers assessed.
(B) Haplotype analysis of the ANE family with polymorphic markers on chromosome 7q31.32-7q32 reveals a homozygous haplotype shared by all patients (boxed in red).
A homozygous T→C transition was identified at cDNA position 1052 of the RBM28 gene (c.T1052C) (Figure 3A). This mutation is predicted to result in the nonconservative substitution of a proline for a leucine residue at amino acid position 351 of the RBM28 protein (p.L351P). By using a PCR-RFLP assay, we found p.L351P to segregate in the family in an autosomal-recessive manner (Figure 3B). We excluded the mutation from a panel of 600 population-matched healthy controls (1200 chromosomes), suggesting that c.T1052C does not represent a common neutral polymorphism (not shown) but rather is a disease-causing mutation.
Figure 3.
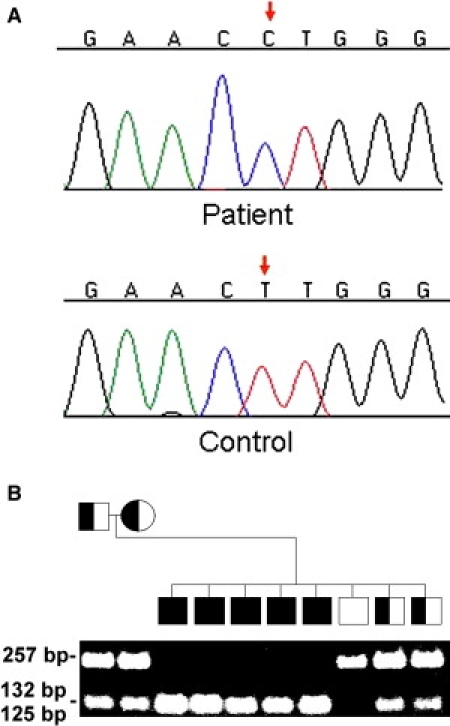
Mutation Analysis
(A) Sequence analysis of RBM28 reveals a homozygous T→C transition at cDNA position 1052 of the RBM28 gene (red arrow, upper panel). The wild-type sequence is given for comparison (lower panel).
(B) PCR-RFLP analysis confirms segregation of the mutation in the family. PCR amplification was performed as described in the text. Mutation c.T1052C creates a recognition site for BSAJI. Thus, affected patients display two 127 bp and 132 bp fragments (appearing as one band) and healthy individuals show a 257 bp fragment only, whereas all fragments are found in heterozygous carriers of the mutation.
The RBM28 gene encodes an 85 kDa protein recently found to be part of the mammalian snRNP complexes and comprising four RNA recognition motifs (RRMs 1–4) as well as an extremely acidic region of 32 amino acids between RRM2 and RRM3.6 RBM28 was observed to be expressed ubiquitously (Figure 4). Mutation p.L351P was found to affect a highly conserved residue located within the RRM3 domain (Figure 5A). The RRM3 motif consists of a four-stranded β sheet flanked by two α helices. L351 is located at the C-terminal end of RRM3 helix 1, directly preceding a very tight loop leading to β strand 2 and containing four contiguous Glu residues. Computational modeling showed that p.L351P is predicted to significantly compromise the integrity of RBM28 structure (Figure 5B).
Figure 4.
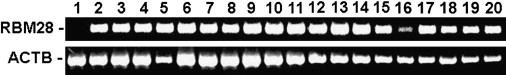
Tissue Expression of RBM28
RBM28 gene expression was assessed with Clontech tissue blot cDNA array. The tissue used in each lane is as follows: lane 1, trachea; lane 2, thymus; lane 3, thyroid; lane 4, testes; lane 5, spleen; lane 6, muscle; lane 7, small intestine; lane 8, prostate; lane 9, heart; lane 10, ovary; lane 11, cervix; lane 12, bladder; lane 13, colon; lane 14, brain; lane 15, lungs; lane 16, liver; lane 17, kidney; lane 18, esophagus; lane 19, adipose tissue; and lane 20, placenta. Expression of RBM28 was compared to that of ACTB.
Figure 5.
