

Abstract
Although elevated levels of C-reactive protein (CRP) independently predict increased risk of development of metabolic syndrome, diabetes, myocardial infarction, and stroke, comprehensive analysis of the influence of genetic variation on CRP is not available. To address this issue, we performed a genome-wide association study among 6345 apparently healthy women in which we evaluated 336,108 SNPs as potential determinants of plasma CRP concentration. Overall, seven loci that associate with plasma CRP at levels achieving genome-wide statistical significance were found (range of p values for lead SNPs within the seven loci: 1.9 × 10−8 to 6.2 × 10−28). Two of these loci (GCKR and HNF1A) are suspected or known to be associated with maturity-onset diabetes of the young, one is a gene-desert region on 12q23.2, and the remaining four loci are in or near the leptin receptor protein gene, the apolipoprotein E gene, the interleukin-6 receptor protein gene, or the CRP gene itself. The protein products of six of these seven loci are directly involved in metabolic syndrome, insulin resistance, beta cell function, weight homeostasis, and/or premature atherothrombosis. Thus, common variation in several genes involved in metabolic and inflammatory regulation have significant effects on CRP levels, consistent with CRP's identification as a useful biomarker of risk for incident vascular disease and diabetes.
Main Text
C-reactive protein (CRP) is a pattern-recognition molecule of innate immunity recognized 80 years ago as an acute-phase reactant and a hallmark of low-grade systemic inflammation. Plasma concentrations of CRP within the low-normal range independently predict incident metabolic syndrome, type 2 diabetes, myocardial infarction, and stroke in otherwise healthy men and women.1–5 Although environmental factors such as obesity, smoking, and hormone-replacement therapy modify CRP levels, variation in CRP is known to be heritable.6 To date, candidate-gene studies have largely focused on the CRP gene itself and explained only a fraction of the estimated variance due to genetics.7–10 It has thus been hypothesized that substantive genetic effects on CRP are present outside the CRP locus and that identification of these additional loci might provide insight into mechanisms underlying relationships between CRP, insulin resistance, metabolic syndrome, and vascular events.
To address these issues, we conducted a genome-wide association study among 6345 apparently healthy women in the Women's Genome Health Study (WGHS), in which we evaluated 336,108 single nucleotide polymorphisms (SNPs) satisfying our quality criteria as potential determinants of plasma CRP level. Details of the rationale, design, and methodology of the WGHS are described elsewhere.11 In brief, participants in the WGHS include American women with no prior history of cardiovascular disease, cancer, or other major chronic illness, who provided a baseline blood sample during the enrollment phase of the Women's Health Study (between 1992 and 1995) and consent for blood-based analyses related to the study of the risk of incident chronic diseases. All baseline blood samples underwent measurement for high-sensitivity C-reactive protein (hsCRP) via a validated immunoturbidometric method (Denka Seiken, Tokyo, Japan). DNA extracted from the baseline blood samples underwent SNP genotyping via the Illumina Infinium II assay for querying of a genome-wide set of 318,237 SNP markers (Human Hap300 panel). An additional focused panel of 45,571 SNP markers was queried as well; these were selected to enhance coverage of genomic regions without regard to allele frequency, in which we had a strong a priori interest owing to the presence of genes believed to be of relevance to metabolic, cardiovascular, and inflammatory diseases or to biologic function in general. The study protocol was approved by the institutional review board of the Brigham and Women's Hospital (Boston, MA, USA).
Before any genetic analyses were performed, SNPs were excluded from analysis that had a low call rate (≤90 percent), and participants were excluded when genotyping failed for >2 percent of SNPs. For all SNPs and samples meeting these criteria, Hardy-Weinberg equilibrium was evaluated with an exact method12 for identification of potential genotyping errors; SNPs with Hardy-Weinberg p values < 10−6 were eliminated from analysis. We also compared the Illumina-based SNP data for each individual participant for a panel of 44 SNPs that had previously been ascertained in this population with alternative genotyping technologies; this step was used as a secondary check to ensure accurate specimen labeling prior to any analyses. In total, 336,108 SNPs with minor allele frequencies > 1 percent passed the criteria for analysis.
We limited our evaluation to 6345 nondiabetic participants in the WGHS who were of European ancestry and were not taking lipid-lowering agents. A principal-component analysis using 1443 ancestry-informative SNPs was performed in order to confirm self-reported ancestry with PLINK.12 In brief, these SNPs were had an Fst higher than 0.4 in HapMap populations (YRB, CEU, CHB+JPT) and had inter-SNP distances of at least 500 kb in order to minimize linkage disequilibrium. Different ethnic groups were clearly distinguished with the first two principal components.
Genotyping was performed in two stages, with an initial group of 4418 study participants (WGHS-1) and then with a second group of 1927 participants (WGHS-2). In all statistical analyses, we adjusted plasma levels of CRP on an a priori basis for age, smoking, body-mass index, hormone therapy, and menopausal status. Because CRP levels rise rapidly with the acute-phase response, we also decided on an a priori basis to perform separate analyses on the 95.2 percent of the cohort with CRP values < 10 mg/l as well as on the full cohort (inclusive of those with CRP levels ≥ 10 mg/l).
