

Abstract
Mitochondrial isolated complex I deficiency is the most frequently encountered OXPHOS defect. We report a patient with an isolated complex I deficiency expressed in skin fibroblasts as well as muscle tissue. Because the parents were consanguineous, we performed homozygosity mapping to identify homozygous regions containing candidate genes such as NDUFA2 on chromosome 5. Screening of this gene on genomic DNA revealed a mutation that interferes with correct splicing and results in the skipping of exon 2. Exon skipping was confirmed on the mRNA level. The mutation in this accessory subunit causes reduced activity and disturbed assembly of complex I. Furthermore, the mutation is associated with a mitochondrial depolarization. The expression and activity of complex I and the depolarization was (partially) rescued with a baculovirus system expressing the NDUFA2 gene.
Introduction
NADH:ubiquinone oxidoreductase, or complex I, is the first and largest complex of the mitochondrial respiratory chain. Its function is to liberate and transfer electrons from NADH to ubiquinone and meanwhile to extract part of their energy to drive proton translocation across the inner mitochondrial membrane and thus to create an electrochemical proton gradient that can be used for the synthesis of ATP. Complex I consists of 45 subunits, of which seven are encoded by the mitochondrial DNA (mtDNA). The complex is L-shaped; it has a membrane arm embedded in the inner mitochondrial membrane and a peripheral arm protruding into the matrix. Complex I can be divided into three functional fractions. The flavoprotein fraction plays a role in FMN and NADH binding, the iron protein fraction is important in reduction-oxidation reactions via iron-sulfur clusters, and the hydrophobic protein fraction is involved in proton translocation.
Isolated complex I deficiency (MIM 252010) is one of the most common defects of the OXPHOS system.1 It accounts for almost 23% of all cases of childhood respiratory-chain deficiency, and the heterogeneous clinical presentation includes Leigh or Leigh-like syndrome [MIM 256000], cardiomyopathy, leukodystrophy, and hepatopathy.2,3 Previous studies identified disease-causing mutations in eleven complex I nuclear structural genes (NDUFS1 [MIM 157655], NDUFS2 [MIM 602985], NDUFS3 [MIM 603846], NDUFS4 [MIM 602694], NDUFS6 [MIM 603848], NDUFS7 [MIM 601825], NDUFS8 [MIM 602141], NDUFV1 [MIM 161015], NDUFV2 [MIM 600532], NDUFA1 [MIM 300078], and NDUFA11).4–14 Most of these genes are evolutionarily highly conserved and play important roles in the catalytic activity of complex I. Mutations described in these genes account for only a small part of all complex I deficiencies, suggesting that disease-causing mutations should be found in other genes such as those encoding the accessory subunits and assembly factors for complex I. The latter mutations have been described in the NDUFAF2 (MIM 609653), NDUFAF1 (MIM 606934), and C6orf66 (MIM 611776) genes.15–17
Although for the 14 core subunits of complex I, primary roles in electron transfer and proton translocation have been established, the exact role of the remaining 31 accessory subunits is not known yet. It has been suggested that some are important for the organization of the biogenesis of complex I. One of the accessory subunits of complex I is NDUFA2, or bovine B8. This subunit is located in the peripheral arm of complex I, and analysis of the structure showed a Fe2S2 ferredoxin fold similar to that found in thioredoxin.18
In this study we describe a patient whose parents were consanguineous and in whom isolated complex I deficiency was detected in both muscle tissue and fibroblasts as a result of a mutation in NDUFA2 (MIM 602137). Homozygosity mapping revealed that the gene NDUFA2 is located in a homozygous region on chromosome 5. Screening of this gene revealed a splice-site mutation resulting in the skipping of exon 2. With cell biological studies we demonstrated the pathogenicity of this mutation. Identifying such new disease-causing mutations will deliver new insights into human complex I deficiency.
Material and Methods
Case Report
The patient, a boy, was the second child of consanguineous Turkish parents (first cousins). His older sister was healthy. Pregnancy and delivery were uneventful. At the fifth day of life, hypertrophic cardiomyopathy was diagnosed. Development was retarded from birth, and at the age of four months, cerebral atrophy and hypoplasia of the corpus callosum were seen on MRI. Some weeks later, atrophy of the N. opticus was diagnosed. At 7.5 months, after a 2 day episode of vomiting and 2 weeks after a varicella infection, the patient suddenly developed severe acidosis, generalized tonic-clonic seizures, and coma. The patient did not gain consciousness again and needed artificial respiration for 43 days. Afterwards, respiration was unstable, and the patient had recurrent episodes of apnea and bradycardia, often accompanied by seizures, which increased in frequency. Finally, MRI showed demyelinization of cortico-spinal tracts and subacute necrotizing encephalomyelopathy as seen in Leigh syndrome. At eleven months, the patient died of cardiovascular arrest, after further episodes of apnea and asystolia.
Cell Culture
Skin fibroblasts were cultured in M199 medium (Life Technologies) supplemented with 10% fetal calf serum and a combination of penicillin and streptomycin in a humified atmosphere of 95% air and 5% CO2 at 37°C. For live-cell imaging, the fibroblasts were cultured as described above but in medium 199 with Earle's salt supplemented with 10% fetal calf serum and penicillin/streptomycin.
Biochemical Analysis
Measurement of mitochondrial respiratory-chain enzymes in skin fibroblasts and in muscle tissue collected at the age of 8 months as previously described,19,20 with the exception of the Figure 3C complex I measurements that were performed with a recently developed assay.21 The values were expressed relative to the activity of cytochrome c oxidase and/or citrate synthase. The oxidation rates of pyruvate in the presence of malate or carnitine and the production rate of ATP from pyruvate were measured in muscle tissue as described before.22
Figure 3.
