

Abstract
Deficiency of carbohydrate sulfotransferase 3 (CHST3; also known as chondroitin-6-sulfotransferase) has been reported in a single kindred so far and in association with a phenotype of severe chondrodysplasia with progressive spinal involvement. We report eight CHST3 mutations in six unrelated individuals who presented at birth with congenital joint dislocations. These patients had been given a diagnosis of either Larsen syndrome (three individuals) or humero-spinal dysostosis (three individuals), and their clinical features included congenital dislocation of the knees, elbow joint dysplasia with subluxation and limited extension, hip dysplasia or dislocation, clubfoot, short stature, and kyphoscoliosis developing in late childhood. Analysis of chondroitin sulfate proteoglycans in dermal fibroblasts showed markedly decreased 6-O-sulfation but enhanced 4-O-sulfation, confirming functional impairment of CHST3 and distinguishing them from diastrophic dysplasia sulphate transporter (DTDST)-deficient cells. These observations provide a molecular basis for recessive Larsen syndrome and indicate that recessive Larsen syndrome, humero-spinal dysostosis, and spondyloepiphyseal dysplasia Omani type form a phenotypic spectrum.
Main Text
Proteoglycans rank among the most important components of connective tissues. They consist of glycosaminoglycan (GAG) polymer chains attached to core proteins. The sugar composition of the glycosaminoglycan (e.g., chondroitin, dermatan, heparan) and the nature of the core protein give rise to a great variety of proteoglycans, macromolecules with different biological roles and tissue distributions, such as aggrecan in cartilage and decorin and biglycan in skin.1 Most GAGs are heavily sulfated, and it is believed that sulfation is crucial to the biological role of GAGs because it forms an array of regularly spaced negative charges.
A mutation in the gene coding for carbohydrate sulfotransferase 3 (CHST3, chondroitin 6-sulfotransferase; MIM 603799) has been reported in a family originating from Oman and was associated with a phenotype described as spondyloepiphyseal dysplasia with spinal misalignment and joint abnormalities (spondyloepiphyseal dysplasia, Omani type; SED-OT; MIM 608637).2,3 Studies in cells cultured from affected individuals confirmed the impairment of 6-sulfation on N-acetyl-D-galactosamine residues in chondroitin GAG chains. The clinical description emphasized the chondrodysplasia aspect rather than the neonatal presentation, and no subsequent patients have been identified.
Larsen syndrome is the name given to a condition characterized by multiple congenital dislocations, and a subset of Larsen syndrome cases (LRS1 [MIM 150250])4–6 is caused by disturbance to the cytoskeleton from dominant mutations in the gene coding for filamin B (FLNB; MIM 150250). However, Larsen syndrome is genetically and molecularly heterogeneous, with a number of cases being inherited as recessive traits, such as the variant that is particularly frequent on Reunion Island (so-called “Ile de la Réunion” variant of Larsen syndrome) (MIM 245600).7,8 These cases have more severe joint disease but lack the characteristic flat face of dominant Larsen syndrome that was emphasized in the original publication.4,5
Humero-spinal dysostosis (HSD [MIM 143095]) was the name given by Kozlowski et al. to two (putative) half-sibs with a combination of dislocating knees, bilateral clubfoot, elbow joint dysplasia, and coronal clefting and superior and inferior notching of the lumbar vertebral bodies.9 The condition has been diagnosed rarely, with only a handful of cases reported, although the phenotype in the reported cases is very homogeneous.9–12 It is likely that similar cases have been simply diagnosed as Larsen syndrome. The inheritance of HSD remained unclear because it was uncertain whether the two original cases were full or half sibs,9 and all other cases have been sporadic.