For all genotype-phenotype association analyses, we assumed an additive model of inheritance and initially conducted univariate linear-regression analyses to test the null hypothesis that adjusted log CRP levels did not differ by individual SNP genotypes.12 To conservatively identify loci that might be associated with CRP levels, we used an initial criterion of statistical significance at a level of p < 5 × 10−8 for single SNP associations in the total sample. All reported associations were also observed at a < 5 × 10−8 level of significance after adjustment of the test statistic by a genomic-control procedure.12 The genomic-control inflation factor was 1.01, signifying no evidence of type 1 error inflation. As shown in the quantile-quantile analysis (Figure 1), p values larger than 0.001 conform to the expected null distribution, again suggesting no inflation of test statistics (red dots). The excess of p values smaller than 0.001 was solely related to the associations at the candidate loci; after further adjustment of CRP residuals for variation at the candidate loci, there was no longer an excess of small p values in genome-wide association testing (blue dots). Finally, a principal-component analysis was performed for the exclusion of potential within-European stratification in 124,931 SNPs chosen to have pairwise linkage disequilibrium r2 < 0.04. The first ten components were then used as covariates in the association analysis; because adjustment by these covariates did not change conclusions regarding participants of European ancestry, the data are presented without further correction for within-European ancestry.
Figure 1.
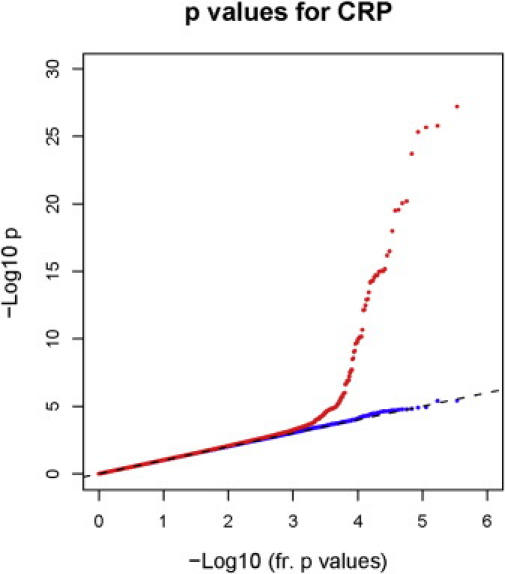
Quantile-Quantile Plot of Actual and Expected p Values for Association with CRP
p values for the association of each individual SNP with plasma CRP levels according to chromosome number and position are shown in Figure 2; the top half of Figure 2 shows data for study participants with CRP levels < 10 mg/l, whereas the bottom half shows data for the full cohort.
Figure 2.
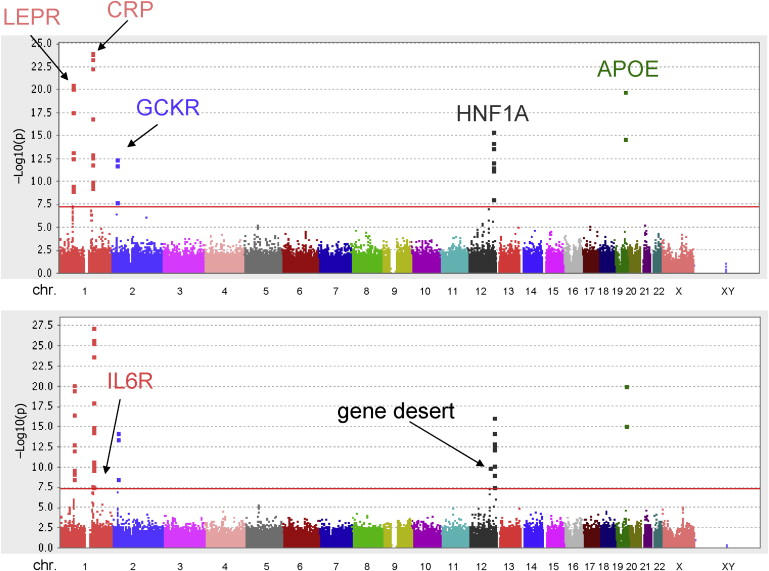
p Values for the Association of Individual SNPs with Plasma CRP Levels According to Chromosome Number and Position
(Top) Data for study participants with CRP levels < 10 mg/l.
(Bottom) Data for the full study cohort. The red horizontal line corresponds to a p value of 10−8.
In the full cohort, 46 SNPs were individually associated with CRP at a genome-wide level of significance (p < 5 × 10−8). All 46 of these SNPs clustered in one of seven chromosomal regions: nine in chromosome locus 1q31.3, associated with the leptin-receptor gene (LEPR [MIM 601007]); 20 in chromosome locus 1q23.2, associated with the CRP gene (MIM 123260); two in chromosome 1q21.3, associated with the interleukin-6 receptor protein gene (IL6R) (MIM 147880); three in chromosome locus 2p23.3, associated with a glucokinase regulatory protein gene (GCKR) (MIM 600842); eight in chromosome locus 12q24.31, associated with an hepatic transcription-factor gene (HNF1A) (MIM 142410) that codes for hepatic nuclear factor 1-alpha; two in chromosome locus 19q13.32, near the apolipoprotein E gene (MIM 107741); and two in a gene-desert region of chromosome 12q23.2. As shown in Table 1, p values for lead SNPs within the seven loci ranged from 1.9 × 10−8 to 6.2 × 10−28. Table 1 also shows the considerable internal consistency in these data for the separate WGHS-1 and WGHS-2 subpopulations of the larger cohort. Replication of the HNF1A finding and replication of the known CRP and APOE findings are also provided in the accompanying manuscript from Reiner et al. With respect to GCKR, IL6R, and LEPR, the Reiner data also support replication, albeit at p values of 0.005, 0.04, and 0.05, respectively.
Table 1.