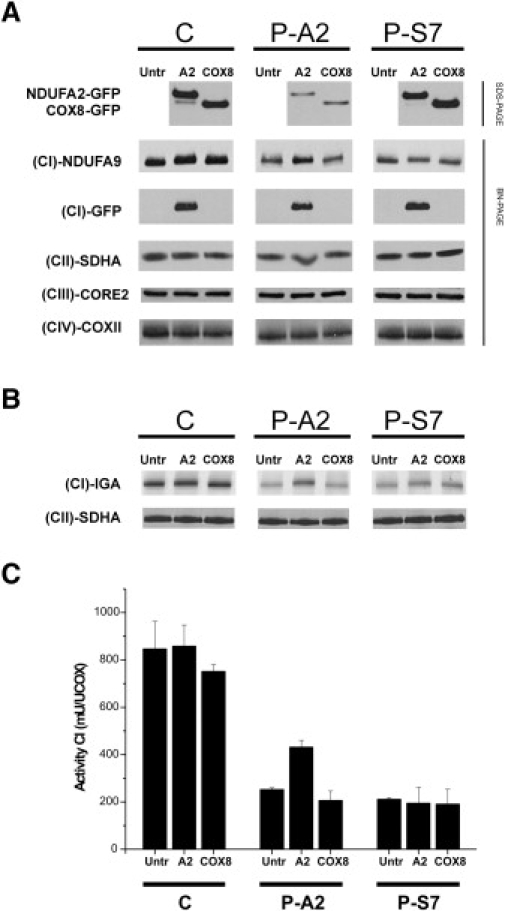
The Decreased Expression and Activity of Complex I Caused by a Mutation in NDUFA2 Can Be Restored by Baculoviral Complementation
SDS-PAGE and BN-PAGE analysis with immunoblotting of complexes I–IV in control fibroblasts, fibroblasts from the patient with a mutation in NDUFA2, and fibroblasts from a patient with a mutation in NDUFS7 confirmed the pathogenicity of the NDUFA2 mutation.
(A) SDS-PAGE analysis followed by immunoblotting confirms expression of the baculovirus transduced with constructs of NDUFA2-GFP or with COX8-GFP (top panel). A BN-PAGE gel was blotted and used for immunodetection with an antibody directed against NDUFA9 to show the expression of complex I. The untreated control cells and the control cells treated with the baculoviruses show the same expression levels. Expression of complex I in the fibroblasts of the index patient with a mutation in NDUFA2 is lower than in the control cells but can be partly rescued if the cells are treated with the baculovirus. Treating the cells of the patient with a mutation in NDUFS7 with the baculovirus did not affect the expression of complex I. In the third panel, detection with an antibody directed against GFP confirms the incorporation of the GFP-tagged subunit. COX8-GFP is located in the bottom of the gel and is not shown. In the next panels, antibodies for complex II, III, and IV showed that the assembly of these complexes was not affected.
(B) In-gel activity assays showed that the activity of complex I is the same in the untreated and treated control cells transduced with the baculoviruses. The NDUFA2-mutated patient cells treated with the baculovirus containing NDUFA2-GFP have higher complex I activity than the untreated cells and than the cells treated with the baculovirus containing COX8-GFP. The treatment of the fibroblasts of the patient with a NDUFS7 mutation showed a slight increase in the activity of complex I because more protein was loaded in the last two lanes (see panel CII-SDHA).
(C) Measurement of the activity of complex I by biochemical analysis showed almost no difference or a slight decrease between the untreated cells and the control cells treated with the baculoviruses. The fibroblasts of the index patient treated with the baculovirus containing NDUFA2-GFP demonstrated an increase in activity compared to the untreated cells and to the cells treated with the baculovirus containing COX8-GFP. The treatment of the cells with the NDUFS7 mutation did not affect the activity of complex I. Values are given as means ± standard deviation.
Abbreviations are as follows: C = control, P-A2 = patient with the mutation in NDUFA2, P-S7 = patient with the mutation p.Val122Met in NDUFS7, Untr = untreated fibroblasts, A2 = fibroblasts treated with the baculovirus containing the NDUFA2 construct, COX8 = fibroblasts treated with the baculovirus containing the COX8 construct, (CI)-NDUFA9 = antibody directed against NDUFA9, (CI)-GFP = antibody for the GFP-tag, (CII)-SDHA = antibody for SDHA to detect complex II, (CIII)-CORE2 = antibody for CORE2 to detect complex III, (CIV)-COXII = antibody for COXII to detect complex IV, and (CI)-IGA = in-gel activity of complex I.
Homozygosity Mapping
Patient DNA was extracted from cultured fibroblasts by standard procedures23 and was used for a genome scan with the ABI PRISM Linkage Mapping Set MD10.A. A total of 382 fluorescently labeled short tandem repeat (STR) polymorphic markers with an average heterozygosity of 0.76 were used. The average distance of the genotyped markers was 10 cM.
Genetic Analysis of the NDUFA2 Gene
Oligonucleotide primers were designed according to public sequences (GenBank accession number NM_002488). The exon-spanning primers F1 (5′-CCTTTCCGCTTCCTGTTTTT-3′) with R1 (5′-GGGGCTTCCCTCAACTTC-3′), F2 (5′-CTGGGCCTGCGTGAGATT-3′) with R2 (5′-GGGCGGTCCCTTCTCTTCT-3′), and F3 (5′-CCATCCAGAATCAGCCAAAA-3′) with R3 (5′-TCCCACACCTAAGCCAAGAC-3′) were applied in a PCR for amplification of the three exons of the NDUFA2 gene. The exon fragments were amplified in a reaction volume of 25 μl containing 100 ng DNA template, 1 unit of Taq DNA polymerase (Invitrogen), 2.5 μl of 10× PCR buffer, 2.0 mM MgCl2, 50 ng of forward and reverse primers, 0.25 mM dNTPs, and 0.75 μl DMSO. After an initial denaturation step of 2 min at 94°C, PCR parameters were 35 cycles for 45 s denaturation at 94°C, 45 s annealing at Ta (annealing temperature; 53°C for the primers F1/R1 and F3/R3 and 60°C for the primers F2/R2), and 1 min extension at 72°C. The cycles were followed by a final extension step of 10 min at 72°C. The amplification products were run on a 1.5% agarose gel. The same oligonucleotides were used for direct sequencing with the BigDye terminator cycle sequencing chemistry on an Applied Biosystems ABI PRISM 3130xl Genetic Analyzer.
The mutation was also checked on the cDNA level. Patient RNA was extracted from cultured fibroblasts by standard procedures. The RNA was reverse transcribed to cDNA by superscript II RNase H− reverse transcriptase with oligo(dT) and random hexamer primers. The primers F (5′-TCGCATCCACTTATGTCAGC-3′) and R (5′-GAGGCTTCAGGCTTTACCAC-3′) were applied in a PCR as described above with a Ta of 58°C and 1.5 mM MgCl2.
SDS-PAGE and Immunoblot Analysis
Mitochondrial lysates were separated on a 10% SDS-PAGE gel. Next, the proteins were transferred to PROTAN nitrocellulose membrane (Schleicher & Schuell) for immunodetection.
