

Abstract
Robust SNP genotyping technologies and data analysis programs have encouraged researchers in recent years to use SNPs for linkage studies. Platforms used to date have been 10 K chip arrays, but the possible value of interrogating SNPs at higher densities has been considered. Here, we present a genome-wide linkage analysis by means of a 500 K SNP platform. The analysis was done on a large pedigree affected with Parkinsonian-pyramidal syndrome (PPS), and the results showed linkage to chromosome 22. Sequencing of candidate genes revealed a disease-associated homozygous variation (R378G) in FBXO7. FBXO7 codes for a member of the F-box family of proteins, all of which may have a role in the ubiquitin-proteosome protein-degradation pathway. This pathway has been implicated in various neurodegenerative diseases, and identification of FBXO7 as the causative gene of PPS is expected to shed new light on its role. The performance of the array was assessed and systematic analysis of effects of SNP density reduction was performed with the real experimental data. Our results suggest that linkage in our pedigree may have been missed had we used chips containing less than 100,000 SNPs across the genome.
Main Text
Genome-wide linkage analyses of Mendelian diseases have traditionally been done with microsatellite markers.1,2 The potential value of using single-nucleotide polymorphisms (SNPs) in such analysis has been more recently proposed.3–7 Their advantages largely stem from their higher density and global distribution in the human genome. Robust SNP genotyping technologies and data analysis programs have encouraged researches to use SNP platforms in linkage studies.8–12 Platforms used to date have been 10 K chip arrays; however, the possible value of interrogating SNPs at a higher density in linkage analysis has been considered.11,13 Here, we present what is to the best of our knowledge the first genome-wide linkage analysis with a 500 K SNP platform. The analysis was performed on a large Iranian pedigree affected with Parkinsonian-pyramidal syndrome (PPS [MIM 260300]; Figure 1). It resulted in the identification of FBXO7 as the likely disease-causing gene. The performance of the array was assessed and systematic analysis of effects of SNP density reduction on information content, maximum LOD scores, and length of linked area was performed. We surmised that for our particular pedigree structure, linkage may well have been missed had we used chips containing less than 100,000 SNPs.
Figure 1.
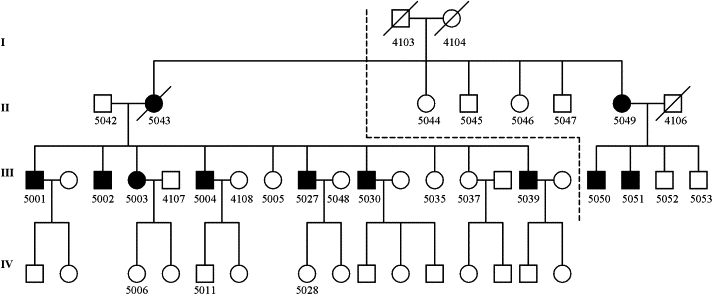
Parkinsonian-Pyramidal Syndrome Pedigree
Affected individuals are shown by shading. All individuals with number ID were included in pedigree file imported into MERLIN. Only living individuals whose ID numbers start with 5 were genotyped on the chips. Individuals 5042 and 5043 and individuals 5049 and 4106 were each reported to be distally related, but consanguinity between the individuals is not indicated because the exact relationship between the individuals could not be ascertained. The dashed line delineates the margin between the two minimized pedigrees.
PPS is a hypokinetic rigid disorder, the most common example of which is Parkinson's disease.14 PPS is a rare disorder that exhibits both Parkinsonian and pyramidal-associated symptoms. Approximately 20 patients have been reported in the literature.14–17 Symptoms, which may be vague in the beginning, start in young adulthood, progress relatively slowly, and may culminate in severe movement incapacity. Response to levadopa is usually dramatic and sustained for many years. Most, but not all, reported cases have been familial and associated with parental consanguinity, suggesting autosomal-recessive inheritance.14,16,18
The research was performed in accordance with the Helsinki Declaration and with approval of the ethics boards of the University of Tehran. All participants consented to participate after being informed of the nature of the research. Living members of the PPS pedigree were examined by two movement-disorder specialists, and many were also examined by specialists in other fields. Ten individuals dispersed in two generations were assessed to be affected with PPS (Figure 1). An 11th individual (5043) was assessed as having been affected based on phenotypic features reported by family members. All other living members of the pedigree were assessed to be normal.
Clinical information on the ten affected individuals is presented in Table 1. All affecteds exhibited equinovarus deformity since childhood, which is usually indicative of a genetic central nervous system disorder.19 None had symptoms associated with cerebellar malfunction. All exhibited Babinski signs, spasticity, and hyperactive DTR. The Babinski signs in all were initially unilateral, and later became bilateral. Spasticity was restricted to lower limbs in most patients. Onset of pyramidal symptoms in the patients was always in the third decade of life. At the time of writing, detectable extrapyramidal symptoms have evolved only in the three most severely affected individuals (5001, 5003, and 5027). These symptoms became evident 5 to 20 years after appearance of pyramidal symptoms. The extrapyramidal symptoms of patient 5027 were rapidly alleviated by L-dopa, and the response has been sustained for 4 years. Patients 5001 and 5003 were not cooperative in receiving treatment. None of the patients exhibited tremor, upgaze paresis, Myerson's sign, or dementia. Brain and spinal MRI, EEG, ocular examination, and EMG were performed on individuals 5001, 5027, and 5030; the results were normal. A complete laboratory metabolic workup on these patients, which included measurement of serum ceruloplasmin and copper level, did not show any biochemical abnormality. Movement anomalies and some other features of affected individuals are evident in video recordings and photograph (see Movies S1–S4 and Figure S1 available online). Reasons by which diseases other than Parkinsonian-pyramidal syndrome were excluded are given in Table S2.