SNPs, Beta Coefficients, and p Values associated with Plasma CRP Levels in the Women's Genome Health Study
|
WGHS-1 (N = 4418) |
WGHS-2 (N = 1927) |
WGHS-Combined (N = 6345) |
||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Loci | SNP | Beta | p value | Beta | p Value | Beta | p Value |
| LEPR | 1q31.3 | rs1892534 | −0.173 | 1.53 × 10−15 | −0.166 | 3.74 × 10−7 | −0.170 | 6.52 × 10−21 |
| rs2889195 | −0.171 | 5.04 × 10−15 | −0.166 | 4.51 × 10−7 | −0.168 | 2.78 × 10−20 | ||
| rs2211651 | −0.170 | 7.45 × 10−15 | −0.168 | 3.32 × 10−7 | −0.168 | 3.10 × 10−20 | ||
| rs12753193 | −0.157 | 6.91 × 10−13 | −0.151 | 5.03 × 10−6 | −0.154 | 3.27 × 10−17 | ||
| rs2186245 | −0.176 | 5.41 × 10−11 | −0.156 | 2.39 × 10−4 | −0.168 | 1.27 × 10−13 | ||
| rs12022410 | 0.126 | 4.03 × 10−9 | 0.137 | 2.43 × 10−5 | 0.128 | 7.48 × 10−13 | ||
| rs7539471 | 0.115 | 5.33 × 10−7 | 0.140 | 5.52 × 10−5 | 0.122 | 1.87 × 10−10 | ||
| rs4291477 | 0.107 | 5.95 × 10−7 | 0.122 | 1.80 × 10−4 | 0.110 | 7.65 × 10−10 | ||
| rs4655537 | 0.112 | 6.42 × 10−7 | 0.110 | 9.14 × 10−4 | 0.110 | 3.28 × 10−9 | ||
| CRP | 1q23.2 | rs3091244 | 0.207 | 1.45 × 10−20 | 0.193 | 7.63 × 10−9 | 0.203 | 6.16 × 10−28 |
| rs1205 | −0.205 | 5.47 × 10−20 | −0.184 | 5.93 × 10−8 | −0.199 | 1.65 × 10−26 | ||
| rs7553007 | −0.207 | 2.68 × 10−20 | −0.179 | 1.35 × 10−7 | −0.199 | 2.16 × 10−26 | ||
| rs2794520 | −0.202 | 2.18 × 10−19 | −0.185 | 4.33 × 10−8 | −0.197 | 4.74 × 10−26 | ||
| rs2027471 | −0.200 | 3.00 × 10−19 | −0.164 | 1.13 × 10−6 | −0.190 | 1.97 × 10−24 | ||
| rs2592887 | −0.162 | 4.78 × 10−14 | −0.151 | 3.65 × 10−6 | −0.159 | 1.04 × 10−18 | ||
| rs1417938 | 0.146 | 2.90 × 10−10 | 0.178 | 4.40 × 10−7 | 0.156 | 9.72 × 10−16 | ||
| rs3116653 | 0.147 | 2.23 × 10−10 | 0.176 | 6.39 × 10−7 | 0.156 | 9.91 × 10−16 | ||
| rs3116636 | 0.147 | 2.59 × 10−10 | 0.176 | 5.69 × 10−7 | 0.155 | 1.06 × 10−15 | ||
| rs3122012 | 0.145 | 4.22 × 10−10 | 0.176 | 6.34 × 10−7 | 0.154 | 1.95 × 10−15 | ||
| rs3116651 | 0.145 | 5.27 × 10−10 | 0.181 | 4.26 × 10−7 | 0.155 | 1.91 × 10−15 | ||
| rs3116656 | 0.144 | 5.45 × 10−10 | 0.175 | 6.97 × 10−7 | 0.153 | 2.78 × 10−15 | ||
| rs1800947 | −0.300 | 3.90 × 10−12 | −0.241 | 2.40 × 10−4 | −0.283 | 5.08 × 10−15 | ||
| rs12093699 | 0.132 | 1.60 × 10−8 | 0.126 | 2.95 × 10−4 | 0.130 | 2.15 × 10−11 | ||
| rs12068753 | 0.286 | 2.17 × 10−10 | 0.149 | 3.72 × 10−2 | 0.249 | 6.70 × 10−11 | ||
| rs3093075 | 0.280 | 4.82 × 10−10 | 0.160 | 2.52 × 10−2 | 0.248 | 7.72 × 10−11 | ||
| rs3093059 | 0.284 | 2.77 × 10−10 | 0.146 | 4.06 × 10−2 | 0.247 | 9.28 × 10−11 | ||
| rs11265260 | 0.289 | 5.64 × 10−10 | 0.150 | 3.63 × 10−2 | 0.249 | 1.99 × 10−10 | ||
| rs3093068 | 0.274 | 1.45 × 10−9 | 0.158 | 2.80 × 10−2 | 0.243 | 2.34 × 10−10 | ||
| rs12744244 | 0.127 | 3.04 × 10−6 | 0.121 | 1.81 × 10−3 | 0.123 | 3.12 × 10−8 | ||
| IL6R | 1q21.3 | rs8192284 | −0.095 | 1.09 × 10−5 | −0.119 | 2.81 × 10−4 | −0.101 | 1.93 × 10−8 |