BN-PAGE Analysis and in-Gel Activity Assay
Blue native-gradient gels (5%–15%) were cast as described previously24 and run with 40 or 80 μg of solubilized mitochondrial protein. After electrophoresis, the gels were further processed for in-gel activity assay or second-dimension 10% SDS-PAGE followed by immunoblot analysis as described by Nijtmans et al.24
Antibodies and ECL Detection
Protein immunodetection was performed with primary antibodies directed against EGFP (a gift from Dr. Frank van Kuppeveld, Nijmegen, The Netherlands), NDUFA9 (Invitrogen), the CII 70 kDa subunit (SDHA; Invitrogen), the CIII CORE2 subunit (UQCRC2; Invitrogen), and the CIV COXII subunit (COXII; Invitrogen). For generation of the NDUFA2-specific antibody, rabbits were immunized with peptide CDQVTRALENVLSGKA that was coupled to keyhole limpet hemocyanin with the Imject maleimide-activated mcKLH kit (Pierce). Secondary antibodies used peroxidase-conjugated anti-mouse or anti-rabbit IgGs (Invitrogen). The signal was generated with ECL Plus (Amersham Biosciences).
Generation of Recombinant Baculoviruses
We created the COX8 leader sequence (first 210 base pairs of COX8A, GenBank accession number NM_004074) and NDUFA2 open-reading-frame sequence (without stop codon) flanked by Gateway ATTB sites (Invitrogen) by PCR and cloned them into pDONR201 by using the Gateway BP Clonase II enzyme mix (Invitrogen) to generate the pEntry vectors.25 We made the Bac-to-Bac system, normally used for protein production in insect cells, suitable for protein expression in mammalian cells by cloning the vesicular stomatitis virus G protein cDNA behind the P10 promoter of the pFastBacDual vector. Next, we introduced the CMV promoter and Gateway destination elements (cassette that contains the chloramphenicol resistance gene and the ccdB gene flanked by attR1 and attR2 sites) into this vector.26 Finally, we cloned the AcGFP1 gene (Clontech) in frame behind the Gateway reading-frame cassette to obtain the destination vector pBac-Dest-GFP. The pEntry vectors were combined with the pBac-Dest-GFP vector in the Gateway LR reaction to yield the baculovirus expression vectors. These vectors were used to generate infectious recombinant baculoviruses by site-specific transposon-mediated insertion into a baculovirus genome (bacmid) propagated in Escherichia coli cells (Bac-to-Bac, Invitrogen). Isolated recombinant bacmids were used for transfecting Spodoptera frugiperda (Sf9) insect cells, and the viruses were harvested after three passages of amplification.
Transduction of Complex I-Deficient Fibroblasts with Baculoviral Constructs
Fibroblasts, grown to ∼50% confluency in 175 cm2 tissue culture flasks (NUNC), were transduced with the appropriate baculovirus in a total volume of 25 ml M199 containing 1.75 mM sodium butyrate (Sigma-Aldrich). After 24 h at 37°C, the virus-containing medium was discarded and replaced with normal growth medium containing sodium butyrate. At 3 days after transduction, cells were harvested for gel electrophoresis or measurement of biochemical complex I activity. Importantly, this transduction protocol was developed on the basis of a careful evaluation of different virus concentrations and different exposure times. We chose the lowest virus concentration, shortest incubation time, and lowest butyrate concentration that gave a considerable transduction (on visual inspection of the GFP signal expressed in ∼70% of the cells) without signs of drastic protein overexpression.
Combined Fluorescence Microscopy of TMRM and NDUFA2-GFP
For fluorescence imaging of tetramethyl rhodamine methyl ester (TMRM) and NDUFA2-GFP, fibroblasts, cultured in the above medium containing, in addition, 25 mM HEPES, were seeded on glass coverslips (diameter 24 mm) and left for 4 hr to attach. Next, cells were transduced with a baculovirus containing the cDNA of NDUFA2-GFP for 24 hr (protocol as described above). After transduction, cells were cultured to ∼70% confluence in the presence of 1.75 mM sodium butyrate for another 48 hr. Immediately before imaging, cells were loaded with 100 nM TMRM (Invitrogen) for 25 min at 37°C. After loading, cells were washed with PBS, and coverslips were mounted in a temperature-controlled incubation chamber attached to the stage of an inverted microscope (Axiovert 200 M, Carl Zeiss, Jena, Germany) equipped with a 63×, 1.25 NA Plan NeoFluar objective lens. During image recording, cells were maintained in a HEPES-Tris (HT) medium (132 mM NaCl, 4.2 mM KCl, 1 mM MgCl2, 5.5 mM D-glucose, 10 mM HEPES, and 1 mM CaCl2 [pH 7.4]) at 37°C. TMRM was excited at 540 nm with an acquisition and illumination time of 100 ms with a monochromator (Polychrome IV, TILL Photonics), and fluorescence light was directed by a 560DRLP dichroic mirror (Omega Optical) through a 565ALP emission filter (Omega) onto a CoolSNAP HQ monochrome CCD camera (Roper Scientific Photometrics). NDUFA2-GFP was excited at 470 nm, and fluorescence light was directed by a 505DRLPXR dichroic mirror (Omega) through the 565ALP emission filter. Importantly, with these settings no bleed-through of the GFP signal into the TMRM image, and vice versa, was observed (for details see also reference27). All hardware was controlled with Metafluor 6.0 software (Molecular Devices Corporation).
Quantitative Data Analysis
After immunoblotting, the signals were quantified with Image Pro Plus 5.1 (Media Cybernetics). The optical density of the bands was determined and corrected for background. Image analysis after the combined fluorescence microscopy of TMRM and NDUFA2-GFP was performed as described previously.27,28 In brief, computer-assisted processing of the acquired TMRM images produced a mitochondria-specific mask. Superimposing this mask on the original image then allowed quantification of the average TMRM fluorescence pixel intensity exclusively in the cell's mitochondrial structures. We inspected sequences of TMRM and GFP fluorescence in parallel to select for GFP-positive and -negative cells. Image analysis was performed with Image Pro Plus 5.1. The average value obtained with the corresponding control cell line (C1) was set at 100%, to which all other values were related. Numerical results were visualized with Origin Pro 7.5 (Originlabs), and values from multiple experiments were expressed as means ± SE (standard error). Statistical significance (Bonferroni corrected) was assessed with a Student's t test.