Table 1.
Clinical Features of Affected Individuals
|
Extrapyramidal Symptoms |
Pyramidal Symptoms |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Rigidity | Bradykinesia | Hypomimia | Monotone Speech | Babinski Signs | Spasticity | Hyperactive DTR | Scissor Gait | Equinovarus Deformity | |
| 5001∗ | + | + | + | + | + | + | + | unable to walk | + |
| 5002 | - | - | - | - | + | + | + | - | + |
| 5003∗ | + | + | + | unable to speak | + | + | + | unable to walk | + |
| 5004 | - | - | - | - | + | + | + | - | + |
| 5027∗ | + | + | + | + | + | + | + | + | + |
| 5030 | - | - | - | - | + | + | + | - | + |
| 5039 | - | - | - | - | + | + | + | - | + |
| 5049 | - | - | - | - | + | + | + | - | + |
| 5050 | - | - | - | - | + | + | + | - | + |
| 5051 | - | - | - | - | + | + | + | - | + |
∗The most severely affected individuals.
Genomic DNA was prepared from venous blood of 10 affected and 14 unaffected members of the pedigree. Genome-wide linkage scan was performed on these DNAs with the GeneChip Human Mapping 500K Array Set according to the manufacturer's recommended protocol (Affymetrix, Santa Clara, CA). Arrays were processed through Affymetrix microfluidics stations, and images were obtained with an Affymetrix Gene Array scanner. Raw microarray feature intensities were processed with GeneChip Genotyping Analysis Software v.4.1.0.26 (GTYPE) to derive SNP genotypes. GTYPE uses the Bayesian Robust Linear Model with Mahalanobis distance classifier (BRLMM) algorithm for genotype calling.
SNP call rate with BRLMM for the 24 individuals tested ranged from 98.12% to 99.62% and was 99.15% on the average. Across all genotype calls of all individuals, 26% (range 25%–27%) were heterozygous. Concordance of the genotypes of the 50 SNPs shared on the two arrays of the 24 individuals genotyped was 99.76%. On the average, 0.58% of 10,524 X chromosome SNPs were genotyped as heterozygous in each of the 14 male individuals genotyped. Often, the same SNP was miscalled in several male individuals. Genotypic information was obtained for the mother (5049) of only four of these male individuals, and these four were siblings. Approximately 7000 homozygous X chromosome SNPs were observed in this female individual, and alternate allele genotype calls for these SNPs in her sons allows for detection of another set of miscalls. It was estimated that 0.1% of these SNPs were miscalled per individual in this part of the pedigree. By extrapolation, the transmission error rate per generation for the entire data set is expected to be approximately 0.7%. In fact, GTYPE detected Mendelian errors in 0.06% of the SNPs dispersed throughout the genome per individual genotyped. By comparison with the more telling SNPs of the X chromosome, the figures suggest that only about 10% of the miscalls on the autosomal chromosomes are detected. GTYPE removed SNPs that were detected as Mendelian errors. It subsequently removed 42% of the remaining SNPs because only one allele for each was observed in our entire pedigree, or because of a call rate less than 90% among all the individuals. The number of remaining SNPs was 286,508.
For genetic analysis, an appropriate GTYPE option was used to automatically export by chromosome genotype calls into MERLIN.20 Because of the large size of the pedigree, it was split into two smaller pedigrees for analysis (Figure 1). Individuals 5042 and 5043 were defined as the ancestors of one of the minimized pedigrees, and individuals 4103 and 4104 as the ancestors of the other. Individual 5043 was included only in the first minimized pedigree. The PedWipe option in MERLIN assessed 0.3% (based on chromosome 22 data) of the calls unlikely to be correct and removed these. MERLIN was then used to assess information content of the arrays and to perform multipoint nonparametric (NPL) and parametric linkage analysis. Inheritance of PPS in affected kindreds has been proposed to be autosomal recessive, but sporadic cases have also been reported and the possibility that this condition may be heterogeneous has been suggested.14–16 The rarity of the condition, the inbreeding reported in our pedigree (especially in generations I, II, and before), and the absence of disease among individuals of generation IV are strongly suggestive of an autosomal-recessive mode of inheritance in this pedigree. On the other hand, the possibility that onset of symptoms had not yet manifested in some young generation IV individuals, ambiguity of consanguinity between parents of affected individuals, and the observation of disease in consecutive generations made it unwise to rule out possible dominant inheritance.