| rs4129267 | −0.096 | 9.33 × 10−6 | −0.117 | 3.54 × 10−4 | −0.101 | 1.97 × 10−8 | ||
| GCKR | 2p23.3 | rs780094 | 0.142 | 3.97 × 10−11 | 0.136 | 3.80 × 10−5 | 0.140 | 6.73 × 10−15 |
| rs1260326 | 0.136 | 2.18 × 10−10 | 0.139 | 3.18 × 10−5 | 0.16 | 3.57 × 10−14 | ||
| rs1260333 | 0.112 | 4.69 × 10−7 | 0.106 | 1.65 × 10−3 | 0.110 | 2.69 × 10−9 | ||
| Unknown | 12q23.2 | rs10778213 | −0.109 | 3.65 × 10−7 | −0.128 | 7.00 × 10−5 | −0.115 | 1.16 × 10−10 |
| rs2292996 | −0.089 | 2.96 × 10−5 | −0.113 | 4.87 × 10−4 | −0.096 | 5.98 × 10−8 | ||
| HNF1A | 12q24.31 | rs7310409 | −0.120 | 4.63 × 10−8 | −0.237 | 1.81 × 10−12 | −0.154 | 6.75 × 10−17 |
| rs735396 | −0.117 | 1.70 × 10−7 | −0.223 | 1.12 × 10−12 | −0.147 | 5.39 × 10−15 | ||
| rs1169300 | −0.112 | 1.81 × 10−6 | −0.230 | 1.64 × 10−10 | −0.146 | 1.15 × 10−13 | ||
| rs2464196 | −0.108 | 4.37 × 10−6 | −0.231 | 1.44 × 10−10 | −0.143 | 3.32 × 10−13 | ||
| rs7953249 | −0.102 | 2.67 × 10−6 | −0.201 | 1.42 × 10−9 | −0.130 | 6.98 × 10−13 | ||
| rs2650000 | −0.093 | 3.64 × 10−5 | −0.198 | 9.90 × 10−9 | −0.123 | 7.13 × 10−11 | ||
| rs1169302 | −0.094 | 1.23 × 10−5 | −0.148 | 6.36 × 10−6 | −0.110 | 9.29 × 10−10 | ||
| rs1169307 | 0.100 | 7.19 × 10−6 | 0.112 | 9.14 × 10−4 | 0.103 | 2.90 × 10−8 | ||
| APOE | 19q13.32 | rs769449 | −0.276 | 8.57 × 10−17 | −0.227 | 1.09 × 10−5 | −0.261 | 8.90 × 10−21 |
| rs2075650 | −0.213 | 4.65 × 10−12 | −0.200 | 2.37 × 10−5 | −0.209 | 6.80 × 10−16 | ||
Adjustments made for age, smoking status, body-mass index, hormone-therapy use, and menopausal status.
In analyses restricted to those study participants with CRP levels < 10 mg/l, virtually identical results were observed for SNPs in the LEPR, CRP, GCKR, HNF1A, and APOE loci (all p values < 10−8). However, in this group with CRP levels generally below concentrations associated with the acute-phase response, the lead SNPs in the IL6R locus and in the gene-desert region had slightly lower levels of statistical significance (p = 1.65 × 10−7 and 1.06 × 10−7, respectively).
Figure 3 presents data that map each SNP according to its physical location at each of the seven loci, as well a plot of the p values in relation to the genetic distance from the lead SNP and the known recombination hotspots according to HapMap. Many of the SNPs evaluated tended to group into easily recognizable linkage blocks, and several SNPs with genome-wide significance fell into blocks that included several genes. For example, the two highly significant SNPs near the APOE locus are in a block that includes the APOC1 [MIM 107710] and TOMM40 [MIM 608061] loci.
Figure 3.
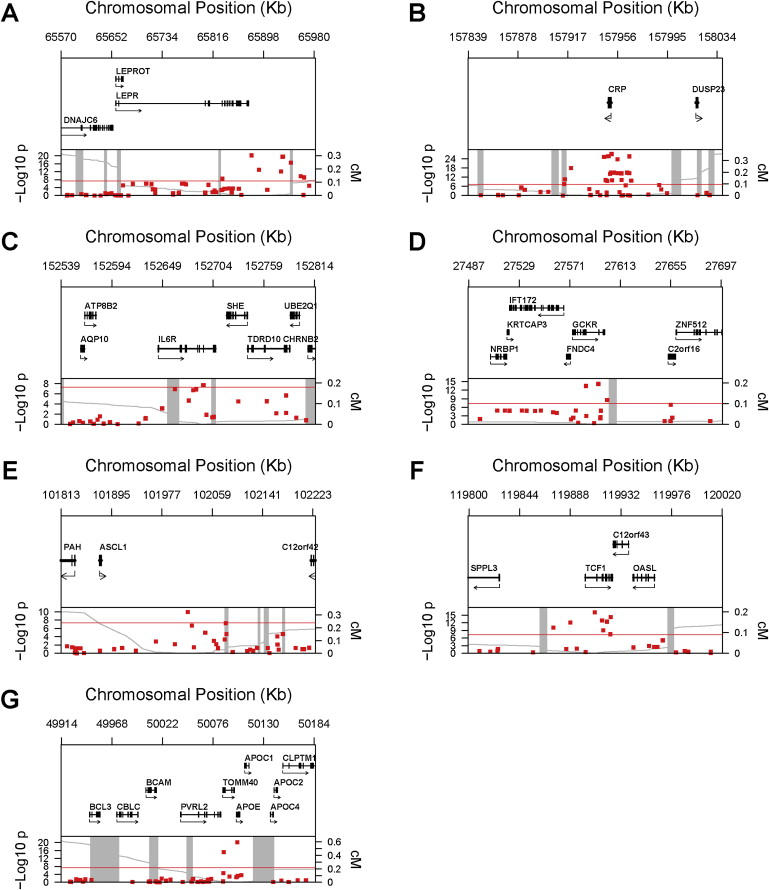
Genomic Context for Each of Seven Loci with Genome-wide Association with CRP Levels
(A) LEPR locus; (B) CRP locus; (C) IL6R locus; (D) GCKR locus; (E) gene-desert region on 12q23.2; (F) HNF1A locus; (G) APOE locus.