Results
Biochemical and Molecular Genetic Studies in the Index Patient
The muscle tissue of the index patient clearly displayed a defect in the mitochondrial energy-generating capacity (Table 1). Further investigation of the activities of the enzymes of the respiratory-chain complex resulted in an isolated complex l deficiency, both in muscle tissue and cultured fibroblasts. The fibroblast and muscle complex I enzymatic activities were 36% and 20% of the lowest control value, respectively (Table 2). We screened the entire mtDNA and excluded the presence of disease-causing mutations. We performed homozygosity mapping to define genomic regions with candidate genes and found six homozygous regions containing structural subunits of complex I. These subunits were NDUFS5, NDUFS4, NDUFA2, NDUFA4, NDUFB6, and NDUFS3. The size of the locus on chromosome 5 between the markers D5S2115 and D5S2011 was 6.46 Mb and contained NDUFA2 and probably around 90 other genes. NDUFS4 and NDUFA2 are both located on chromosome 5. Screening of the NDUFS4 gene did not reveal a pathogenic mutation. Amplification of the three NDUFA2 exons with exon-spanning primers and sequence analysis of genomic DNA of the patient demonstrated the presence of a homozygous G-to-A substitution (c.208+5 G > A) after exon 2 (Figure 1A). The mother was found to be heterozygous for the mutation, and the mutation was not found in 107 Turkish controls or 25 patients with isolated complex I deficiency who did not harbor a mutation in the mtDNA. Leigh syndrome was diagnosed in five of these patients. We used three different splice-prediction websites to investigate the possible effect of the mutation on splicing of the NDUFA2 gene in more detail. The programs predicted that the mutation would result in a less efficient splice site, impairing splicing of exon 2. PCR analysis of the mRNA and subsequent sequencing of the mutant fragment showed that exon 2 was almost completely missing (Figures 1B and 1C). A small amount of wild-type product was present, probably because the splice site could still be used, but less efficiently. The mutation induced alternative splicing and thereby introduced a frame-shift resulting in a truncated protein.
Table 1.
Pyruvate Oxidation Rates and ATP Production Rate from Pyruvate
| Oxidation of | Patient | Control Range |
|---|---|---|
| [1-14C]pyruvate + malate | 0.91a | 3.61–7.48a |
| [1-14C]pyruvate + carnitine | 2.19a | 2.84–8.24a |
| ATP + CrP − production from pyruvate | 8b | 42–81b |
nmol CO2/hr · mU citrate synthase.
nmol ATP/hr · mU citrate synthase.
Table 2.
Respiratory-Chain Enzyme Activities in Skin Fibroblasts and Muscle Tissue of the Index Patient
| Complex | Muscle Tissue |
Skin Fibroblasts |
||
|---|---|---|---|---|
| Patient | Control Range | Patient | Control Range | |
| I | 14a | 70–251a | 0.04b | 0.11–0.26b |
| II | 78a | 67–177a | 0.17b | 0.07–0.18b |
| III | 2459a | 2200–6610a | 1.55b | 1.27–2.62b |
| IV | 1281a | 810–3120a | 0.90c | 0.68–1.19c |
mU/U citrate synthase.
mU/mU cytochrome c oxidase.
mU/mU citrate synthase.
Figure 1.
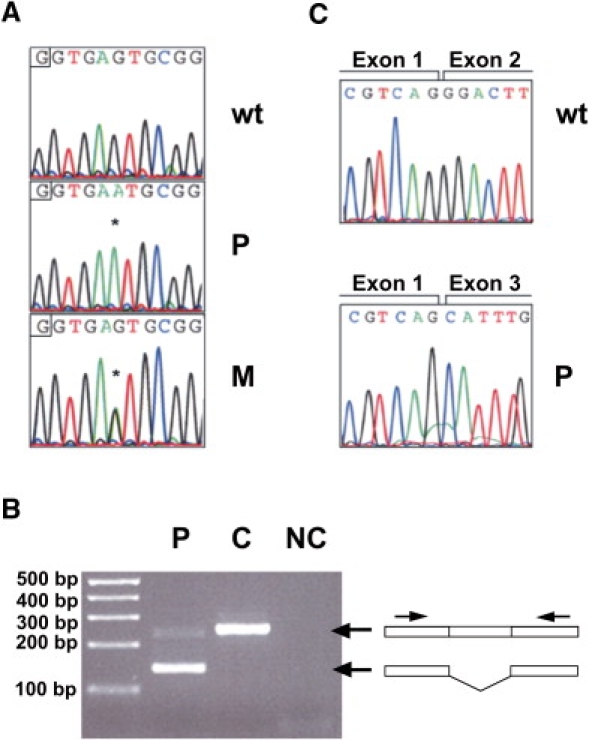
Genetic Analysis of the Mutation in NDUFA2
(A) Electropherograms showing the normal sequence of NDUFA2 (top) and the nucleotide change in fibroblasts of the patient (middle) and the mother of the patient (bottom). Asterisks represent the nucleotide substitution.
(B) PCR analysis of the mRNA showed a product of approximately 250 base pairs with control RNA, whereas PCR analysis with patient RNA resulted in a fragment of about 150 base pairs. Notably, a small amount of wild-type product was still present.
(C) Sequence analysis of the PCR fragments of (B) showed a normal exon boundary between exons 1 and 2 in the control (top) and skipping of exon 2 in the patient (bottom).
Abbreviations are as follows: wt = wild-type, p = index patient, M = mother of patient, C = control, and NC = negative control.
Complementation of the Complex I Deficiency by a Baculovirus System Expressing NDUFA2 Protein
To study the effect of the mutation on the expression of complex I, we performed SDS-PAGE analysis followed by immunoblotting with a mitochondria-enriched fraction from cultured patient fibroblasts. Figure 2A shows a marked decrease in protein level of NDUFA2 in the patient compared to the control. The immunoblot did not present the band derived from the mRNA lacking exon 2, probably because the mutant protein is not stable and will be degraded. Immunodetection with an antibody against NDUFA9 also displays a decrease of this subunit, which suggests a decrease in the amount of complex I. No increase in CII-SDHA was observed. These results are in agreement with the biochemical data. Next, we performed BN-PAGE analysis and in-gel activity measurement or immunoblotting to study the effect of the mutation on the activity and amount of fully assembled complex I. The in-gel activity (CI-IGA) assay showed a lower activity of complex I in the patient fibroblasts compared to those of the control (Figure 2B). Incubation with an antibody directed against NDUFA9 resulted in an extra band when patient fibroblasts were used, which suggests the formation of catalytically inactive subcomplexes. Furthermore, incubation with an antibody against NDUFA2 demonstrated less NDUFA2, which was only present in the holo complex I and not in the subcomplexes, than in the control. To test whether the assembly of complex I is disturbed, we performed two-dimensional BN/SDS-PAGE analysis followed by immunoblotting with antibodies directed against NDUFS3, NDUFB6, and NDUFA2 (Figure 2C). The figure shows the accumulation of subcomplexes in the patient fibroblasts, and less NDUFA2 was visible as well. These results indicate a disturbance in the assembly and/or stability of complex I.
Figure 2.