Merlin calculated a nonparametric logarithm of odds (LOD) score by use of the Kong and Cox exponential model.21 MERLIN also generated parametric LOD scores under assumption of disease allele frequency of 0.0001 and full penetrance. Because of limitations of computer memory, data of three unaffected individuals in generation IV (individuals 5006, 5011, and 5028) and the unaffected mother of one of these (individual 5048) were not included in the original analysis. The information content and results of scans for all the chromosomes under the NPL, autosomal-recessive, and autosomal-dominant models based on data of 20 individuals are shown in Figures S2–S5. After having focused on chromosome 22, data on all 24 individuals were included for the analysis of this relatively small chromosome. The average information content for each of the 23 chromosomes was notable and ranged from 0.948 to 0.996. Maximum potential LOD score calculated for our pedigree structure by MERLIN under NPL was 4.21 (Z = 9.806; p = 10−5).20 NPL analysis resulted in LOD scores of greater-than-suggestive LOD score 2.1 (p < 0.001) on chromosomes 3, 5, 7, 11, 14, 15, and 22, and LOD scores greater-than-significant LOD score of 3.3 (p < 0.00005) on chromosomes 5, 11, 14, and 22.22 Because the peaks of chromosomes 5, 11, and 14 each included at most 6 SNPs and covered a maximal range of 0.2 cM, it was assumed they were probably due to erroneous genotypings not detected by the various error-checking protocols. On chromosome 22, a broad region extending from 4.17 to 28.18 cM was associated with an average LOD score of 2.90, and another region extending from 34.40 to 41.93 cM (28,934,667 bp–34,951,655 bp) was associated with an average LOD score of 4.08 (average p = 0.00003) (Figure 2A). Within the latter region, the maximum predicted LOD score of 4.21 was observed in region extending from 36.58 to 39.98 cM. Parametric analysis under an autosomal-dominant model resulted in only negative LOD scores on all chromosomes except 22, on which an average score of 0.81 was achieved at region extending from 34.40 to 35.97 cM (maximum LOD = 1.57 at 34.57 cM) (Figure 2A). Finally, analysis under an autosomal-recessive model resulted in positive LOD scores on chromosomes 5 (average LOD of peak = 3.00), 15 (average LOD = 1.18), and 22. The region on chromosome 22 was broad and extended from 30.48 to 56.24 cM; the average associated LOD score was 2.825 (Figure 2A). Within this region, there was a peak extending from 34.50 cM to 36.88 cM (29,055,302 bp–31,414,345 bp), with an average associated LOD score of 3.32. The maximum score achieved within the region was 3.39. The peak region on chromosome 22 as compared to the peak region of chromosome 5 was considered a better candidate region because a significantly overlapping region of chromosome 5 was not observed under the NPL model. Furthermore, it was observed that the genotypes of the affected individuals for many of the SNPs in the chromosome 5 region were heterozygous. For example, at the SNP position associated with the maximum LOD score (3.38), seven of the ten affected individuals genotyped were heterozygous. Twenty simulations for each of the inheritance models by means of the chromosome 22 genotypes resulted in maximum LOD scores of 1.54, −5.87, and −1.08, respectively, for the NPL, autosomal-dominant, and autosomal-recessive models (not shown). The information content across the length of chromosome 22 approached 1, and the average was 0.964 (Figure 2B). Visual inspection, in fact, showed that all affected individuals and only affected individuals shared a homozygous genotype for all 209 SNPs within the region (2.38 cM) having the average LOD value of 3.32. It was surmised that the locus most likely associated with the disease status was on chromosome 22 and that its mode of inheritance in the pedigree was autosomal recessive. Interestingly, results of haplotyping of chromosome 22 by MERLIN revealed that one of the recombination events that delimited the linked region occurred in five meioses. This event occurred between rs134176 and rs738996, suggesting a recombination hotspot. Reanalysis of chromosome 22 data by means of clusters at r2 values of 0.4 produced NPL, autosomal-dominant, and autosomal-recessive LOD plots nearly identical to those produced without clustering (not shown).
Figure 2.
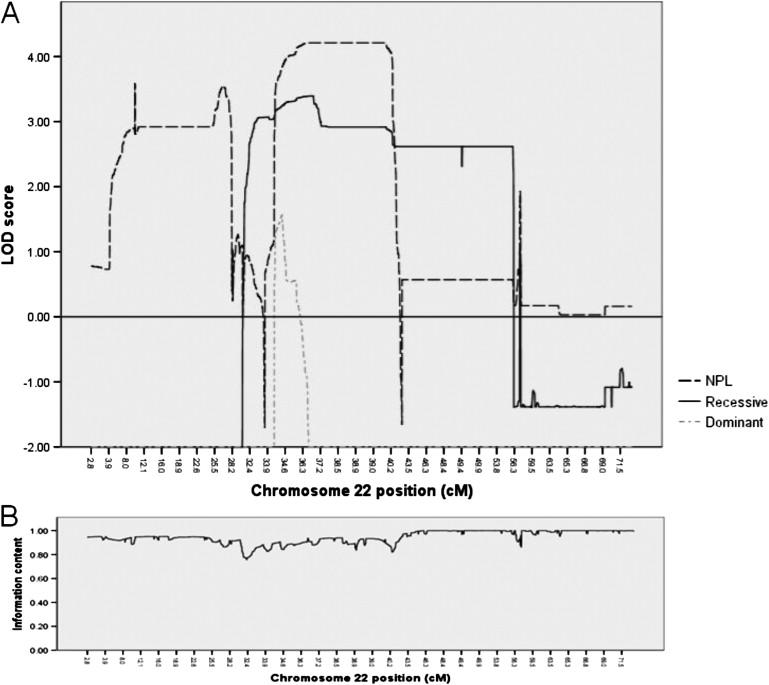
LOD Plots and Information Content for Chromosome 22
(A) The LOD plots under NPL, autosomal-dominant, and autosomal-recessive models of disease inheritance are shown.