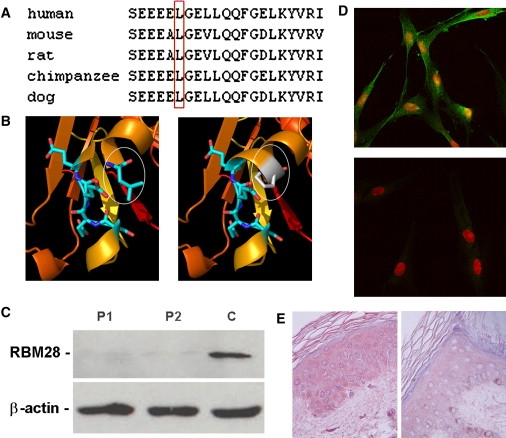
Consequences of p.L351P Mutation in RBM28
(A) ClustalW analysis of the RBM28 protein region encompassing the mutation site demonstrates that L351 (boxed in red) is conserved across species.
(B) The sequence of the N-terminal part of RBM28 was modeled with the Phyre homology recognition engine server with the crystal structure of the N-terminal region of the yeast RNA splicing factor Prp2426 (PDB code 2GHP). Four hundred and twenty residues could be modeled in this fashion with an extremely high degree of confidence (E = 10−24). The Prp24 structure includes three RNA recognition motifs (RRM), consisting of a four-stranded β sheet flanked by two α helices. Wild-type L351 (white ellipse, left panel) is located on the C-terminal end of helix 1 of RRM3 and is expected to destabilize the protein α-helix 1 when mutated to proline (white ellipse, right panel).
(C) Protein was extracted from fibroblast cell cultures established from two ANE patients (P1 and P2) and a control healthy individual (C) and immunoblotted against anti-RBM28 antibodies. Membranes were reblotted with anti-β-actin antibodies to control for protein loading.
(D) Cultured fibroblasts obtained from a patient (lower panel) and a control individual (upper panel) were stained with an anti-RBM28 antibody (green) and propidium iodide (red) and were examined with confocal microscopy. Merged images are presented. Note increased focal expression of RBM28 in the nucleus of control cells and markedly decreased expression of the protein in patient cells.
(E) Skin-biopsy sections obtained from a patient (right panel) and a control individual (left panel) were stained with antibodies directed against RBM28. Immunostaining is markedly decreased in the skin of the patient as compared with control skin (original magnification, 400×).
To assess the consequences of p.L351P in vivo, we ascertained RBM28 expression in fibroblasts derived from ANE patients and normal control individuals. Although the mutation was found to lack any significant effect on mRNA expression as assessed by quantitative RT-PCR (not shown), we found that RBM28 was barely detectable by immunoblotting in fibroblasts derived from patient skin (Figure 5C), suggesting that p.L351P results in protein destabilization and degradation as predicted by computational modeling (Figure 5B). At the cellular level, immunofluorescence staining of control fibroblasts revealed focal expression of RBM28 in the nucleus mainly and, less conspicuously, in the cytoplasm. In contrast, RBM28 was barely detectable in patient fibroblasts (Figure 5D). Moreover, immunostaining of patient epidermis revealed decreased expression of RBM28 in the patient skin (Figure 5E). These data suggested a loss-of-function effect of the mutation, although in the absence of a better understanding of RBM28 function, the remote possibility of a gain-of-function effect cannot be entirely dismissed.
Ribosome Depletion in ANE Syndrome
Because the RBM28 yeast ortholog Nop4p has been shown to be essential for proper ribosome biogenesis,13 we assessed the effect of RBM28 deficiency on ribosome distribution at the ultrastructural level. We observed strikingly abnormal structure of the rough endoplasmic reticulum (RER) in patient cells as compared with control fibroblasts (Figure 6A). In addition, ribosome density was significantly lower in cells derived from patient skin as compared with control cells (Figures 6A and 6B). In contrast, shape and size of nucleolar fibrillar centers, dense fibrillar components, and granular components in patient fibroblasts appeared to be not different from those of control subjects (data not shown).
Figure 6.