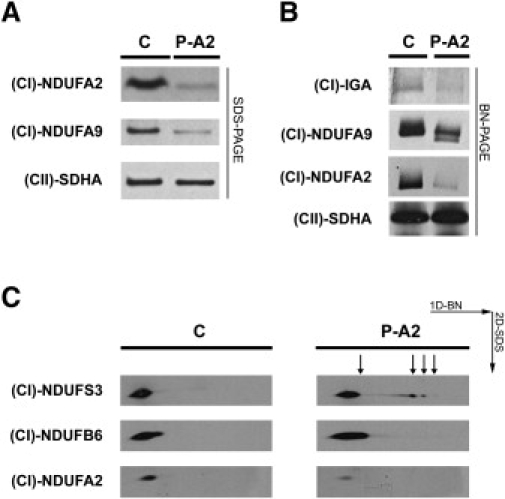
The Mutation in NDUFA2 Results in Lower Expression and Activity of Complex I and in an Accumulation of Subcomplexes in the Patient Fibroblasts
(A) SDS-PAGE analysis and immunoblotting with an antibody directed against NDUFA2 shows that the expression of NDUFA2 is decreased in the patient fibroblasts. The expression of subunit NDUFA9 is also reduced, which suggests a reduction in the expression of complex I.
(B) BN-PAGE analysis followed by an in-gel activity assay shows decreased complex I activity in the patient with a mutation in NDUFA2 versus the control. Immunoblotting and incubation with an antibody directed against NDUFA9 shows a lower expression of complex I and the formation of an extra band in the fibroblasts of the patient with the NDUFA2 mutation. The extra band suggests the formation of a subcomplex. Incubation with an antibody against NDUFA2 shows less NDUFA2, which is present in the holo complex I, when patient fibroblasts were used.
(C) Immunoblots of two-dimensional BN/SDS-PAGE show the accumulation of subcomplexes in the patient with the NDUFA2 mutation versus the control. There is also less NDUFA2 detectable in the patient fibroblasts (third panel).
Abbreviations are as follows: C = control, P-A2 = patient with the mutation in NDUFA2, (CI)-NDUFA2 = antibody directed against NDUFA2, (CI)-NDUFA9 = antibody for NDUFA9 to detect whole complex I, (CII)-SDHA = antibody for SDHA to detect complex II, (CI)-IGA = in-gel activity of complex I, (CI)-NDUFS3 = antibody directed against NDUFS3, and (CI)-NDUFB6 = antibody directed against NDUFB6.
To confirm the pathogenic role of NDUFA2, we used a baculovirus expression system containing the NDUFA2 gene in frame with a GFP tag or COX8-GFP as a control. The viruses were used for the transduction of control fibroblasts, fibroblasts of the index patient with a mutation in NDUFA2, and fibroblasts of an unrelated patient with a mutation in the NDUFS7 gene (p.Val122Met).9 The GFP tag allowed us to distinguish between endogenous and transduced protein, and this tag did not interfere with the incorporation of NDUFA2 into complex I (data not shown). The untreated fibroblasts and the fibroblasts treated with the baculovirus were used for SDS-PAGE and BN-PAGE analysis. The SDS-PAGE demonstrated proper expression of the transduced NDUFA2-GFP fusion protein (Figure 3A, first row). Next, we performed BN-PAGE of the transduced cells to investigate the expression of complex I. Immunoblot detection with an antibody directed against NDUFA9 showed almost no difference in the expression of complex I between the untreated control fibroblasts and the cells treated with the baculovirus containing the NDUFA2 or COX8 construct (Figure 3A, second row). In contrast, when fibroblasts of the index patient were treated with the NDUFA2 construct, the expression of complex I was increased in comparison to expression in untreated cells and cells treated with the COX8 construct, whereas fibroblasts of the patient with a mutation in the NDUFS7 gene showed almost no difference in complex I expression in untreated cells versus cells treated with the constructs. To check whether the wild-type NDUFA2 subunit was indeed incorporated, we performed immunoblotting with the antibody directed against GFP (Figure 3A, third row). All NDUFA2-GFP was incorporated in complex I in the fibroblasts of the patient with the NDUFA2 mutation, whereas in the control and the unrelated patient monomeric (unincorporated) NDUFA2-GFP protein could also be detected (data not shown). The band of GFP attached to COX8 was visible at the bottom of the gel (data not shown). Incubation with antibodies for complex II, III, and IV showed that the assembly of these complexes was not affected by the NDUFA2 mutation (Figure 3A, rows four to six). We performed an IGA assay to measure the activity of complex I (Figure 3B). Transduction of the control fibroblasts with baculovirus did not affect the activity of complex I. The activity of the untreated cells of the index patient was lower than that of the untreated control cells, but it was partially rescued in the cells treated with the baculovirus containing the construct with NDUFA2. Transduction of the baculovirus into the cells of the patient with a mutation in NDUFS7 did not affect the activity of complex I. To confirm these observations, we also measured the activity of complex I by biochemical analysis (Figure 3C). In the controls cells, the activity of complex I did not detectably change after treatment with the baculoviruses. However, in the fibroblasts of the patient with the NDUFA2 mutation, the activity of complex I was greater in the cells treated with the baculovirus containing NDUFA2-GFP than in the untreated cells and the cells treated with the baculovirus with COX8-GFP. No increase in activity was seen in the fibroblasts of the patient with the NDUFS7 mutation after treatment with the baculovirus containing NDUFA2-GFP. All of these results together confirm that the complex I deficiency expressed in fibroblasts is complemented by a baculovirus system expressing NDUFA2.
Mutation in NDUFA2 Is Associated with Mitochondrial Depolarization in Living Cells, which Can Be Restored by Baculoviral Complementation
The mitochondrial membrane potential (Δψ) is one of the most important indicators of mitochondrial function and metabolic activity. It is directly coupled to proper action of the respiratory chain. Using a recently developed method to sensitively measure Δψ in cultured human skin fibroblasts,27,28 we here show that the accumulation of TMRM in the mitochondrial matrix is significantly decreased in fibroblasts of the index patient compared to four healthy subjects (Figure 4A). In addition, we demonstrate that mitochondrial TMRM fluorescence is also reduced in the fibroblasts of the patient with the NDUFS7 mutation, which suggests that the decrease in Δψ is an important feature of complex I deficiency. Although this reduction appears to be relatively small, it is important to note that treatment of healthy fibroblasts with 100 nM rotenone, which caused a marked reduction in complex I activity,29 decreased the TMRM signal by ∼10%.28
Figure 4.