(B) Information content for chromosome 22.
Annotations provided by GTYPE and the NCBI database were used to identify genes within chromosomal regions that showed linkage. The critical region of 34.50–36.88 cM on chromosome 22 contains 34 annotated genes. To prioritize the region to be examined, we reasoned that the region containing the causative gene should cause a drop in LOD score under the dominant model, because at least nine unaffected individuals were expected to carry one copy of the disease-causing allele. Furthermore, we expected the disease-causing gene to be included in the region associated with a high LOD score under the NPL model. The right border of the linked region under the dominant model was at 35.97 cM, and the overlap between the critical region under the recessive model and the region having an average LOD score of 4.08 under the NPL model extended from 36.00 cM to 36.88 cM (Figure 3). Four genes are annotated in this region of chromosome 22: ret finger protein-like 3 (RFPL3), C22orf28 (alias HSPC117), bactericidal/permeability-increasing protein-like 2 (BPIL2), and F-box protein 7 (FBXO7). Part of synapsin III (SYN3) also lies within the region, and TIMP metallopeptidase inhibitor 3 (TIMP3) lies just downstream. C22orf28, FBXO7 (MIM 605648), SYN3 (MIM 602705), and TIMP3 (MIM 188826) were considered to be good biological candidate genes for the disease phenotype of our pedigree.23–28
Figure 3.

Prioritization of Linkage to Region of 0.9 cM on Chromosome 22
Regions on chromosome 22 showing maximum linkage to disease phenotype under NPL (34.40–41.93 cM), autosomal-dominant (34.40–35.97 cM), and autosomal-recessive (34.49–36.88 cM) models are shown as overlapping regions. The solid gray bar (36.00–36.88 cM) indicates region prioritized for selection of candidate genes.
The exons and flanking intronic sequences of these four genes were amplified from the DNA of patient 5001 by PCR and subsequently sequenced with the ABI Big Dye terminator chemistry and an ABI Prism 3700 instrument (Applied Biosystems, Foster City, CA). Sequences were analyzed with the Sequencher software (Gene Codes Corporation, Ann Arbor, MI) (Table S1). The frequencies of alleles at variant positions in different populations were obtained from the HapMap site when available. Predicted effects of variant sequences on splicing were determined by comparison with known canonical splice site motifs by means of NNsplice 0.9 and genescan. For determination of extent of conservation of amino acids altered due to nucleotide variations found, the amino acid sequences of homologous proteins from other species were obtained from NCBI and aligned with the ClustalW 1.83 software.
Five sequence variations in the homozygous state were observed in C22orf28, six in FBXO7, three in SYN3, and none in TIMP3 (Table 2). Except for g.18550C→G in FBXO7, the observed variations were considered not to be associated with disease status. They were assessed not to affect splicing, and most were also reported at relatively high frequencies in other populations (0.1–0.9).
Table 2.
Sequence Variations
| Gene | Gene Locationa | cDNA Locationb | Effect on Proteinc | rs Numberd | Allele Frequency Ranged,e |
|---|---|---|---|---|---|
| C22orf28 | g.3571A→G | c.172+77A→G | - | rs2076044 | 0.15–0.53 |
| g.3580C→T | c.172+86C→T | - | rs2076043 | 0.67–0.90 | |
| g.3923C→T | c.173-84C→T | - | rs5754073 | 0.67–0.90 | |
| g.17233T→G | c.1179+10T→G | - | rs2072818 | 0.48–0.79 | |
| g.20160A→T | c.1410+151T→G | - | rs5994562 | 0.09–0.54 | |
| FBXO7 | g.677T→G | c.122+272T→G 1 | p.L12R 2 | rs8137714 | - |
| c.35T→G 2 | |||||
| g.736C→A | c.122+331C→A 1 | - | rs8140067 | 0.1–22 | |
| c.37+57C→A 2 | |||||
| g.4484G→A | c.343G→A 1 | p.M115I 1 | rs11107 | 0.36–0.68 | |
| c.233G→A 2 | p.M36I 2 | ||||
| g.16292T→C | c.872-75T→C1 | - | rs738982 | 0.36–0.68 | |
| c.635-75T→C2 | |||||
| g.16444C→T | c.949C→T 1 | p.L317L 1 | rs9726 | 0.36–0.68 | |
| c.712C→T 2 | p.L238L 2 | ||||
| g.18550C→G | c.1132C→G 1 | p.R378G 1 | ss99938574 | NA | |
| c.895C→G 2 | p.R299G 2 | ||||
| SYN3 | g.75518C→A | c.461+209C→A IIIa,IIIc | - | rs183588 | 0–0.12 |
| g.477951G→C | c.1230+128G→C IIIa,IIIc | - | rs135123 | 0.1–0.19 | |
| g.493006C→T | c.∗1C→T IIIa | - | ss99938576 | NA | |
| c.∗118C→T IIIc | - | ||||
Putative disease-associated variation is shown in bold. NA, not available.
Gene positions in C22orf28, FBXO7, and SYN3 are, respectively, with reference to sequences NT_011520.11 (nucleotides 12198804-12174138), NT_011520.11 (nucleotides 12261276-12285387), and NT_011520.11 (nucleotides 12793252-12299109).
cDNA positions in C22orf28 are with reference to sequence NM_014306.3. cDNA positions in FBXO7 are with reference to sequence NM_012179.3 for isoform 1 and and sequence NM_001033024.1 for isoform 2. cDNA positions in SYN3 are with reference to sequence NM_003490.2 for isoform IIIa and sequence NM_133633.1 for isoform IIIc.