We report here on the identification of CHST3 mutations in six individuals born with joint dislocations: Three had been diagnosed with Larsen syndrome, three with HSD. All six of our patients are the only affected people in their families, and only one family is known to be consanguineous. However, two other patients originate from geographically isolated regions and have homozygous mutations, indicating possible distant consanguinity (Table 1). At least three patients were delivered by Cesarean section on account of breech presentation. Birth length was below the third percentile in all patients where recorded, but only one child was noted to have short limbs on ultrasound (patient 5). Birth lengths ranged from 41.5 cm to 44 cm. All patients had a height significantly below the third percentile at last examination. Our lone adult patient (patient 4), whose clinical and radiographic features at age 2 yr have been published as an example of HSD,10 achieved a final height of 134 cm. Congenital joint dislocations were present in all children and were the presenting feature for five out of six patients (Figure 1, Table 1). Six of six patients had congenital clubfeet and knee dislocations, with three having genu recurvatum. Four of six had hip luxation, and six of six had radial head dislocation. All children have had multiple orthopedic procedures on the knees and feet, with variable success. All patients have residual joint restriction, and the older patients, presently aged 31 yr (patient 4), 17 yr (patient 2), and 10.5 yr (patient 1), all needed aids (crutches, wheelchair) for ambulation. Spinal radiographs of the three older patients (patients 1, 2, and 4) show severe degeneration of the intervertebral disks with thoracic kyphosis (not shown). Intellect, hearing, and vision were normal in all. No children had cleft palate, and none had the typical facial features of dominant Larsen syndrome: The facial features in childhood were normal but reminiscent of diastrophic dysplasia with small mouth, small alae nasi, and high nasal bridge. The parents are all of normal height and had no joint abnormalities. All human-subject studies have been carried out in accordance with IRB requirements of the participating institutions.
Table 1.
Overview of Genetic Data and Results on the Families Reported in This Study
| Patient No. | Ethnicity | Parental Consanguinity | Unaffected Sibs | Maternal CHST3 Allele | Paternal CHST3 Allele | Short Stature at Birth | Breech Presentation | Postnatal Short Stature (percentile) | Elbow Joint Subluxation or Limited Extension | Hip Dysplasia or Dislocation | Knee Luxation or Genu Recurvatum | Club feet |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Turkish-German | yes | 1 brother | c.776T→C, L259P | c.776T→C, L259P | + | + | yes; 100.2 cm at 10 yr (<3rd) | + | + | + | + |
| 2 | Swiss (Valais) | no, but same small village | 1 brother | c.664C→T, R222W | c.664C→T, R222W | not recorded | no | yes; 125 cm at 16 yr (<3rd) | + | + | + | + |
| 3 | Sardinian | no | none (only child) | c.920T→C, L307P | c.920T→C, L307P | + | + | yes; 99 cm at 6 yr (<3rd) | + | + | + | + |
| 4 | Spanish (Central Spain) | no | 1 sister | c.1086 delG, G363A fsX393 | c.603C→A, Y201X | not recorded | no | yes: 134 cm adult height (<3rd) | + | + | + | + |
| 5 | Spanish | no | 1 sister | c.617-618 TC→CA, F206X | IVS2 −1G→C and c.141G→C (adjacent nucleotides), missplicing? | + | + | yes, 90 cm at 5 yr (<3rd) | + | no | + | + |