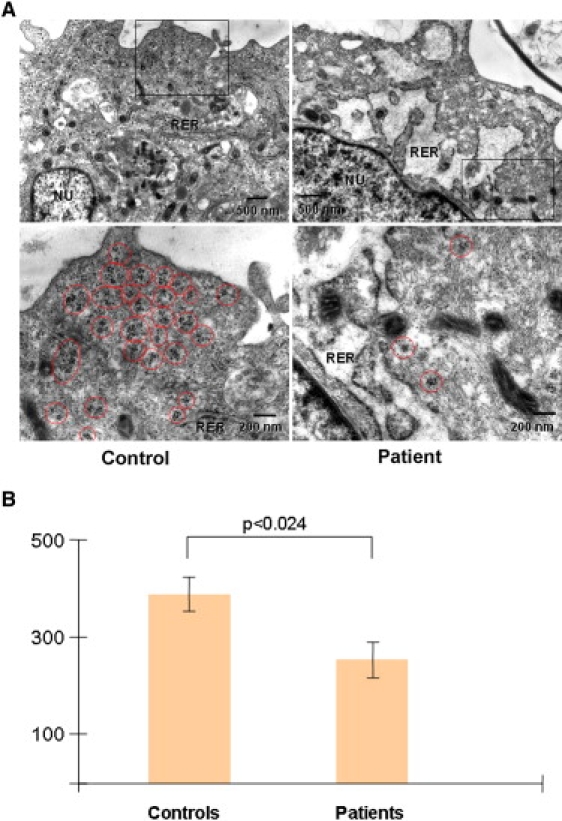
Ultrastructural Abnormalities Associated with RBM28 Deficiency
(A) Transmission electron microscopy (TEM) of cultured fibroblasts in a patient (right panels) and a healthy control individual (left panels) revealed dilated cisterns of the rough endoplasmic reticulum (RER, upper panels) and decreased number of free ribosomes (red circles, lower panels) in the patient cells. The rectangles in the upper panels correspond to the magnified electron micrographs shown in the lower panels.
(B) Free cytoplasmic ribosomes were counted at a TEM magnification of 40000× in control and patient fibroblasts in six independent samples (>1500 ribosomes counted in each group). Results are given as number of free ribosomes per μm2 ± SE. Statistical differences were assessed with a paired Student's t test.
Discussion
In the present study, we describe the clinical manifestations and delineate the molecular basis of ANE syndrome, a complex pleiotropic disorder. The absence of structural abnormalities on brain MRI and the type of endocrine defects found in patients distinguishes ANE syndrome from other rare phenotypes previously reported including Woodhouse-Sakati syndrome (MIM 241080) and Johnson-McMillin syndrome (MIM 147770).14–17 A homozygous missense mutation in RBM28 was found to segregate with the disease and to result in loss of protein expression in patient fibroblasts and epidermis. In spite of its ubiquitous pattern of expression, RBM28 deficiency results in a disease phenotype confined to a limited number of tissues, possibly reflecting functional redundancy among snRNA binding proteins, as previously shown for mutations affecting other nucleolar proteins.18
The precise role played by RBM28 is still poorly understood. RBM28 has been found in association with snRNAs, suggesting that it may be involved in snRNP maturation or be part of the spliceosomal machinery.6 However, its yeast ortholog, Nop4p, has been shown to be required during the biogenesis and assembly of ribosomal subunits.13 Actually, a deleterious mutation in the RRM3 domain of Nop4p was found to result in ribosome depletion, due to defective assembly of the 60S subunit,13 an observation that is in line with the effect of RBM28 deficiency in human cells (Figure 5C). Other nucleolar snRNA-associated proteins, such as Prp43p, a DEAH-box spliceosome disassembly factor, have similarly been found to be essential for both ribosome biogenesis as well as spliceosome dynamics.19 Although inhibition of ribosome biogenesis may directly cause decreased cell proliferation and thus explain the developmental abnormalities observed in ANE syndrome, a recent study suggested that nucleolar stress may result in p53 stabilization, ensuing in cellular loss and tissue dysplasia.20
Regardless of the exact function of RBM28 during the formation of mature ribosomes, the present study clearly indicates that this protein is at least required for the correct development of three major physiological systems: the hair follicle, the hypothalamic-hypophyseal axis, and the nervous system. Although the role of RNP complexes during neural ontogenesis has been well established,21 the essential function played by RBM28 during hair-follicle formation is puzzling. The absence of mature hair follicles and the conspicuous presence of dermal cysts on histopathology in ANE syndrome skin are reminiscent of the features seen in mice lacking β-catenin epidermal expression22 and in humans affected with atrichia with papular lesions (APL; MIM 209500).11 APL is caused by defective function of hairless, a protein responsible for promoting WNT signaling,23 which in turn results in stabilization of β-catenin. Interestingly, immunohistochemistry performed on skin biopsies obtained from ANE syndrome patients revealed markedly decreased expression of β-catenin (not shown). Moreover, recent data indicate that β-catenin plays a key role in determining pituitary development;24 these observations may help understanding endocrine functional (Table 1) and structural (Figure 1H) anomalies found in ANE syndrome. Taken together, these data suggest a hitherto unrecognized role for RBM28 during hair formation, probably related to the hairless/WNT/β-catenin signaling pathway.