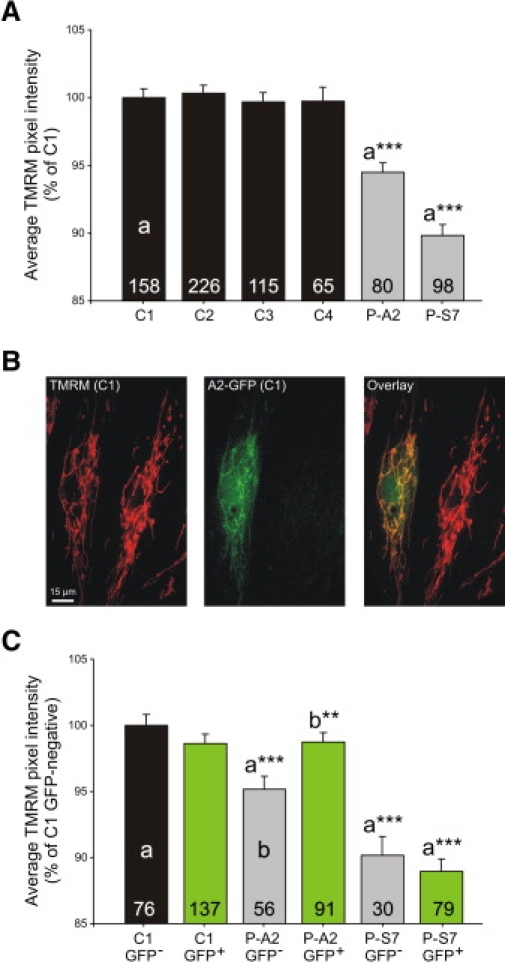
Mutation in NDUFA2 in Living Cells Is Associated with Mitochondrial Depolarization, which Can Be Restored by Baculoviral Complementation
(A) Average TMRM fluorescence pixel intensity per cell (indicative of the cell's mitochondrial membrane potential) of control fibroblasts (C1, C2, C3, C4; black bars), of fibroblasts of the patient with the mutation in NDUFA2 (P-A2 cells; gray bar), and of fibroblasts from a patient with a mutation in NDUFS7 (P-S7; gray bar). Values are given as means ± standard error. The mean value obtained with the C1 cells was set at 100%, to which all other values were related. a∗∗∗, significantly different from C1 cells (p < 0.001).
(B) Combined imaging of TMRM and NDUFA2-GFP fluorescence in baculovirus-transduced C1 cells. The left image depicts TMRM fluorescence (red color), the middle image shows the GFP signal of the same cells (green color), and the right image shows an overlay of these two images. The yellow color demonstrates colocalization of TMRM and GFP signal, indicating that only the left cell is transduced. Importantly, no bleed-through from the GFP signal into the TMRM image was present, and vice versa.
(C) Quantification of the average TMRM pixel fluorescence intensity in C1, P-A2, and P-S7 cells after transduction with a baculovirus containing the cDNA of NDUFA2-GFP. After imaging, cells were selected according to their GFP signal (GFP-negative, black and gray bars; GFP-positive, green bars), and for each subgroup, the average TMRM pixel intensity per cell was calculated. Values are given as means ± standard error. The mean value obtained with the GFP-negative C1 cells was set at 100%, to which all other values were related. a∗∗∗, significantly different from GFP-negative C1 cells (p < 0.01); b∗∗, significantly different from GFP-negative P-A2 cells (p < 0.01). Numerals in (A) and (C) represent the number of individual cells analyzed.
Abbreviations are as follows: C1-4 = control 1-4, P-A2 = patient with the mutation in NDUFA2, P-S7 = patient with the mutation p.Val122Met in NDUFS7.
Next, we investigated whether baculoviral transduction of the patient fibroblasts with the NDUFA2-GFP construct could normalize the observed depolarization of Δψ. Here, we used the advantage of combined measurement of TMRM and GFP fluorescence, enabling us to assess Δψ in non-transduced (GFP-negative) and transduced (GFP-positive) cells present on the same coverslip (Figure 4B). The left image shows that TMRM is virtually exclusively present in the mitochondria (red color), whereas the middle image shows that NDUFA2-GFP fluorescence is most intense in these organelles but also clearly visible in the nucleus and cytosol. This image furthermore shows that the right cell does not detectably express NDUFA2-GFP and that there was virtually no bleed-through of TMRM fluorescence during recording of the GFP image. Importantly, we carefully verified that there was also no bleed-through of GFP fluorescence in the TMRM image (for further details, see reference27).
As shown in Figure 4C, successful transduction of healthy fibroblasts (C1, GFP-positive cells) did not alter the TMRM signal (there was no significant difference between GFP-negative and GFP-positive cells). On the other hand, the TMRM signal was fully normalized in GFP-positive cells of the index patient, whereas the signal was still significantly decreased in their GFP-negative counterparts. Importantly, no significant changes could be observed for the NDUFS7 patient, demonstrating that the effect of the NDUFA2 transduction is specific for the cells of the index patient. Taken together, these results convincingly demonstrate that the mutation in the NDUFA2 gene is responsible for the observed depolarization of Δψ in these patient fibroblasts.
Of note, our GFP measurements revealed that not all cells were transduced, which might explain why biochemical complex I activity measurements and blue native-gel electrophoresis followed by either immunoblotting or in-gel activity measurement did not show full recovery of complex I amount and activity in the patient fibroblasts.
Discussion
To our knowledge, this is the first report describing a mutation in the NDUFA2 gene in a patient with an isolated complex I deficiency expressed in skin fibroblasts and muscle tissue. A mutation in the splice donor site after exon 2 resulted in skipping of that exon and, subsequently, in a truncated protein of 48 instead of 99 amino acids. Several lines of evidence suggest that this mutation is the underlying cause of the complex I deficiency and, therefore, of the disease in the patient: (1) the mutation involved a nucleotide change in a conserved region of the 5′ splice site, which is important for correct splicing;30 (2) the mother was heterozygous for the mutation and the mutation was not present in 107 controls of the same ethnic background; (3) the functional consequence of the mutation, skipping of exon 2, was confirmed at the cDNA level; (4) patient fibroblasts showed a decrease in complex I amount and activity as revealed by immunoblotting and biochemical and in-gel activity measurements; (5) the assembly of complex I was disturbed in these cells; (6) the mitochondrial membrane potential, as determined in intact cells, was depolarized in intact patient fibroblasts; and most importantly, (7) transduction with a baculoviral vector containing the wild-type NDUFA2 gene specifically restored both complex I amount and activity and mitochondrial membrane potential in these patient fibroblasts.
