

Abstract
Bacterial cell-surface display systems coupled with quantitative screening methods offer the potential to expand protein engineering capabilities. To more fully exploit this potential, a unique bacterial surface display scaffold was engineered to display peptides more efficiently from the surface exposed C- and N-termini of a circularly permuted outer membrane protein. Using directed evolution, efficient membrane localization of a circularly permuted OmpX (CPX) display scaffold was rescued, thereby improving the presentation of diverse passenger peptides on the cell surface. Random and targeted mutagenesis directed towards linkers joining the native N- and C-termini of OmpX coupled with screening by FACS yielded an enhanced CPX (eCPX) variant which localized to the outer membrane as efficiently as the non-permuted parent. Interestingly, enhancing substitutions coincided with a C-terminal motif conserved in outer membrane proteins. Surface localization of various passenger peptides and mini-proteins was expedited using eCPX relative to that achieved with the parent scaffold. The new variant also permitted simultaneous display and labeling of distinct peptides on structurally adjacent C- and N-termini, thus enabling display level normalization during library screening and the display of bidentate or dimeric peptides. Consequently, the evolved scaffold, eCPX, expands the range of applications for bacterial display. Finally, this approach provides a route to improve the performance of cell-surface display vectors for protein engineering and design.
Keywords: bacterial display, circular permutation, directed evolution, OmpX, peptide display
Introduction
Display methodologies have proven invaluable to the discovery, production and optimization of proteins and peptides in a variety of biotechnological applications. Various approaches including phage display (Smith, 1985), mRNA (Wilson et al., 2001) and DNA display (Yonezawa et al., 2003), ribosome display (Mattheakis et al., 1994; Hanes and Pluckthun, 1997), eukaryotic virus display (Muller et al., 2003; Bupp and Roth, 2002), yeast display (Boder and Wittrup, 1997) and bacterial display (Lu et al., 1995) have been developed to screen diverse molecular repertoires for desired activities. In particular, bacterial display libraries have enabled antibody affinity maturation (Daugherty et al., 2000), the discovery of protein-binding peptides (Bessette et al., 2004), the isolation of cell-specific ligands (Nakajima et al., 2000; Dane et al., 2006) and the identification of optimal protease substrates (Boulware and Daugherty, 2006). One of the key advantages of bacterial surface display is the ability to use flow cytometry for quantitative screening of the libraries, allowing for real-time analysis of binding affinity and specificity to optimize the screening process (Wittrup, 2001). Additionally, ease of genetic manipulation, high transformation efficiency and rapid growth rate make Escherichia coli a well-suited host for display. A broad range of bacterial surface display systems have been developed allowing for insertional or terminally fused peptides and proteins to be displayed on the cell surface. Several outer membrane proteins and cellular appendage proteins have been used to present polypeptides as insertional fusions (Charbit et al., 1986; Taschner et al., 2002; Bessette et al., 2004). Ice nucleation protein (Jung et al., 1998), intimins (Christmann et al., 1999) and LppOmpA (Francisco et al., 1992) have been used to display proteins on the C-terminus of a transmembrane scaffold while N-terminal display has been accomplished using autotransporters IgA1 protease (Maurer et al., 1997) and EstA (Becker et al., 2005).
Recently, a unique bacterial display scaffold was developed that allows for both N- and C-terminal display from a circularly permuted variant of outer membrane protein OmpX (CPX) (Rice et al., 2006). This scaffold enabled display of peptides on both termini, but with reduced efficiency when compared to that obtained using insertions into OmpX. Reduced membrane localization of CPX may result from slower folding rates and reduced stability that has been described previously for circularly permuted proteins (Heinemann and Hahn, 1995). Regardless, reduced display efficiency makes it necessary to employ longer induction times to achieve sufficient display for screening via fluorescence-activated cell sorting (FACS). Importantly, inefficient display can also create an undesired selection pressure resulting in growth biases, reduced viability or differing levels of passenger localization on the cell surface. As a result, library screening based on cell fluorescence can favor passengers most efficiently localized to the surface, rather than passengers improved for the properties of interest (e.g. binding affinity).