In conclusion, the present data provide strong evidence for RBM28 involvement in the development of the neural system, the pituitary gland, and the hair follicle, as well as demonstrate its importance for proper ribosome biogenesis. Hence, ANE syndrome can be added to the short but steadily expanding list of human inherited diseases associated with ribosomal abnormalities.25
Supplemental Data
Two tables are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GDB database, http://www.gdb.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/sites/entrez?db=Omim
Phyre homology recognition engine server, http://www.sbg.bio.ic.ac.uk/phyre/
Superlink platform, http://bioinfo.cs.technion.ac.il/superlink-online/
Acknowledgments
We are grateful to the family members for their enthusiastic and generous participation in our study. We wish to thank Shirley Horn-Saban for SNP genotyping services, Vered Friedman and Rita Fuhrer-Mor for their help with nucleic acid analysis, and Gabriele Richard and Leonard D. Shultz for helpful discussions. This study was supported in part by grants provided by the Rappaport Institute for Research in the Medical Sciences.
References
- 1.Boisvert F.M., van Koningsbruggen S., Navascues J., Lamond A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007;80:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- 2.Andersen J.S., Lam Y.W., Leung A.K., Ong S.E., Lyon C.E., Lamond A.I., Mann M. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 3.Scherl A., Couté Y., Déon C., Callé A., Kindbeiter K., Sanchez J.C., Greco A., Hochstrasser D., Diaz J.J. Functional proteomic analysis of human nucleolus. Mol. Biol. Cell. 2002;13:4100–4109. doi: 10.1091/mbc.E02-05-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ropers H.H. New perspectives for the elucidation of genetic disorders. Am. J. Hum. Genet. 2007;81:199–207. doi: 10.1086/520679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonarakis S.E., Beckmann J.S. Mendelian disorders deserve more attention. Nat. Rev. Genet. 2006;7:277–282. doi: 10.1038/nrg1826. [DOI] [PubMed] [Google Scholar]
- 6.Damianov A., Kann M., Lane W.S., Bindereif A. Human RBM28 protein is a specific nucleolar component of the spliceosomal snRNPs. Biol. Chem. 2006;387:1455–1460. doi: 10.1515/BC.2006.182. [DOI] [PubMed] [Google Scholar]
- 7.Silberstein M., Tzemach A., Dovgolevsky N., Fishelson M., Schuster A., Geiger D. Online system for faster multipoint linkage analysis via parallel execution on thousands of personal computers. Am. J. Hum. Genet. 2006;78:922–935. doi: 10.1086/504158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett-Lovsey R.M., Herbert A.D., Sternberg M.J., Kelley L.A. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–625. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- 9.Kantaputra P.N., Limwongse C., Tochareontanaphol C., Mutirangura A., Mevatee U., Praphanphoj V. Contiguous gene syndrome of holoprosencephaly and hypotrichosis simplex: Association with an 18p11.3 deletion. Am. J. Med. Genet. A. 2006;140:2598–2602. doi: 10.1002/ajmg.a.31386. [DOI] [PubMed] [Google Scholar]
- 10.Kazantseva A., Goltsov A., Zinchenko R., Grigorenko A.P., Abrukova A.V., Moliaka Y.K., Kirillov A.G., Guo Z., Lyle S., Ginter E.K. Human hair growth deficiency is linked to a genetic defect in the phospholipase gene LIPH. Science. 2006;314:982–985. doi: 10.1126/science.1133276. [DOI] [PubMed] [Google Scholar]