Fourteen of the complex I subunits are highly conserved across species, from mammals to bacteria.31,32 It is assumed that the orthologs of the bacterial subunits form the minimal functional complex of higher organisms.33 These subunits are essential for the catalysis of electron transfer from NADH to ubiquinone and for the generation of the proton gradient. Several mutations have been described in these subunits, causing complex I deficiency. However, the core subunits are not the only locations of disease-causing mutations. Mutations have also been described in genes encoding NDUFS4, NDUFS6, NDUFA1, and NDUFA11, which belong, together with NDUFA2, to the accessory subunits.
Not much is yet known about the role of complex I accessory subunits, but the presence of a disease-causing mutation in NDUFA2 supports the notion that at least some of them are crucial for proper complex I function. With 94% identity among mammals, NDUFA2 is one of the most conserved accessory subunits.34 The structure of its bovine ortholog, B8, shows a four-stranded mixed β sheet with three α helices packed against one side. Its overall structure is very similar to that of thioredoxins, which has led to the idea that this subunit might link cellular redox state and complex I assembly and/or activity.18 The data presented in this study show that proper assembly of enzymatic active complex I is indeed impaired in fibroblasts of the index patient. Our data furthermore show a parallel decrease in the amount and activity of the fully assembled complex, suggesting that the mutation primarily decreases the expression of the fully assembled complex rather than its intrinsic activity. In addition to its structure, homology to other proteins might also give an indication of the function of this accessory subunit. Gabaldon et al. described a paralogous relationship of NDUFA2 with the mitochondrial ribosomal proteins L43 and S25,35 which might suggest a possible connection between complex I and mitochondrial translation.
To asses the cellular consequence of the reduction in complex I amount and activity in the fibroblasts of the index patient, we performed live-cell imaging of the mitochondrial membrane potential (Δψ). Δψ is a key indicator of mitochondrial function and cell viability and is maintained by the complexes I to IV of the mitochondrial respiratory chain. The mutation in NDUFA2 caused a small but significant depolarization of Δψ in the patient cells; such a depolarization might influence the function of the mitochondria. We also observed that Δψ was significantly depolarized in a patient cell line with a mutation in NDUFS7, indicating that this depolarization might be a hallmark of complex I deficiency. Importantly, mitochondrial depolarization was restored upon baculoviral complementation of fibroblasts from the index patient, whereas the NDUFS7 cell line did not show any improvement upon this treatment. This demonstrates that mitochondrial depolarization was indeed a specific consequence of the NDUFA2 mutation in the fibroblasts of the index patient.
Complex I deficiency is one of the most frequent defects of the OXPHOS system.36 At present, the exact cause of the disease is still unknown in many patients with complex I deficiency. Therapeutic strategies for the often devastating disorders due to complex I malfunction are hardly available, and it is therefore of eminent importance to identify possible genetic causes for prenatal diagnostics and genetic counseling. Here, we report on a patient who had isolated complex I deficiency and who carried a homozygous mutation in the nuclear gene encoding the accessory complex I subunit NDUFA2, and we demonstrate with the use of baculoviral complementation technology that this mutation is responsible for a disturbed assembly of complex I and that this disturbed assembly leads to a decrease in the amount of enzymatic active complex and, at the level of the intact cell, a reduction in mitochondrial membrane potential.
Web Resources
The URLs for data presented herein are as follows:
Acknowledgments
This work was supported by the European Community's sixth Framework Programme for Research, Priority 1 “Life sciences, genomics and biotechnology for health,” contract numbers LSHM-CT-2004-005260 (MITOCIRCLE) and LSHM-CT-20040503116 (EUMITOCOMBAT). The authors would like to thank Dr. Werner Koopman (Department of Membrane Biochemistry, Radboud University Nijmegen Medical Center, Nijmegen, The Netherlands) for technical assistance in imaging studies and Angelien Heister (Department of Human Genetics, Radboud University Nijmegen Medical Center, Nijmegen, The Netherlands) for performance of homozygosity mapping.
References
- 1.Smeitink J., van den Heuvel L., DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001;2:342–352. doi: 10.1038/35072063. [DOI] [PubMed] [Google Scholar]
- 2.Pitkanen S., Feigenbaum A., Laframboise R., Robinson B.H. NADH-coenzyme Q reductase (complex I) deficiency: Heterogeneity in phenotype and biochemical findings. J. Inherit. Metab. Dis. 1996;19:675–686. doi: 10.1007/BF01799845. [DOI] [PubMed] [Google Scholar]
- 3.Loeffen J.L., Smeitink J.A., Trijbels J.M., Janssen A.J., Triepels R.H., Sengers R.C., van den Heuvel L.P. Isolated complex I deficiency in children: Clinical, biochemical and genetic aspects. Hum. Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 4.Loeffen J., Smeitink J., Triepels R., Smeets R., Schuelke M., Sengers R., Trijbels F., Hamel B., Mullaart R., van den Heuvel L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998;63:1598–1608. doi: 10.1086/302154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loeffen J., Elpeleg O., Smeitink J., Smeets R., Stockler-Ipsiroglu S., Mandel H., Sengers R., Trijbels F., van den Heuvel L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. 2001;49:195–201. doi: 10.1002/1531-8249(20010201)49:2<195::aid-ana39>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 6.Benit P., Slama A., Cartault F., Giurgea I., Chretien D., Lebon S., Marsac C., Munnich A., Rotig A., Rustin P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 2004;41:14–17. doi: 10.1136/jmg.2003.014316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van den Heuvel L., Ruitenbeek W., Smeets R., Gelman-Kohan Z., Elpeleg O., Loeffen J., Trijbels F., Mariman E., de Bruijn D., Smeitink J. Demonstration of a new pathogenic mutation in human complex I deficiency: A 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am. J. Hum. Genet. 1998;62:262–268. doi: 10.1086/301716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirby D.M., Salemi R., Sugiana C., Ohtake A., Parry L., Bell K.M., Kirk E.P., Boneh A., Taylor R.W., Dahl H.H. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J. Clin. Invest. 2004;114:837–845. doi: 10.1172/JCI20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Triepels R.H., van den Heuvel L.P., Loeffen J.L., Buskens C.A., Smeets R.J., Rubio Gozalbo M.E., Budde S.M., Mariman E.C., Wijburg F.A., Barth P.G. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann. Neurol. 1999;45:787–790. doi: 10.1002/1531-8249(199906)45:6<787::aid-ana13>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 10.Schuelke M., Smeitink J., Mariman E., Loeffen J., Plecko B., Trijbels F., Stockler-Ipsiroglu S., van den Heuvel L. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat. Genet. 1999;21:260–261. doi: 10.1038/6772. [DOI] [PubMed] [Google Scholar]