| 6 | Spanish | no | 1 sister | c.1086 delG, G363AfsX393 | c.1114G→A, E372K | + | no | yes; 77cm at 2.5 yr (<3rd) | + | no | + | + |
Figure 1.
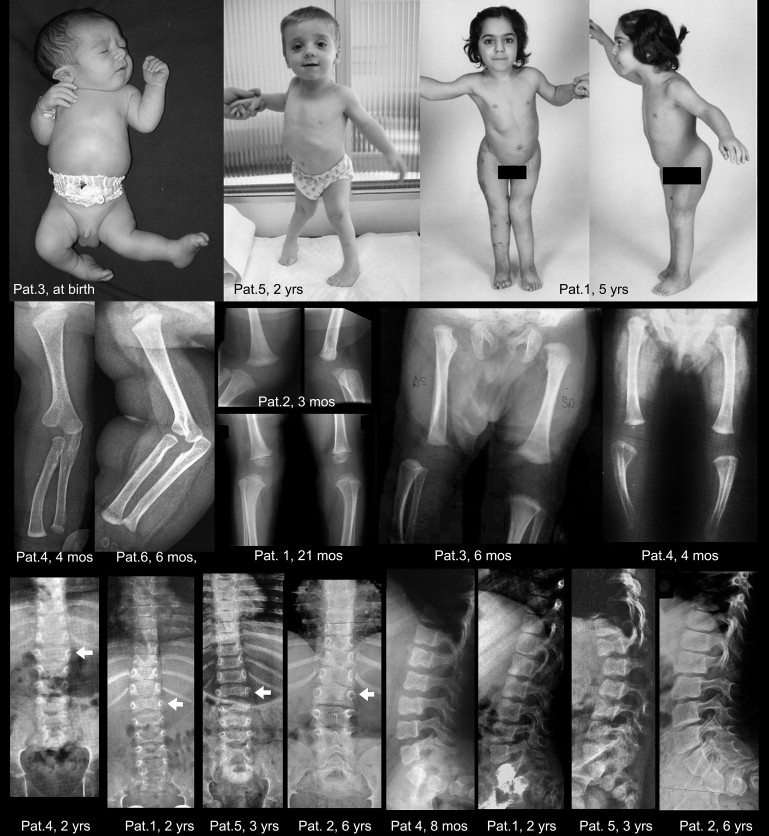
Synopsis of Clinical Aspect and Main Radiographic Features
The first row shows congenital dislocation of the left knee and clubfoot in patient 3, diagnosed with Larsen syndrome, at a few days of age (the appearance is identical to that of the HSD patient reported by Hall11). At birth, the left leg was extended in such a way that the foot rested against the thoracic wall. The central photograph shows marked valgus deformity of the knees and bilateral clubfoot in patient 5 at age 2 yr. The right panels show patient 1, a girl, at age 5 yr, after several surgical procedures had been carried out to stabilize the dislocated knees and the dysplastic hips. Note also the inability to extend the elbows completely. The second row shows the bifid humerus and radial dislocation, as well as the knee dislocation or subluxation with delayed development of the knee epiphyses (patient identification and ages are within the panels). The third row shows the diagnostically useful spinal changes: In the AP projection, there is marked widening of the interpedicular space from T12 to L1 (the arrows mark the vertebral body of L1); the interpedicular space then decreases progressively from L3 to L5. In the lateral projection, notching or scalloping of the upper and lower endplates of the lumbar vertebral bodies can be seen; these changes have been emphasized in the original descriptions of humero-spinal dysostosis9,10. The changes are more pronounced in the younger patients.
Table 1 and Figure 2 show the CHST3 mutations identified in the six patients. Formally, nine mutations on eight different alleles (five missense mutations, three premature-termination mutations, and one splice-site mutation) were identified. All parents were shown to be heterozygous for one of the mutations found in their child. Patients 1, 2, and 3 were homozygous for point mutations leading to amino acid substitutions. The genetic data are in accordance with the pedigree, with family 1 being consanguineous, family 2 originating from the same small village, and family 3 originating from the island of Sardinia, where there is a relatively high degree of consanguinity. One mutation (c.1086 delG) was seen in two different families. In patient 5, an unusual mutation was found that involved two adjacent nucleotides (GG→CC), the last nucleotide of IVS2 (i.e., the second nucleotide of the canonical splice acceptor site), and the first nucleotide of exon 3 (the second base of codon 47). Formally, the two single-nucleotide substitutions can be described as IVS2−1G→C and c.141G→C (R74S); subcloning the PCR product always showed cosegregation of the two changes, and analysis of the parents showed that the patient had inherited both mutations from his father. Thus, the adjacent nucleotide substitutions were in cis and probably originated from a single mutational event. Unfortunately, no material was available to determine whether the mutations resulted in missplicing or in amino acid substitution R47S (or in a combination of the two); mutation of two nucleotides within a splicing-consensus sequence is highly likely to affect splicing, and thus we will not consider R47S as a potential amino acid substitution in the further discussion.
Figure 2.