Upper panel: Genes from RefSeq release 25. Only one isoform is shown when multiple splicing variants are known.
Lower panel: SNPs are plotted according to their physical location with the y axis, indicating p values for association with CRP (red dots). Also shown is the genetic distance in cM from the lowest p value SNP for each locus (light-gray line), along with the position of recombination hotspots (light-gray vertical bars). Recombination rates and hotspots are based on HapMap data.32,33
Table 2 presents results of backward-selection models using the Akaike Information Criterion to seek nonredundant SNPs contributing to CRP level at each of the seven loci; in this analysis, all potential SNPs with p values of 10−4 or smaller within 500kB of the locus SNPs with p < 10−7 were considered in order to avoid inadvertent exclusion of modest effects. This procedure yielded a subset of nonredundant SNPs accounting for the common genetic variance in CRP at each locus after adjustment for clinical and environmental factors. Also shown in Table 2 are the crude median levels of CRP (mg/l) for homozygous minor-allele carriers, heterozygotes, and homozygous major-allele carriers of each of the nonredundant SNPs that influenced CRP levels within each of the seven loci. In each instance, carriers of the SNP of interest expressed markedly different ranges of CRP; for example, median levels of plasma CRP were 1.40, 1.88, and 2.09 mg/l for homozygous minor allele carriers, heterozygotes, and homozygous major allele carriers, respectively, for SNP rs1892534 in the LEPR gene (p = 6.5 × 10−21). In aggregate, after adjustment for the clinical and environmental factors, 10.1 percent of the residual variation in CRP was explained by the common polymorphism observed in these seven loci. When examined on a per-locus basis, the greatest contributor to this residual variation in our data was attributable to polymorphism in the CRP gene (3.4 percent). For comparison, 1.6 percent was attributable to polymorphism in the LEPR gene, 1.5 percent to polymorphism in the apolipoprotein E gene, 1.1 percent to polymorphism in the GCKR gene, 1.1 percent to polymorphism in the HNF1A gene, 0.8 percent to polymorphism in the gene-desert region, and 0.6 percent to polymorphism in the IL6R gene. We repeated the model selection, using a more stringent backward-forward procedure with the Bayes Information Criterion, and observed similar locus-wide proportion of variance explained, but with a smaller number of critical SNPs retained in the regression models. These SNPs are underlined in Table 2.
Table 2.
Nonredundant SNPs in Each of Seven Loci that Contribute to Adjusted CRP Levels in Backward-Selection Models
| Median hsCRP Level (mg/l) |
Locus-wideVariance |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | SNP | Chromosome | Position | MAF | HW | A1-A2 | A1A1 | A1A2 | A2A2 | Explained (%) |
| LEPR | rs1892534 | 1 | 65878531 | 0.384 | 0.11 | A-G | 1.40 | 1.88 | 2.09 | 1.6 |
| rs12753193 | 1 | 65942266 | 0.375 | 0.11 | G-A | 1.41 | 1.88 | 2.08 | ||
| rs7539471 | 1 | 65957990 | 0.315 | 0.25 | A-G | 2.24 | 1.91 | 1.79 | ||
| rs12409877 | 1 | 65716459 | 0.392 | 0.16 | A-G | 1.69 | 1.82 | 2.07 | ||
| rs5010905 | 1 | 65971810 | 0.260 | 0.03 | G-A | 2.15 | 1.97 | 1.80 | ||
| CRP | rs3091244 | 1 | 157951288 | 0.360 | 0.94 | T/A-G∗ | 2.41 | 2.03 | 1.66 | 3.4 |
| rs1800947 | 1 | 157950061 | 0.065 | 0.92 | G-C | 0.73 | 1.61 | 1.95 | ||
| rs3122012 | 1 | 157955946 | 0.302 | 0.95 | G-A | 2.25 | 2.04 | 1.74 | ||
| rs3093059 | 1 | 157951759 | 0.059 | 0.91 | G-A | 2.05 | 2.34 | 1.84 | ||
| rs4131568 | 1 | 157988679 | 0.334 | 0.37 | A-G | 2.11 | 1.98 | 1.77 | ||
| rs3116654 | 1 | 157962384 | 0.126 | 0.22 | G-A | 1.22 | 1.65 | 1.99 | ||
| rs863013 | 1 | 157466643 | 0.330 | 0.60 | A-C | 1.66 | 1.82 | 2.01 | ||
| IL6R | rs4129267 | 1 | 152692887 | 0.398 | 0.20 | A-G | 1.65 | 1.95 | 1.98 | 0.6 |
| rs12750774 | 1 | 152783100 | 0.304 | 0.29 | A-G | 1.63 | 1.84 | 2.01 | ||
| GCKR | rs780094 | 2 | 27594740 | 0.401 | 0.07 | A-G | 2.17 | 1.92 | 1.74 | 1.1 |
| rs1260333 | 2 | 27602127 | 0.453 | 0.74 | A-G | 2.11 | 1.86 | 1.81 | ||
| rs780106 | 2 | 27535101 | 0.382 | 0.04 | C-A | 1.81 | 1.86 | 1.98 | ||
| rs1647266 | 2 | 27546988 | 0.382 | 0.04 | G-A | 1.81 | 1.87 | 1.98 | ||
| rs13013484 | 2 | 27842324 | 0.270 | 0.67 | G-A | 1.62 | 1.82 | 1.98 | ||
| Unknown | rs10778213 | 12 | 102019280 | 0.468 | 0.71 | G-A | 1.75 | 1.85 | 2.08 | 0.8 |
| rs4433630 | 12 | 102174160 | 0.487 | 0.69 | A-G | 1.80 | 187 | 2.05 | ||
| HNF1A | rs7310409 | 12 | 119909243 | 0.388 | 0.28 | A-G | 1.66 | 1.78 | 2.15 | 1.1 |
| rs1169300 | 12 | 119915607 | 0.295 | 0.45 | A-G | 1.78 | 1.74 | 2.07 | ||
| rs2464196 | 12 | 119919809 | 0.294 | 0.43 | A-G | 1.80 | 1.73 | 2.07 | ||
| rs1169302 | 12 | 119916684 | 0.425 | 0.49 | C-A | 1.68 | 1.86 | 2.09 | ||
| APOE | rs769449 | 19 | 50101841 | 0.115 | 0.51 | A-G | 1.38 | 1.46 | 2.00 | 1.5 |
| rs157580 | 19 | 50087105 | 0.392 | 0.13 | G-A | 2.05 | 1.94 | 1.76 | ||
Adjustments made for age, smoking status, body-mass index, hormone-therapy use, and menopausal status.