Protein positions in F-box only protein 7 are with reference to sequence NP_036311.3 for isoform 1 and sequence NP_001028196.1 for isoform 2. Superscripts of cDNA and amino acid variations refer to respective isoforms.
NCBI Build 36.2 was used to obtain all reference sequences.
Range of frequency of the varied alleles observed in pedigree reported for populations of HapMap project. No sequence variations were observed in sequenced regions of gene TIMP3.
FBXO7 codes two cDNA and protein isoforms. The isoform 1 protein is longer than the isoform 2, but amino acid 92 to amino acid 522 of the former correspond exactly to amino acids 7 to 443 of the latter. Because references to FBXO7 in the literature are generally with respect to isoform 1 of the cDNA and protein, we will henceforth make references with respect to this isoform.29–31 The c.1132C→G change results in the nonconservative amino acid substitution of glycine for arginine at position 378 (R378G) in the F-box protein 7. Arginine at position 378 is completely conserved in all F-box protein 7 proteins sequenced, including the protein in species as distally related as humans and the purple sea urchin (Table S3). Furthermore, it lies within a well-conserved region of the protein and within a completely conserved -RDF- motif. By sequencing, the variant allele was also observed in the homozygous state in three other affected individuals (5003, 5049, and 5051) and in the heterozygous state in one unaffected obligate carrier (5005). By an ARMS-PCR target mutation analysis assay, it was shown that the remaining six available affected pedigree members each carried two mutant alleles and that eight obligate carriers each carried one mutant allele. These data showed segregation of R367G with the disease phenotype in an autosomal-recessive pattern in the pedigree. The mutant allele was not observed in the chromosomes of 800 control individuals. 600 of the controls were unrelated healthy Iranian individuals over the age of 60 without self-reported movement or neurological disorders, and 200 were unrelated Iranians diagnosed with Parkinson's disease.
Our results suggest that the mutation causing the R378G substitution in the FBXO7 protein is the cause of the disease phenotype exhibiting pyramidal and extrapyramidal anomalies in our pedigree. The condition was inherited in an autosomal-recessive fashion. FBXO7 codes for a member of the F-box family of proteins. F-box proteins all contain an F-box motif, a motif consisting of ∼40 amino acids that is functionally defined as one that can interact with the Skp1 component of the SCF (Skp1, Cdc53/Cullin1, F-box) protein complex.30,32–34 FBXO7 also contains a proline-rich region (PRR) that is responsible for binding discs, large homolog 7 (DLG7, alias HURP), a known substrate of FBXO7.30 The R378G mutation lies at or near the right border of the F-box motif and approximately 45 amino acids upstream of the left border of the PRR motif and may disrupt the function of either or both of these (Figure 4). Interactions of FBXO7 with proteins other than DLG7 have also been reported and it has been suggested that some of the effects of FBXO7 are not mediated by ubiquitin-mediated degradation of the proteins with which it interacts.35 And, of course, ubiquinitation may affect cellular functions other than proteolysis.36,37
Figure 4.

Schematic View of F-Box Only Protein 7, Isoform 1
Numbers on top indicate amino acid residue positions of left and right borders of F-box and proline-rich regions. The position of putative disease associated variation is shown below.
The role of FBXO7 in neurons is not known. However, mutations affecting proteins involved in the ubiquitin-proteolytic pathway have been associated with various neurodegenerative diseases, including Parkinson's and Alzheimer's diseases, signifying the importance of this pathway in neuron function.24 Mutations in FBXO7 have not been observed in any of the aforementioned diseases. Functional studies need to be performed to confirm and expand on the genetic findings reported here.
The analysis performed on our pedigree produced data that allowed evaluation of the performance of the Affymetrix 500 K chips for linkage analysis. Although GTYPE seems to detect only a small fraction of the SNP miscalls, the number of miscalls in our analysis was sufficiently low not to preclude detection of linkage in our pedigree. As reported by others, the PedWipe option in MERLIN was very effective in removing faulty peaks caused by one or few SNPs (not shown). Consideration of effect of linkage disequilibrium (LD) on LOD scores is an issue when high-density marker platforms are used. However, this consideration is most important in sib pair analysis and multipedigree analysis, especially when parental genotypes are not available.4,38–41 The effect is least important in linkage analysis by means of a single pedigree, as is the case in the present study. MERLIN allows for consideration of linkage disequilibrium by use of the Cluster option. Clustering effectively identifies adjacent markers that are in LD as a single marker and thus minimizes any possible effect LD may have on parameter being queried, i.e., LOD score. As expected, clustering had minimal effects on the LOD plots of our analysis.