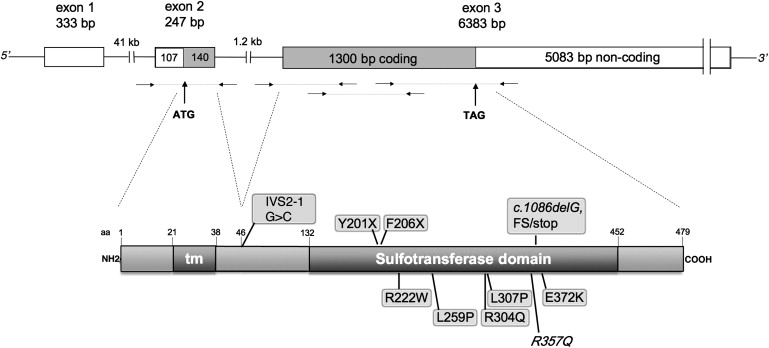
Scheme of the CHST3 Gene and Protein
The upper part is a schematic representation of the CHST3 exon-intron structure and of the coding regions. Immediately below, the small horizontal arrows indicate the position of primer couples used to amplify genomic DNA. The lower part is a schematic representation of the CHST3 protein with its transmembrane domain and the intraluminal sulfotransferase domain as inferred from data on the Expasy site. Above the protein are mutations leading to premature stop codons as well as the putative splicing mutation (see Table 1 and Supplemental Material and Methods, available online); below the proteins are amino acid substitutions, including the R304Q substitution reported by Thiele et al.,2 and the known polymorphism R357Q.
Several different lines of evidence suggest that the amino acid substitutions observed (R222W, L259P, L307P, and E372K) are pathogenic. First, all mutations segregated as expected for a pathogenic mutation, with parents being heterozygous for (only) one mutation in each family. Second, all five substitutions affect amino acids that are highly conserved (data not shown). Third, the substitutions are nonconservative; R222W abolishes a charged residue, E372K substitutes a negatively charged with a positively charged residue, and L259P and L307P substitute the aliphatic leucine with a helix-breaking proline. Fourth, none of the respective sequence changes were seen in any of a panel of over 70 unaffected controls (140 unrelated chromosomes). Finally, in the two patients who consented to donate skin biopsies (patient 1, L259P; patient 2, R222W; both at homozygosity), evidence of severely impaired CHST3 activity was obtained in fibroblast cultures (see below). In these two cell lines, cDNA fragments harboring the mutations were amplified by quantitative RT-PCR using primers spanning from exon 2 to exon 3 with the same efficiency as in control lines (data not shown). Mutations Y201X and F206X predict truncation of the protein including a large portion of the sulfotransferase domain (Figure 1), whereas the frameshift deletion c.1086 delG would remove approximately one-fourth of the sulfotransferase domain and probably destabilize either the mRNA (by nonsense-mediated decay) or the protein (by adding a stretch of 30 missense amino acids). The individuals in our small series appeared to have a phenotype of similar severity and do not support the concept of a phenotypic gradient correlated with specific mutations.
The consequence of the L259P and R222W mutations on CHST3 function was tested in cultured skin fibroblasts from patient 1 and 2 by determining the sulfation pattern of chondroitin sulfate chains. Cells were labeled with [3H]glucosamine, a precursor for N-acetyl-D-galactosamine, and the sulfation of chondroitin sulfate PGs was measured by HPLC disaccharide analysis after digestion with chondroitinase ABC and ACII. The main disaccharide constituents of chondroitin sulfate—ΔDi-0S, ΔDi-4S, and ΔDi-6S—were measured. Labeling in standard medium showed a 4- to 5-fold reduction of the relative amount of ΔDi-6S in the patients' fibroblasts compared to the controls (Figure 3). We previously observed that in cells from patients with mutations in the sulfate transporter, DTDST (SLC26A2), there was a relationship between the rate of PG synthesis and the degree of total GAG sulfation.13 Thus, we stimulated GAG synthesis in fibroblast cultures by incubation with β-D-xyloside, a compound that acts as a GAG-chain initiator.14 In presence of 1 mM β-D-xyloside, the overall incorporation of labeled glucosamine was higher (data not shown) and the relative amount of ΔDi-0S decreased in all the cell lines, indicating that overall GAG synthesis and sulfation was stimulated, as we had previously observed;13 in the patients' cells, the relative amount of ΔDi-6S was dramatically reduced, demonstrating that both the L259P and R222W mutations severely impair or abolish CHST3 function (Figure 3).
Figure 3.