- 11.Benit P., Beugnot R., Chretien D., Giurgea I., de Lonlay-Debeney P., Issartel J.P., Corral-Debrinski M., Kerscher S., Rustin P., Rotig A. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum. Mutat. 2003;21:582–586. doi: 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Moreira D., Ugalde C., Smeets R., Rodenburg R.J., Lopez-Laso E., Ruiz-Falco M.L., Briones P., Martin M.A., Smeitink J.A., Arenas J. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann. Neurol. 2007;61:73–83. doi: 10.1002/ana.21036. [DOI] [PubMed] [Google Scholar]
- 13.Benit P., Chretien D., Kadhom N., de Lonlay-Debeney P., Cormier-Daire V., Cabral A., Peudenier S., Rustin P., Munnich A., Rotig A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet. 2001;68:1344–1352. doi: 10.1086/320603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger I., Hershkovitz E., Shaag A., Edvardson S., Saada A., Elpeleg O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008;63:405–408. doi: 10.1002/ana.21332. [DOI] [PubMed] [Google Scholar]
- 15.Ogilvie I., Kennaway N.G., Shoubridge E.A. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest. 2005;115:2784–2792. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunning C.J., McKenzie M., Sugiana C., Lazarou M., Silke J., Connelly A., Fletcher J.M., Kirby D.M., Thorburn D.R., Ryan M.T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;26:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saada A., Edvardson S., Rapoport M., Shaag A., Amry K., Miller C., Lorberboum-Galski H., Elpeleg O. C6ORF66 is an assembly factor of mitochondrial complex I. Am. J. Hum. Genet. 2008;82:32–38. doi: 10.1016/j.ajhg.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brockmann C., Diehl A., Rehbein K., Strauss H., Schmieder P., Korn B., Kuhne R., Oschkinat H. The oxidized subunit B8 from human complex I adopts a thioredoxin fold. Structure. 2004;12:1645–1654. doi: 10.1016/j.str.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Smeitink J., Sengers R., Trijbels F., van den Heuvel L. Human NADH:ubiquinone oxidoreductase. J. Bioenerg. Biomembr. 2001;33:259–266. doi: 10.1023/a:1010743321800. [DOI] [PubMed] [Google Scholar]
- 20.Janssen A.J., Smeitink J.A., van den Heuvel L.P. Some practical aspects of providing a diagnostic service for respiratory chain defects. Ann. Clin. Biochem. 2003;40:3–8. doi: 10.1258/000456303321016114. [DOI] [PubMed] [Google Scholar]
- 21.Janssen A.J., Trijbels F.J., Sengers R.C., Smeitink J.A., van den Heuvel L.P., Wintjes L.T., Stoltenborg-Hogenkamp B.J., Rodenburg R.J. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- 22.Janssen A.J., Trijbels F.J., Sengers R.C., Wintjes L.T., Ruitenbeek W., Smeitink J.A., Morava E., van Engelen B.G., van den Heuvel L.P., Rodenburg R.J. Measurement of the energy-generating capacity of human muscle mitochondria: Diagnostic procedure and application to human pathology. Clin. Chem. 2006;52:860–871. doi: 10.1373/clinchem.2005.062414. [DOI] [PubMed] [Google Scholar]
- 23.Miller S.A., Dykes D.D., Polesky H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nijtmans L.G., Henderson N.S., Holt I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- 25.Vogel R.O., Dieteren C.E., van den Heuvel L.P., Willems P.H., Smeitink J.A., Koopman W.J., Nijtmans L.G. Identification of mitochondrial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J. Biol. Chem. 2007;9:7582–7590. doi: 10.1074/jbc.M609410200. [DOI] [PubMed] [Google Scholar]
- 26.El-Sheikh A.A., van den Heuvel J.J., Koenderink J.B., Russel F.G. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J. Pharmacol. Exp. Ther. 2007;320:229–235. doi: 10.1124/jpet.106.110379. [DOI] [PubMed] [Google Scholar]
- 27.Distelmaier F., Koopman W.J., Testa E.R., de Jong A.S., Swarts H.G., Mayatepek E., Smeitink J.A., Willems P.H. Live cell quantification of mitochondrial membrane potential at the single organelle level. Cytometry A. 2008;73:129–138. doi: 10.1002/cyto.a.20503. [DOI] [PubMed] [Google Scholar]
- 28.Komen J.C., Distelmaier F., Koopman W.J., Wanders R.J., Smeitink J., Willems P.H. Phytanic acid impairs mitochondrial respiration through protonophoric action. Cell. Mol. Life Sci. 2007;64:3271–3281. doi: 10.1007/s00018-007-7357-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koopman W.J., Verkaart S., Visch H.J., van der Westhuizen F.H., Murphy M.P., van den Heuvel L.W., Smeitink J.A., Willems P.H. Inhibition of complex I of the electron transport chain causes O2-mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005;288:C1440–C1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- 30.Cartegni L., Chew S.L., Krainer A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 31.Carroll J., Fearnley I.M., Shannon R.J., Hirst J., Walker J.E. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol. Cell. Proteomics. 2003;2:117–126. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Murray J., Zhang B., Taylor S.W., Oglesbee D., Fahy E., Marusich M.F., Ghosh S.S., Capaldi R.A. The subunit composition of the human NADH dehydrogenase obtained by rapid one-step immunopurification. J. Biol. Chem. 2003;278:13619–13622. doi: 10.1074/jbc.C300064200. [DOI] [PubMed] [Google Scholar]
- 33.Fearnley I.M., Walker J.E. Conservation of sequences of subunits of mitochondrial complex I and their relationships with other proteins. Biochim. Biophys. Acta. 1992;1140:105–134. doi: 10.1016/0005-2728(92)90001-i. [DOI] [PubMed] [Google Scholar]
- 34.Hirst J., Carroll J., Fearnley I.M., Shannon R.J., Walker J.E. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim. Biophys. Acta. 2003;1604:135–150. doi: 10.1016/s0005-2728(03)00059-8. [DOI] [PubMed] [Google Scholar]
- 35.Gabaldon T., Rainey D., Huynen M.A. Tracing the evolution of a large protein complex in the eukaryotes, NADH:ubiquinone oxidoreductase (Complex I) J. Mol. Biol. 2005;348:857–870. doi: 10.1016/j.jmb.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 36.Janssen R.J., Nijtmans L.G., van den Heuvel L.P., Smeitink J.A. Mitochondrial complex I: Structure, function and pathology. J. Inherit. Metab. Dis. 2006;29:499–515. doi: 10.1007/s10545-006-0362-4. [DOI] [PubMed] [Google Scholar]