Having identified the putative disease-causing gene, we attempted to ascertain the value of using a dense SNP map in our analysis under NPL and recessive models by systematically reducing the number of SNPs (Figures 5 and 6). A program was written in PERL for SNP reduction that allowed SNP selections starting at designated SNP and multiplier. The multiplier determines the number of SNPs to be removed between consecutive SNPs to be kept. Linkage analysis was repeated only for chromosome 22 SNPs while reducing the number of SNPs by factors ranging between 2 and 100. The average information content was reduced notably (Figure S6) and became uneven (not shown) across the length of chromosome 22 at greater than 20-fold reductions. In both models, the maximum LOD score progressively decreased with reduction in density of SNPs, but the effect was more pronounced in the NPL model (Figure 5A). Finally, length of linked region at greater than 20-fold reductions under the NPL model increased notably and became more sporadic (Figure 5B). A similar pattern was observed under the recessive model (Figure 5C). As SNP number reduction increased and cut off below maximum LOD decreased, it became increasingly probable that the critical region containing FBXO7 would not be included in selected region under the NPL model (Figure 5B). In addition to these effects, at reductions greater than 20-fold, spurious peaks in other regions of chromosome 22 were observed (not shown).
Figure 5.
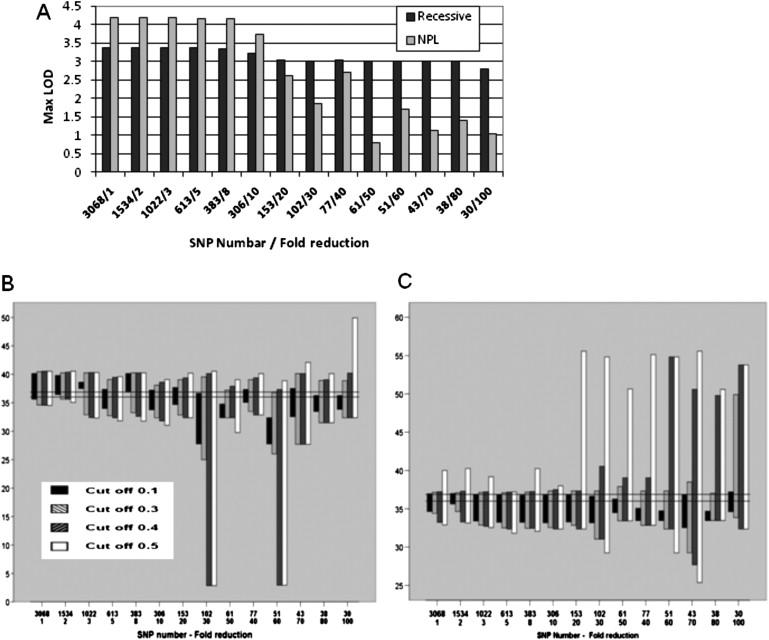
Effects of Reduction of Chromosome 22 SNP Numbers
(A) Effect on maximum LOD scores under NPL and recessive models of inheritance.
(B and C) Effect on position and length of linked area with cut offs of 0.1, 0.3, 0.4, and 0.5 below maximum LOD scores under NPL (B) and recessive (C) models.
The y axis shows cM along chromosome 22. The horizontal grid shows the 36.00–36.88 cM region.
Figure 6.
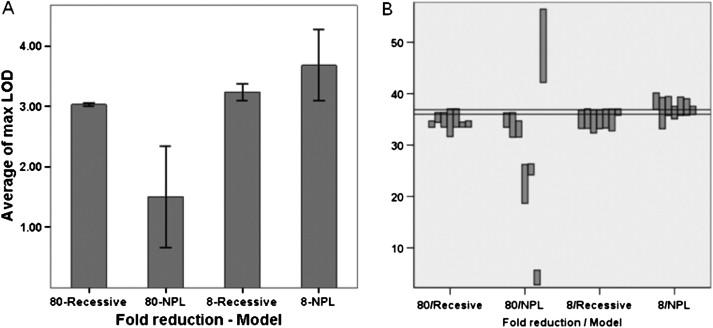
Effects of Change In-Frame of SNP Reductions
(A) Effect on maximum LOD score. Bars indicate ± 1 standard deviation.
(B) Effect on length of linked region. The linked region is defined by cut off of 0.1 below maximum LOD score.
The y axis shows cM along chromosome 22. The horizontal grid shows the 36.00–36.88 cM region.
The 80-fold and 8-fold reduction of SNPs produce, respectively, platforms comparable to 10 K and 100 K chips. In order to consider possible consequence of using different combination of SNPs, these two levels of reductions were each performed in several frames (Figure 6). All seven possible frames for the 8-fold reduction and the first seven frames for the 80-fold reduction were tested. The complement of SNPs included had minimal effects on information content (not shown). The average maximum LOD score for the 8-fold reductions under the NPL and recessive models were 3.68 ± 0.59 and 3.2 ± 0.14, respectively (Figure 6A). The trend of less variation in maximum LOD scores under the recessive model was more notable at 80-fold reductions of SNP numbers. The average maximum LOD scores under NPL and recessive models were, respectively, 1.5 ± 0.84 and 3.0 ± 0.02. Interestingly, the selected 36.00 cM to 36.88 cM region lies within the linked region of all 8-fold reduction frames under the recessive model and within seven of the 8-fold reductions under the NPL model. Among the ten 80-fold reductions tested, the gene lies within the linked area in only two frames under the recessive model and in none under the NPL model. Linked regions were notably sporadic with 80-fold reductions under the NPL model (Figure 6B).
Based on these observations, we conclude that for our pedigree, it may have proved unproductive to use chips containing less than 100,000 SNPs spread across the genome. The conclusion may not be valid for all linkage analysis with single pedigrees, but should be taken into consideration because the costs associated with the use of dense chips has decreased. In linkage analysis with a larger number of pedigrees, the fraction of SNPs that are uninformative because of only one allele being observed throughout the pedigrees is expected to decrease, and chips of less density may perform adequately in such studies.