Also shown are the crude median blood levels of hsCRP observed among homozygous carriers of the minor allele (A1A1), heterozygotes (A1A2), and homozygous carriers of the major allele (A2A2) for each selected SNP, and the loci-wide variance in CRP is explained.
MAF denotes “minor-allele frequency.”
HW indicates significance of deviation from Hardy-Weinberg equilibrium.
Selection criteria and methods to identify lead SNPs described in text.
Tri-allelic SNP. The two less-common alleles (T/A) were pooled.
Despite adjustment of CRP concentration for body-mass index prior to genetic analysis, we sought further evidence to exclude the possibility of residual confounding by this factor. In this regard, none of the lead SNPs reported here had even a modest level of significance for association with body-mass index (range of p values: 0.02 to 0.70).
We believe these genetic data linking inflammation and CRP to loci known functionally to relate to metabolic syndrome, insulin resistance, weight homeostasis, and premature atherothrombosis to be of considerable interest given epidemiologic data linking CRP concentrations to early diabetogenesis and atherogenesis. Although recent work in diabetic and nondiabetic populations has linked polymorphism in the GCKR gene to plasma-triglyceride and glucose levels,13,14 we are not aware of prior data linking polymorphism in this same genetic locus to CRP level. However, the product of this gene is a regulatory protein that inhibits glucokinase in liver and pancreatic-islet cells, and mutations in the GCKR gene can result in defects in sensitivity of beta cells to glucose as a result of reduced glucose phosphorylation and limited hepatic storage of glucose as glycogen. As such, through its regulatory effect on the GCK protein, the GCKR gene has been considered a possible susceptibility-gene candidate for one type of maturity-onset diabetes of the young (MODY-2, MIM 606391).15,16 In this light, it is also of interest that a second loci described here as a genetic determinant of CRP, hepatic transcription factor-1 that codes for hepatic nuclear factor 1-alpha, is known to be functionally linked to a different type of MODY (MODY-3, MIM 606391), a form of non-insulin-dependent diabetes with an autosomal-dominant inheritance pattern with a young age of onset (typically < 25 years of age) and primary defects of insulin secretion.15 Moreover, given that the human CRP promoter is known to be activated by direct interaction with the HNF-1 protein,17 our finding in this genome-wide association study—that HNF1A polymorphism has a significant impact on CRP levels—would appear to have a strong functional basis.
We also believe our data regarding polymorphism in the leptin receptor protein to be compelling, not only because we replicate prior work demonstrating leptin to be an inflammatory modulator via genetic links to CRP18 but also because these data provide insights linking CRP to weight homeostasis. In humans, prior work has shown leptin levels to correlate with blood CRP levels and with vascular risk.19 Further, leptin at physiologic or pharmacologic doses increases plasma CRP, whereas reductions in leptin during weight loss and exercise are accompanied by reductions in plasma CRP. Interestingly, Chen et al. have recently demonstrated that leptin resistance can be induced through a direct binding of CRP to leptin and that infusion of human CRP into ob/ob mice blocks the clinical effects of leptin on satiety and weight reduction;20 this observation hints at a putative positive-feedback role for CRP in the pathophysiology of obesity. Regulation of adiposity also yields changes in adipose-derived cytokines such as IL-6, which have previously been shown in this cohort to correlate positively with CRP levels and to independently predict risk of future vascular events and diabetes.21 In this regard, our finding in the full cohort that polymorphism in the IL6R gene is an additional determinant of CRP expression is consistent with data linking IL-6 to hepatic production of CRP and suggesting a role for IL-6 in both incident diabetes and atherothrombosis.22 Moreover, the association reported here between IL6R and CRP is also consistent with recent data from the Health ABC study, in which IL6R was found to be significantly associated with plasma IL-6 levels.23
With regard to SNPs within or near the CRP gene and APOE locus, our data are consistent with prior work from candidate-gene studies that have linked CRP concentration to each of these genetic regions.6–10,24–26 Although we did not explicitly ascertain the SNPs that determine the canonical E2, E3, and E4 genotypes in the APOE region, these are known to associate with risk of myocardial infarction and premature atherothrombosis.27 Additional evidence links polymorphism in this region to dysfunction of bacterial lipid-antigen presentation in innate immunity28 and to the development of metabolic syndrome.29
The data described here suggest that most of the common genetic polymorphism that contributes to CRP level is closely related to genic pathways known to have an impact on insulin resistance, weight gain, beta-cell function, diabetes, and early atherogenesis. This is an intriguing observation, given that diet, exercise, glucose control, and statin therapy—the basic interventions for patients at high vascular risk due to metabolic syndrome—all reduce CRP levels. Recent work has identified polymorphisms within the CRP gene that associate with circulating CRP levels and with incident vascular events, data that support a potential causal role for CRP itself.30 Moreover, direct CRP inhibitors with apparent vascular-protective effects have recently been described.31 However, it remains controversial as to whether CRP itself is a target for therapy, and other data (albeit with limited power to define an informative null result) have not linked CRP polymorphism to incident events. On the basis of the inheritance patterns observed here, continued investigations into the potential mechanisms whereby CRP reduction might impact each of these interrelated disease states appear warranted.