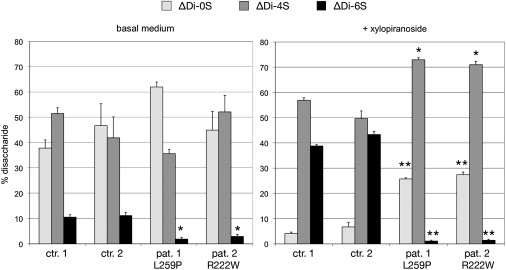
Disaccharide Composition of Chondroitin Sulfate Synthesized by Fibroblast Cultures
Fibroblasts from two controls and from patients 1 and 2 were labeled simultaneously with the radiolabeled glycosaminoglycan sugar precursor, [6-3H]glucosamine, and the chondroitin sulfate synthesized was harvested, purified, and cleaved into its constituent disaccharides, ΔDi-0S, ΔDi-4S and ΔDi-6S. These were separated by high-performance liquid chromatography (HPLC) and measured (see Supplemental Material and Methods). In patients' cells, the proportion of ΔDi-6S (black bars) is markedly reduced at basal condition; under stimulation of GAG synthesis with beta-D-xylopiranoside, the difference between controls and patients increases further. The bars show the percentage of each of the three disaccharides (and the standard error of the mean) in each experiment. The statistical significance of the difference between the value of each disaccharide in the patient cell lines and the mean of the same disaccharide in the two control lines was calculated with Student's t test; ∗ p < 0.01; ∗∗ p < 0.001.
The clinical and radiographic features of our patients with recessive Larsen syndrome and HSD are different but complementary to those reported by Rajab et al. in the older individuals in the Omani kindred.3 At birth, the prominent clinical and skeletal features are short stature of prenatal onset, dislocation or subluxation of the knee joints, clubfeet, dysplasia of the distal humerus, and elbow joint dysplasia with radial subluxation and limitation of extension. Most cases have hip dislocation. The face is unlike the dish-like face observed in classic Larsen syndrome but is characteristic with small mouth, small and overfolded ears, and small alae nasi. It resembles the face seen in diastrophic dysplasia. Most affected individuals had several surgical procedures to stabilize the hip and knee joints. Later in childhood, progressive degenerative arthritis of the hips leads to painful contractures; early and severe degeneration of intervertebral disks leads to short trunk, kyphoscoliosis, and eventually vertebral fusion; and brachydactyly appears because of progressive relative shortening of the metacarpals. Although some of the original patients with HSD had dysplasia of heart valves,9,11 only one of our patient (patient 2) had cardiac findings, namely, mild subvalvular aortic stenosis. Intelligence has been normal in all cases; myopia and cleft palate have not been observed. The diagnostically useful radiographic findings are shown in Figure 1. Changes recognizable at birth are misalignment or frank dislocation of the knee joints; bifid distal humerus with subluxation of the radial head; posterior and inferior scalloping or coronal clefting of the lumbar vertebral bodies; and pronounced widening of the interpedicular distance of L1 and L2 with reduction of the distance from L3 toward L5. The hip joints have been dysplastic or dislocated in five of our six cases. In childhood, signs of a moderate dysplasia develop with secondary changes in the hip and knee joint, shortening of the metacarpals (without frank brachydactyly), and progressive degeneration of the intervertebral disks leading to lumbar lordosis and thoracic kyphosis or kyphoscoliosis with reduction of the intervertebral spaces and, eventually, fusion of vertebral bodies.