Acknowledgments
We thank the patients and their family members for consenting to participate in this study, Kaveh Moini for writing the SNP filtering program, and Ali Katanforoush and Alireza Saleh for computer access. We acknowledge the Iran National Science Foundation; the Center of Excellence in Biomathematics, School of Mathematics, University of Tehran; and Iranian Studies for Parkinson's Disease Grant (5187 Iran PD genetics) for funding this research. All authors declare absence of conflict of interests.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Berkeley Drosophila Genome Project, http://www.fruitfly.org/seq_tools/splice.html
ClustalW2, http://www.ebi.ac.uk/clustalw
GENSCAN, http://genes.mit.edu/GENSCAN.html
International HapMap Project, http://www.hapmap.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
SNP filter program, http://www.fos.ut.ac.ir/elahehelahi/Programs/tabid/2964/Default.aspx
References
- 1.Sheffield V.C., Weber J.L., Buetow K.H., Murray J.C., Even D.A., Wiles K., Gastier J.M., Pulido J.C., Yandava C., Sunden S.L. A collection of tri-and tetranucleotide repeat markers used to generate high quality, high resolution human genomewide linkage maps. Hum. Mol. Genet. 1995;4:1837–1844. doi: 10.1093/hmg/4.10.1837. [DOI] [PubMed] [Google Scholar]
- 2.Weissenbach J., Gyapay G., Dib C., Vignal A., Morissette J., Millasseau P., Vaysseix G., Lathrop M. A second-generation linkage map of the human genome. Nature. 1992;359:794–801. doi: 10.1038/359794a0. [DOI] [PubMed] [Google Scholar]
- 3.Goddard K.A., Wijsman E.M. Characteristics of genetic markers and maps for cost-effective genome screens using diallelic markers. Genet. Epidemiol. 2002;22:205–220. doi: 10.1002/gepi.0177. [DOI] [PubMed] [Google Scholar]
- 4.Kruglyak L. The use of a genetic map of biallelic markers in linkage studies. Nat. Genet. 1997;17:21–24. doi: 10.1038/ng0997-21. [DOI] [PubMed] [Google Scholar]
- 5.Matise T.C., Sachidanandam R., Clark A.G., Kruglyak L., Wijsman E., Kakol J., Buyske S., Chui B., Cohen P., de Toma C. A 3.9-centimorgan-resolution human single-nucleotide polymorphism linkage map and screening set. Am. J. Hum. Genet. 2003;73:271–284. doi: 10.1086/377137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson A.F., Sorant A.J. Equivalence of single-and multilocus markers: power to detect linkage with composite markers derived from biallelic loci. Am. J. Hum. Genet. 2000;66:1610–1615. doi: 10.1086/302889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woods C.G.V.E.M., Bond J., Roberts E. A new method for autozygosity mapping using single nucleotide polymorphisms (SNPs) and excludeAR. J. Med. Genet. 2004;41:e101. doi: 10.1136/jmg.2003.016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abd El-Aziz M.M., El-Ashry M.F., Chan W.M., Chong K.L., Barragan I., Antiñolo G.A., Pang C.P., Bhattacharya S.S. A novel genetic study of Chinese families with autosomal recessive retinitis pigmentosa. Ann. Hum. Genet. 2006;71:281–294. doi: 10.1111/j.1469-1809.2006.00333.x. [DOI] [PubMed] [Google Scholar]
- 9.Middleton F.A., Pato M.T., Gentile K.L., Morley C.P., Zhao X., Eisener A.F., Brown A., Petryshen T.L., Kirby A.N., Medeiros H. Genomewide linkage analysis of bipolar disorder by use of a high-density single-nucleotide–polymrphism (SNP) genotyping assay: a comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am. J. Hum. Genet. 2004;74:886–897. doi: 10.1086/420775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paterson A.D., Wang X.-Q.L.K., Magistroni R., Song X., Kappel J., Klassen J., Cattran D., George-Hyslop P.S., Pei Y. Genome-wide linkage scan of a large family with IgA nephropathy localizes a novel susceptibility locus to chromosome 2q36. J. Am. Soc. Nephrol. 2007;18:2408–2415. doi: 10.1681/ASN.2007020241. [DOI] [PubMed] [Google Scholar]
- 11.Sellick G.S., Longman C., Tolmie J., Newbury-Ecob R., Geenhalgh L., Hughes S., Whiteford M., Garrett C., Houlston R.S. Genomewide linkage searches for Mendelian disease loci can be efficiently conducted using high-density SNP genotyping arrays. Nucleic Acids Res. 2004;32:e164. doi: 10.1093/nar/gnh163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vance C., Al-Chalabi A., Ruddy D., Smith B.N., Hu X., Sreedharan J., Siddique T., Schelhaas H.J., Kusters B., Troost D. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 13.Evans D.M., Cardon L.R. Guidelines for genotyping in genomewide linkage studies: single-nucleotide–polymorphism maps versus microsatellite maps. Am. J. Hum. Genet. 2004;75:687–692. doi: 10.1086/424696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nisipeanu P.D., Korczyn A. The Parkinsonian-Pyramidal Syndrome. In: Joseph A.B., Young R.R., editors. Movement Disorders in Neurology and Neuropsychiatry. Blackwell Science; Malden, MA: 1999. pp. 247–250. [Google Scholar]