Acknowledgments
This study was supported by grants from the National Heart, Lung, and Blood Institute and the National Cancer Institute (Bethesda, MD, USA), the Donald W Reynolds Foundation (Las Vegas, NV, USA), the Doris Duke Charitable Foundation, and the Fondation Leducq (Paris, France), with collaborative scientific and genotyping support provided by Amgen. P.M.R. is listed as a coinventor on patents held by the Brigham and Women's Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease.
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Ridker P.M., Cushman M., Stampfer M.J., Tracy R.P., Hennekens C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 2.Pradhan A.D., Manson J.E., Rifai N., Buring J.E., Ridker P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 3.Laaksonen D.E., Niskanen L., Nyyssonen K., Punnonen K., Tuomainen T.P., Valkonen V.P., Salonen R., Salonen J.T. C-reactive protein and the development of the metabolic syndrome and diabetes in middle-aged men. Diabetologia. 2004;47:1403–1410. doi: 10.1007/s00125-004-1472-x. [DOI] [PubMed] [Google Scholar]
- 4.Ridker P.M., Rifai N., Rose L., Buring J.E., Cook N.R. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N. Engl. J. Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 5.Han T.S., Sattar N., Williams K., Gonzalez-Villalpando C., Lean M.E., Haffner S.M. Prospective study of C-reactive protein in relation to the development of diabetes and metabolic syndrome in the Mexico City Diabetes Study. Diabetes Care. 2002;25:2016–2021. doi: 10.2337/diacare.25.11.2016. [DOI] [PubMed] [Google Scholar]
- 6.Hage F.G., Szalai A.J. C-reactive protein gene polymorphisms, C-reactive protein blood levels, and cardiovascular disease risk. J. Am. Coll. Cardiol. 2007;50:1115–1122. doi: 10.1016/j.jacc.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 7.Kathiresan S., Larson M.G., Vasan R.S., Guo C.Y., Gona P., Keaney J.F., Jr., Wilson P.W., Newton-Cheh C., Musone S.L., Camargo A.L. Contribution of clinical correlates and 13 C-reactive protein gene polymorphisms to interindividual variability in serum C-reactive protein level. Circulation. 2006;113:1415–1423. doi: 10.1161/CIRCULATIONAHA.105.591271. [DOI] [PubMed] [Google Scholar]
- 8.Crawford D.C., Sanders C.L., Qin X., Smith J.D., Shephard C., Wong M., Witrak L., Rieder M.J., Nickerson D.A. Genetic variation is associated with C-reactive protein levels in the Third National Health and Nutrition Examination Survey. Circulation. 2006;114:2458–2465. doi: 10.1161/CIRCULATIONAHA.106.615740. [DOI] [PubMed] [Google Scholar]
- 9.Carlson C.S., Aldred S.F., Lee P.K., Tracy R.P., Schwartz S.M., Rieder M., Liu K., Williams O.D., Iribarren C., Lewis E.C. Polymorphisms within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels. Am. J. Hum. Genet. 2005;77:64–77. doi: 10.1086/431366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller D.T., Zee R.Y., Suk Danik J., Kozlowski P., Chasman D.I., Lazarus R., Cook N.R., Ridker P.M., Kwiatkowski D.J. Association of common CRP gene variants with CRP levels and cardiovascular events. Ann. Hum. Genet. 2005;69:623–638. doi: 10.1111/j.1529-8817.2005.00210.x. [DOI] [PubMed] [Google Scholar]
- 11.Ridker P.M., Chasman D.I., Zee R.Y., Parker A., Rose L., Cook N.R., Buring J.E., for the Women's Genome Health Study Working Group. Rationale, design, and methodology of the Women's Genome Health Study: a genome-wide association study of more than 25000 initially healthy American women. Clin. Chem. 2008;54:249–255. doi: 10.1373/clinchem.2007.099366. [DOI] [PubMed] [Google Scholar]
- 12.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Saxena R., Voight B.F., Lyssenko V., Burtt N.P., de Bakker P.I., Chen H., Roix J.J., Kathiresan S., Hirschhorn J.N., Daly M.J. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 14.Sparso T., Anderson G., Nielsen T., Burgdorf K.S., Gjesing A.P., Nielsen A.L., Albrechtsen A., Rasmussen S.S., Jorgensen T., Borch-Johnsen K. The GCKR rs780094 polymorphism is associated with elevated fasting serum triacylglycerol, reduced fasting and OGTT-related insulinaemia, and reduced risk of type 2 diabetes. Diabetologia. 2008;51:70–75. doi: 10.1007/s00125-007-0865-z. [DOI] [PubMed] [Google Scholar]
- 15.Fajans S.S., Bell G.I., Polonsky K.S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med. 2001;345:971–980. doi: 10.1056/NEJMra002168. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Herrero C.M., Galan M., Vincent O., Flandez B., Gargallo M., Delgado-Alvarez E., Blazquez E., Navas M.A. Functional analysis of human glucokinase gene mutations causing MODY2: exploring the regulatory mechanisms of glucokinase activity. Diabetologia. 2007;50:325–333. doi: 10.1007/s00125-006-0542-7. [DOI] [PubMed] [Google Scholar]