The differential diagnosis of multiple dislocations in a newborn is large, with Larsen syndrome being one of the most common entities. The original description of Larsen syndrome emphasized the congenital joint dislocations and a characteristic flat face.4,5 In clinical practice, “Larsen syndrome” is a diagnostic label that is applied to conditions that are probably heterogeneous. Similarly, individuals diagnosed with HSD in the literature and in our series were given that diagnosis as infants or young children. All patients with recessive Larsen syndrome and HSD had congenital joint dislocations but lacked the flat face typical of classic, dominant Larsen, and they had pronounced vertebral changes that are instead not typical of dominant Larsen syndrome.4,5,15 Larsen syndrome is frequently caused by dominant mutations in the filamin B (FLNB) gene,6,15 but it has long been known that a recessive variant of Larsen syndrome exists with clinical features that are similar to, but distinct from, those of dominant Larsen syndrome.7,8 CHST3 deficiency is the first molecular basis for the recessive variant of Larsen syndrome. Although the incidence of CHST3-associated Larsen syndrome remains to be determined, there are reports of Larsen syndrome that would fit with CHST3 deficiency: Weisenbach and Melegh reported two sibs with “Larsen syndrome with vertebral changes”; the clinical and radiographic changes are identical of those reported in the present paper.16 Joseph and Varghese reported a patient with humeral dysplasia and dislocations and discussed the possibility that the condition may represent a variant of Larsen syndrome.17
Several features may help in distinguishing CHST3-deficient recessive Larsen syndrome and HSD from classic Larsen syndrome caused by dominant FLNB mutations. Pre- and postnatal short stature are not typical of dominant Larsen syndrome. Although congenital dislocations are present in both conditions, in CHST3 deficiency there is no generalized joint laxity; the dislocations are produced by primary joint dysplasia rather than joint laxity. The development of severe spinal changes distinguishes this condition from classic Larsen syndrome. Radiographically, the most distinctive features are a wide interpedicular distance at L1, shortening and clefting of the lumbar vertebrae in infancy, the presence of a bifid distal humerus with dislocation or subluxation of the radial heads, and the development of short metacarpals and phalanges. Supernumerary carpal bones are not seen, and bone age is not significantly advanced. The single feature most useful to recognize CHST3 deficiency seems to be the dysostotic changes in the thoracolumbar spine—namely, widening of the interpedicular distance at L1 seen in the antero-posterior projection and the short and cleft vertebral bodies in the lateral projection—that were present in our patients. These skeletal changes were mentioned by both Kozlowski et al.9 and Cortina et al.10 in their descriptions of HSD, and they are also recognizable in Figure 1 of the report on SED-OT by Rajab et al.3 The affected individuals in the SED-OT kindred reported by Rajab et al. were studied as older children and adults; congenital dislocations were not mentioned, the vertebral clefting was no longer present, kyphoscoliosis and bone dysplastic changes were perceived as more prominent, and the condition was described as a form of spondyloepiphyseal dysplasia.3 It is possible that had these patients been identified as newborns, they might have had features compatible with HSD or recessive Larsen syndrome. Only identification of a larger cohort of patients will allow us to determine the extent of overlap or of identity of these three conditions.
The sulfate group transferred to N-acetylgalactosamine by CHST3 is the same that is removed in the lysosomes by GalNAc-6-sulfatase (GALNS), the enzyme deficient in mucopolysaccharidosis type IV A (MPS IV A; Morquio syndrome [MIM 253000]). Thus, there is a clinical phenotype associated with either sense of the reaction, highlighting the biological importance of chondroitin 6-sulfation in man. Interestingly, the mouse differs from the human in this respect: neither chondroitin-6-sulfotransferase (Chst3)-deficient or GalNAc-6-sulfatase (Galns)-deficient mice develop a skeletal phenotype.18,19 In contrast, mice with defects in the sulfate transporter Slc26a2 (Dtd mouse)20 or in chondroitin-4-sulfotransferase 121 have strong skeletal phenotypes. Chondroitin 6-sulfate seems to be important for joint development and homeostasis in humans but not in the mouse.
Fibroblasts from patients with mutations in either the DTDST sulfate transporter (SLC26A2; MIM 606718) or the CHST3 sulfotransferase show defective sulfation of chondroitin proteoglycans that becomes more evident when GAG-chain synthesis is stimulated with β-D-xyloside.13 Unlike in DTDST mutant cells, where both 4- and 6-sulfation are affected, the sulfation defect in CHST3-deficient cells is limited to the 6-position on galactosamine in chondroitin, whereas the 4-position is highly sulfated (Figure 3). The phenotypic difference between DTDST conditions and CHST3 deficiency must be ascribed either to different function of the 4-sulfation versus 6-sulfation on chondroitin sulfate or to the degree of sulfation of other glycosaminoglycans, affected in DTDST but not in CHST3 deficiency. Even in the presence of two putative CHST3 null mutations (Table 1, patient 4), the degree of chondrodysplasia is much milder than what is seen in the severe DTDST disorders (achondrogenesis type 1B and atelosteogenesis type 2; MIM 600972 and 256050).22 By contrast, dislocation and degeneration of articulations (at the hips, knees, and spine) seem to be more prominent in CHST3 deficiency. Thus, when compared to DTDST deficiency, CHST3 deficiency has a similar or more pronounced effect on joint formation and articular homeostasis but a less severe effect on endochondral growth of skeletal elements. This seems to support the hitherto unproven hypothesis that the sulfation defect in DTDST dysplasias can involve proteoglycans other than chondroitin sulfate, such as heparan sulfates, and influence FGFR signaling pathways in the growth plate.23
In conclusion, CHST3 deficiency is a molecular basis for two conditions with congenital joint dislocations such as Larsen syndrome and HSD. Although the diagnostic features for recessive Larsen syndrome, HSD, and SED-OT are different, our results indicate that they constitute a relatively narrow phenotypic spectrum and in fact may be different age-related descriptions of the same condition. The proportion of Larsen syndrome cases caused by CHST3 deficiency remains to be determined. The possibility of molecular diagnosis will be useful in the management and in the counseling of affected patients and their families. Recessive Larsen syndrome is a second sulfation disorder that is analogous to a dominant FLNB disorder, the first one being atelosteogenesis 2 (DTDST),22 which resembles atelosteogenesis type 1/3 (FLNB; MIM 108720 and 108721);6 a molecular link between filamin B and proteoglycan sulfation seems likely and should be investigated.