- 15.Kalita J., Misra U., Das B. Sporadic variety of pallido-pyramidal syndrom. Neurol. India. 2003;51:383–384. [PubMed] [Google Scholar]
- 16.Panagariya A., Sharma B., Dev A. Pallido-Pyramidal syndrome: a rare entity. Indian J. Med. Sci. 2007;61:156–157. [PubMed] [Google Scholar]
- 17.Sirvastava T., Goyal V., Singh S., Shukla G., Behari M. Pallido-pyramidal syndrome with blepharospasm and good response to levadopa. J. Neurol. 2005;252:1537–1538. doi: 10.1007/s00415-005-0852-6. [DOI] [PubMed] [Google Scholar]
- 18.Tranchant C., Boulay C., Warter J. Le syndrome pallido-pyrimidal: une entité méconnue. Rev. Neurol. 1991;147:308–310. [PubMed] [Google Scholar]
- 19.Jankovic J., Tolosa E. Lippincott Williams & Wilkins; Philadelphia: 2007. Parkinson's Disease & Movement Disorders. [Google Scholar]
- 20.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 21.Kong A., Cox N. Allele-sharing models: LOD scores and accurate linkage tests. Am. J. Hum. Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lander E., Kruglyak L. Genetic dissection of complex traits: guidlines for interpreting and reporting linkage results. Nature. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 23.Apte S., Olsen B., Murphy G. The gene structure of tissue inhibitor of metalloproteinases (TIMP)-3 and its inhibitory activities define the distinct TIMP gene family. J. Biol. Chem. 1995;270:14313–14318. doi: 10.1074/jbc.270.24.14313. [DOI] [PubMed] [Google Scholar]
- 24.Ciechanover A., Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 25.Hosaka M., Südhof T. Synapsin III, a novel synapsin with an unusual regulation by Ca2+ J. Biol. Chem. 1998;273:13371–13374. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- 26.Jin J., Cardozo T., Lovering R.C., Elledge S.J., Pagano M., Harper J.W. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanai Y., Dohmae N., Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 28.Kao H., Porton B., Czernik A., Feng J., Yiu G., Häring M., Benfenati F., Greengard P. A third member of the synapsin gene family. Proc. Natl. Acad. Sci. USA. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang Y.-F., Cheng C.-M., Chang L.-K., Jong Y.-J., Yuo C.-Y. The F-box protein Fbxo7 interacts with human inhibitor of apoptosis protein cIAP1 and promotes cIAP1 ubiquitination. Biochem. Biophys. Res. Commun. 2006;342:1022–1026. doi: 10.1016/j.bbrc.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 30.Hsu J.-M., Lee Y.-C.G., Yu C.-T.R., Huang C.-Y.F. Fbx7 functions in the SCF complex regulating Cdk1-Cyclin B-phosphorylated hepatoma up-regulated protein (HURP) proteolysis by a proline-rich region. J. Biol. Chem. 2004;279:32592–32602. doi: 10.1074/jbc.M404950200. [DOI] [PubMed] [Google Scholar]
- 31.Laman H. Fbxo7 gets proactive with cyclin D/cdk6. Cell Cycle. 2006;5:279–282. doi: 10.4161/cc.5.3.2403. [DOI] [PubMed] [Google Scholar]
- 32.Bai C., Sen P., Mathias N., Hofmann K., Ma L., Goebl M., Harper J., Elledge S. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 33.Cenciarelli C., Chiaur D.S., Guardavaccaro D., Parks W., Vidal M., Pagano M. Identification of a family of human F-box proteins. Curr. Biol. 1999;9:1177–1179. doi: 10.1016/S0960-9822(00)80020-2. [DOI] [PubMed] [Google Scholar]
- 34.Winston J.T., Koepp D.M., Zhu C., Elledge S.J., Harper J.W. A family of mammalian F-box proteins. Curr. Biol. 1999;9:1180–1182. doi: 10.1016/S0960-9822(00)80021-4. [DOI] [PubMed] [Google Scholar]
- 35.Laman H., Funes J.M., Ye H., Henderson S., Galinanes-Garcia L., Hara E., Knowles P., McDonald N., Boshoff C. Transforming activity of Fbxo7 is mediated specifically through regulation of cyclin D/cdk6. EMBO J. 2005;24:3104–3116. doi: 10.1038/sj.emboj.7600775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haglund K., Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shcherbik N., Haines D. Ub on the move. J. Cell. Biochem. 2004;93:11–19. doi: 10.1002/jcb.20130. [DOI] [PubMed] [Google Scholar]
- 38.Huang Q., Shete S., Amos C.I. Ignoring linkage disequilibrium among tightly linked markers induces false-positive evidence of linkage for affected sib pair analysis. Am. J. Hum. Genet. 2004;75:1106–1112. doi: 10.1086/426000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaid D.J., McDonnell S.K., Wang L., Cunningham J.M., Thibodeau S.N. Caution on pedigree haplotype inference with software that assumes linkage equilibrium. Am. J. Hum. Genet. 2002;71:992–995. doi: 10.1086/342666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Consortium T.A.G.P. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levinson D.F., Holmans P. The effect of linkage disequilibrium on linkage analysis of incomplete pedigrees. BMC Genet. 2005;6:S6. doi: 10.1186/1471-2156-6-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.