- 17.Toniatti C., Demartis A., Monaci P., Nicosia A., Ciliberto G. Synergistic trans-activation of the human C-reactive protein promoter by transcription factor HNF-1 binding at two distinct sites. EMBO J. 1990;9:4467–4475. doi: 10.1002/j.1460-2075.1990.tb07897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y.Y., Gottardo L., Mlynarski W., Frazier W., Nolan D., Duffy J., Marescotti M.C., Gervino E.V., Johnstone M.T., Mantzoros C.S. Genetic variability at the leptin receptor (LEPR) locus is a determinant of plasma fibrinogen and C-reactive protein levels. Atherosclerosis. 2007;191:121–127. doi: 10.1016/j.atherosclerosis.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 19.Wallace A.M., McMahon A.D., Packard C.J., Kelly A., Shepherd J., Gaw A., Sattar N. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS) Circulation. 2001;104:3052–3056. doi: 10.1161/hc5001.101061. [DOI] [PubMed] [Google Scholar]
- 20.Chen K., Li F., Li J., Cai H., Strom S., Bisello A., Kelley D.E., Friedman-Einat M., Skibinski G.A., McCrory M.A. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat. Med. 2006;12:425–432. doi: 10.1038/nm1372. [DOI] [PubMed] [Google Scholar]
- 21.Ridker P.M., Hennekens C.H., Buring J.E., Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 22.Kado S., Nagase T., Nagato N. Circulating levels of interleukin-6, its soluble receptor and interleukin-6/interleukin-6 receptor complexes in patients with type 2 diabetes mellitus. Acta Diabetol. 1999;36:67–72. doi: 10.1007/s005920050147. [DOI] [PubMed] [Google Scholar]
- 23.Reich D., Patterson N., Ramesh V., De Jager P.L., McDonald G.J., Tandon A., Choy E., Hu D., Tamraz B., Pawlikowska L. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am. J. Hum. Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Judson R., Brain C., Dain B., Windemuth A., Ruano G., Reed C. New and confirmatory evidence of an association between APOE genotype and baseline C-reactive protein in dyslipidemic individuals. Atherosclerosis. 2004;177:345–351. doi: 10.1016/j.atherosclerosis.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Austin M.A., Zhang C., Humphries S.E., Chandler W.L., Talmud P.J., Edwards K.L., Leonetti D.L., McNeely M.J., Fujimoto W.Y. Heritability of C-reactive protein and association with apolipoprotein E genotypes in Japanese Americans. Ann. Hum. Genet. 2004;68:179–188. doi: 10.1046/j.1529-8817.2004.00078.x. [DOI] [PubMed] [Google Scholar]
- 26.Chasman D.I., Kozlowski P., Zee R.Y., Kwiatkowski D.J., Ridker P.M. Qualitative and quantitative effects of APOE genetic variation on plasma C-reactive protein, LDL-cholesterol, and apoE protein. Genes Immun. 2006;7:211–219. doi: 10.1038/sj.gene.6364289. [DOI] [PubMed] [Google Scholar]
- 27.Bennet A.M., Di Angelantonio E., Ye Z., Wensley F., Dahlin A., Ahlbom A., Keavney B., Collins R., Wiman B., de Faire U., Danesh J. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298:1300–1311. doi: 10.1001/jama.298.11.1300. [DOI] [PubMed] [Google Scholar]
- 28.van den Elzen P., Garg S., Leon L., Brigl M., Leadbetter E.A., Gumperz J.E., Dascher C.C., Cheng T.Y., Sacks F.M., Illarionov P.A. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature. 2005;437:906–910. doi: 10.1038/nature04001. [DOI] [PubMed] [Google Scholar]
- 29.Sima A., Iordan A., Stancu C. Apolipoprotein E polymorphism–a risk factor for metabolic syndrome. Clin. Chem. Lab. Med. 2007;45:1149–1153. doi: 10.1515/CCLM.2007.258. [DOI] [PubMed] [Google Scholar]
- 30.Lange L.A., Carlson C.S., Hindorff L.A., Lange E.M., Walston J., Durda J.P., Cushman M., Bis J.C., Zeng D., Lin D. Association of polymorphisms in the CRP gene with circulating C-reactive protein levels and cardiovascular events. JAMA. 2006;296:2703–2711. doi: 10.1001/jama.296.22.2703. [DOI] [PubMed] [Google Scholar]
- 31.Pepys M.B., Hirschfield G.M., Tennent G.A., Gallimore J.R., Kahan M.C., Bellotti V., Hawkins P.N., Myers R.M., Smith M.D., Polara A. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature. 2006;440:1217–1221. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- 32.McVean G.A., Myers S.R., Hunt S., Deloukas P., Bentley D.R., Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 33.Winckler W., Myers S.R., Richter D.J., Onofrio R.C., McDonald G.J., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–111. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]