Acknowledgments
We are grateful to Dr. Stephen Robertson for FLNB mutation analysis in patient 2. This work was supported by the German BMBF Rare Diseases network SKELNET (project GFGN01141901), by the European Community (FP6, “EuroGrow” project, LSHM-CT-2007-037471), by the Swiss National Foundation (grant no. 320000-116506), and by Telethon-Italy (grant no.GGP06076).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Expasy/UniProt/Swiss-Prot Browser, http://www.expasy.org/uniprot/Q7LGC8/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway/
References
- 1.Raman R., Sasisekharan V., Sasisekharan R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 2005;12:267–277. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 2.Thiele H., Sakano M., Kitagawa H., Sugahara K., Rajab A., Hohne W., Ritter H., Leschik G., Nurnberg P., Mundlos S. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. USA. 2004;101:10155–10160. doi: 10.1073/pnas.0400334101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajab A., Kunze J., Mundlos S. Spondyloepiphyseal dysplasia Omani type: A new recessive type of SED with progressive spinal involvement. Am. J. Med. Genet. A. 2004;126:413–419. doi: 10.1002/ajmg.a.20606. [DOI] [PubMed] [Google Scholar]
- 4.Larsen L.J., Schottstaedt E.R., Bost F.C. Multiple congenital dislocations associated with characteristic facial abnormality. J. Pediatr. 1950;37:574–581. doi: 10.1016/s0022-3476(50)80268-8. [DOI] [PubMed] [Google Scholar]
- 5.Latta R.J., Graham C.B., Aase J., Scham S.M., Smith D.W. Larsen's syndrome: A skeletal dysplasia with multiple joint dislocations and unusual facies. J. Pediatr. 1971;78:291–298. doi: 10.1016/s0022-3476(71)80014-8. [DOI] [PubMed] [Google Scholar]
- 6.Krakow D., Robertson S.P., King L.M., Morgan T., Sebald E.T., Bertolotto C., Wachsmann-Hogiu S., Acuna D., Shapiro S.S., Takafuta T. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat. Genet. 2004;36:405–410. doi: 10.1038/ng1319. [DOI] [PubMed] [Google Scholar]
- 7.Laville J.M., Lakermance P., Limouzy F. Larsen's syndrome: Review of the literature and analysis of thirty-eight cases. J. Pediatr. Orthop. 1994;14:63–73. doi: 10.1097/01241398-199401000-00014. [DOI] [PubMed] [Google Scholar]
- 8.Bonaventure J., Lasselin C., Mellier J., Cohen-Solal L., Maroteaux P. Linkage studies of four fibrillar collagen genes in three pedigrees with Larsen-like syndrome. J. Med. Genet. 1992;29:465–470. [PMC free article] [PubMed] [Google Scholar]
- 9.Kozlowski K.S., Celermajer J.M., Tink A.R. Humero-spinal dysostosis with congenital heart disease. Am. J. Dis. Child. 1974;127:407–410. doi: 10.1001/archpedi.1974.02110220105015. [DOI] [PubMed] [Google Scholar]
- 10.Cortina H., Vidal J., Vallcanera A., Alberto C., Muro D., Dominguez F. Humero-spinal dysostosis. Pediatr. Radiol. 1979;8:188–190. doi: 10.1007/BF00973833. [DOI] [PubMed] [Google Scholar]
- 11.Hall B.D. Humero-spinal dysostosis: Report of the fourth case with emphasis on generalized skeletal involvement, abnormal craniofacial features, and mitral valve thickening. J. Pediatr. Orthop. B. 1997;6:11–14. [PubMed] [Google Scholar]