- 11.Sprecher E. Genetic hair and nail disorders. Clin. Dermatol. 2005;23:47–55. doi: 10.1016/j.clindermatol.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 12.Wali A., John P., Gul A., Lee K., Chishti M.S., Ali G., Hassan M.J., Leal S.M., Ahmad W. A novel locus for alopecia with mental retardation syndrome (APMR2) maps to chromosome 3q26.2-q26.31. Clin. Genet. 2006;70:233–239. doi: 10.1111/j.1399-0004.2006.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun C., Woolford J.L. The yeast nucleolar protein Nop4p contains four RNA recognition motifs necessary for ribosome biogenesis. J. Biol. Chem. 1997;272:25345–25352. doi: 10.1074/jbc.272.40.25345. [DOI] [PubMed] [Google Scholar]
- 14.Cushman L.J., Torres-Martinez W., Weaver D.D. Johnson-McMillin syndrome: Report of a new case with novel features. Birth Defects Res. A. Clin. Mol. Teratol. 2005;73:638–641. doi: 10.1002/bdra.20178. [DOI] [PubMed] [Google Scholar]
- 15.Schneider S.A., Bhatia K.P. Dystonia in the Woodhouse Sakati syndrome: A new family and literature review. Mov. Disord. 2008;23:592–596. doi: 10.1002/mds.21886. [DOI] [PubMed] [Google Scholar]
- 16.Al-Semari A., Bohlega S. Autosomal-recessive syndrome with alopecia, hypogonadism, progressive extra-pyramidal disorder, white matter disease, sensory neural deafness, diabetes mellitus, and low IGF1. Am. J. Med. Genet. A. 2007;143:149–160. doi: 10.1002/ajmg.a.31497. [DOI] [PubMed] [Google Scholar]
- 17.Devriendt K., Legius E., Fryns J.P. Progressive extrapyramidal disorder with primary hypogonadism and alopecia in sibs: A new syndrome? Am. J. Med. Genet. 1996;62:54–57. doi: 10.1002/(SICI)1096-8628(19960301)62:1<54::AID-AJMG11>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 18.Choesmel V., Bacqueville D., Rouquette J., Noaillac-Depeyre J., Fribourg S., Crétien A., Leblanc T., Tchernia G., Da Costa L., Gleizes P.E. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood. 2007;109:1275–1283. doi: 10.1182/blood-2006-07-038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Combs D.J., Nagel R.J., Ares M., Stevens S.W. Prp43p is a DEAH-box spliceosome disassembly factor essential for ribosome biogenesis. Mol. Cell. Biol. 2006;26:523–534. doi: 10.1128/MCB.26.2.523-534.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones N.C., Lynn M.L., Gaudenz K., Sakai D., Aoto K., Rey J.P., Glynn E.F., Ellington L., Du C., Dixon J. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008;14:125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8:340–345. doi: 10.1038/sj.embor.7400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huelsken J., Vogel R., Erdmann B., Cotsarelis G., Birchmeier W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 23.Thompson C.C., Sisk J.M., Beaudoin G.M. Hairless and Wnt signaling: Allies in epithelial stem cell differentiation. Cell Cycle. 2006;5:1913–1917. doi: 10.4161/cc.5.17.3189. [DOI] [PubMed] [Google Scholar]
- 24.Olson L.E., Tollkuhn J., Scafoglio C., Krones A., Zhang J., Ohgi K.A., Wu W., Taketo M.M., Kemler R., Grosschedl R. Homeodomain-mediated beta-catenin-dependent switching events dictate cell-lineage determination. Cell. 2006;125:593–605. doi: 10.1016/j.cell.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 25.Liu J.M., Ellis S.R. Ribosomes and marrow failure: Coincidental association or molecular paradigm? Blood. 2006;107:4583–4588. doi: 10.1182/blood-2005-12-4831. [DOI] [PubMed] [Google Scholar]
- 26.Bae E., Reiter N.J., Bingman C.A., Kwan S.S., Lee D., Phillips G.N., Butcher S.E., Brow D.A. Structure and interactions of the first three RNA recognition motifs of splicing factor prp24. J. Mol. Biol. 2007;367:1447–1458. doi: 10.1016/j.jmb.2007.01.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.