- 12.Sparks M.J., Gaines P.A., Levick R.K. Case report: Humero-spinal dysostosis. Clin. Radiol. 1994;49:829–831. doi: 10.1016/s0009-9260(05)81977-4. [DOI] [PubMed] [Google Scholar]
- 13.Rossi A., Kaitila I., Wilcox W.R., Rimoin D.L., Steinmann B., Cetta G., Superti-Furga A. Proteoglycan sulfation in cartilage and cell cultures from patients with sulfate transporter chondrodysplasias: Relationship to clinical severity and indications on the role of intracellular sulfate production. Matrix Biol. 1998;17:361–369. doi: 10.1016/s0945-053x(98)90088-9. [DOI] [PubMed] [Google Scholar]
- 14.Sobue M., Habuchi H., Ito K., Yonekura H., Oguri K., Sakurai K., Kamohara S., Ueno Y., Noyori R., Suzuki S. beta-D-xylosides and their analogues as artificial initiators of glycosaminoglycan chain synthesis. Aglycone-related variation in their effectiveness in vitro and in ovo. Biochem. J. 1987;241:591–601. doi: 10.1042/bj2410591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bicknell L.S., Farrington-Rock C., Shafeghati Y., Rump P., Alanay Y., Alembik Y., Al-Madani N., Firth H., Karimi-Nejad M.H., Kim C.A. A molecular and clinical study of Larsen syndrome caused by mutations in FLNB. J. Med. Genet. 2007;44:89–98. doi: 10.1136/jmg.2006.043687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weisenbach J., Melegh B. Vertebral anomalies in Larsen's syndrome. Pediatr. Radiol. 1996;26:682–683. doi: 10.1007/BF01356836. [DOI] [PubMed] [Google Scholar]
- 17.Joseph B., Varghese R.A. Congenital distal humeral dysplasia: A case report. Pediatr. Radiol. 2003;33:7–10. doi: 10.1007/s00247-002-0660-4. [DOI] [PubMed] [Google Scholar]
- 18.Uchimura K., Kadomatsu K., Nishimura H., Muramatsu H., Nakamura E., Kurosawa N., Habuchi O., El-Fasakhany F.M., Yoshikai Y., Muramatsu T. Functional analysis of the chondroitin 6-sulfotransferase gene in relation to lymphocyte subpopulations, brain development, and oversulfated chondroitin sulfates. J. Biol. Chem. 2002;277:1443–1450. doi: 10.1074/jbc.M104719200. [DOI] [PubMed] [Google Scholar]
- 19.Tomatsu S., Orii K.O., Vogler C., Nakayama J., Levy B., Grubb J.H., Gutierrez M.A., Shim S., Yamaguchi S., Nishioka T. Mouse model of N-acetylgalactosamine-6-sulfate sulfatase deficiency (Galns−/−) produced by targeted disruption of the gene defective in Morquio A disease. Hum. Mol. Genet. 2003;12:3349–3358. doi: 10.1093/hmg/ddg366. [DOI] [PubMed] [Google Scholar]
- 20.Forlino A., Piazza R., Tiveron C., Della Torre S., Tatangelo L., Bonafè L., Gualeni B., Romano A., Pecora F., Superti-Furga A. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: Morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum. Mol. Genet. 2005;14:859–871. doi: 10.1093/hmg/ddi079. [DOI] [PubMed] [Google Scholar]
- 21.Kluppel M., Wight T.N., Chan C., Hinek A., Wrana J.L. Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development. 2005;132:3989–4003. doi: 10.1242/dev.01948. [DOI] [PubMed] [Google Scholar]
- 22.Rossi A., Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum. Mutat. 2001;17:159–171. doi: 10.1002/humu.1. [DOI] [PubMed] [Google Scholar]
- 23.Wallis G.A. The importance of being sulphated. Curr. Biol. 1995;5:225–227. doi: 10.1016/s0960-9822(95)00044-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.