A variety of proteins have been circularly permuted to create novel proteins of desired topologies. For instance, Guntas et al. coupled a circularly permuted β-lactamase with maltose binding protein to create a switch in which β-lactam hydrolysis activity is increased in the presence of maltose (Guntas et al., 2005). Also, fluorescent proteins have been circularly permuted to create molecular sensors that respond to Ca2+ (Baird et al., 1999). In these instances, the point of circular permutation can have important consequences for protein function. Random circular permutations often yield inactive or unfolded variants (Graf and Schachman, 1996), and those variants retaining activity often fold more slowly and are less stable than the non-permuted parent (Lindqvist and Schneider, 1997). With the aim of rescuing the reduced surface localization of CPX relative to the non-permuted OmpX, we used targeted combinatorial mutagenesis and directed evolution to identify CPX variants that would localize passenger peptides to the cell surface with an efficiency equivalent to that of native OmpX. This strategy yielded a more robust display scaffold to present diverse peptides as C- and N-terminal fusions on the surface of bacteria.
Results
Circularly permuted OmpX libraries
We previously reported a new type of protein scaffold for polypeptide display that allows for both N- and C-termini to be exterior to the cell, using circularly permuted outer membrane protein OmpX (CPX) (Rice et al., 2006). The CPX protein scaffold consists of the native OmpX signal sequence, which is cleaved after translocation; a sequence with an embedded SfiI restriction site (GQSGQ) after which peptides may be inserted; a flexible linking sequence (GGQSGQ); amino acids S54–F148 of the mature OmpX; a GGSG linker joining the native C- and N-termini; and finally, amino acids A1–S53 of the mature OmpX. To assess the extent of display, a disulfide-constrained streptavidin-binding peptide (SApep) with the amino acid sequence AECHPQGPPCIEGRK (Giebel et al., 1995) was fused to the N-terminus of CPX, allowing cell labeling using a fluorescently conjugated streptavidin probe and measurement of display levels using cytometry. CPX yielded a reduced level of peptide display when compared to cells displaying the same peptide presented as an insertion within the corresponding region of OmpX (Fig. 1). The reduced display ability requires longer induction periods to allow for sufficient fluorescence labeling and library screening, thereby causing cell stress that can result in growth biases during library screening (Daugherty et al., 1999).
Fig. 1.

Display of a streptavidin-binding peptide using OmpX, CPX and eCPX. Flow cytometric analysis of E. coli displaying a streptavidin-binding peptide (AECHPQGPPCIEGRK) (Giebel et al., 1995) as an insertion into OmpX, an N-terminal fusion to CPX or an N-terminal fusion to eCPX after varying durations of induction. The cells were induced at room temperature for time increments between 0 and 90 min then labeled with 100 nM SA–PE.
The CPX scaffold was constructed using an arbitrarily chosen flexible linker (GGSG) to join the native N- and C-termini. Thus, we reasoned that alternative linkers and point mutations within CPX could enhance the display of peptides on the cell surface. To improve the display characteristics of CPX, various regions of the transmembrane protein were targeted for mutagenesis. An optimized linker region joining the native N- and C-termini was identified by generating and screening four libraries allowing for three (3X), four (4X), five (5X) and six (6X) random residues to be inserted in place of the GGSG linker using the degenerate codon NNK. Each library was screened separately using FACS for clones exhibiting a high level of fluorescence after 50 min of induction, indicating increased display of SApep binding to streptavidin–R-phycoerythrin (SA–PE). Under these conditions, the parent CPX scaffold yielded display levels only slightly greater than background autofluorescence, making the selection of improved mutants efficient. After sorting, the display level of several clones was measured using cytometry, and their sequences were determined by DNA sequencing (Table I). Isolated clones exhibited 3- to 15-fold improved display compared to CPX after only a 30 min induction period. The identified linker sequences exhibited a preference for basic residues, and glycine was present at the first position of the linker in 14 of 16 clones characterized. In addition, four of the clones had unintended mutations preceding the native C-terminus, two with the substitution A165V and two others with G166S. Interestingly, these four clones were among the most improved display scaffolds isolated. The average display level of the selected clones from each library increased with increasing linker length, and was highest for 5- and 6-mer linker clones.
Table I.
Mutations improving CPX surface localization
| Clone | Positions | Linker | Relative display level | |
|---|---|---|---|---|
| 165 | 166 | |||
| CPX | A | G | GGSG | 2.3a/1.5b |
| Three residue linker library | ||||
| CPX-3X-1 | A | G | GRK | 8.9a |
| CPX-3X-2 | A | G | GRK | 8.3a |
| CPX-3X-3 | A | G | GTK | 7.1a |
| CPX-3X-4 | A | G | GKK | 10a |
| Four residue linker library | ||||
| CPX-4X-1 | A | G | GSKR | 18a |
| CPX-4X-2 | A | G | GRQK | 14a |
| CPX-4X-3 | A | G | SWPN | 15a |
| CPX-4X-4 | V | G | PRKS | 22a |
| Five residue linker library | ||||
| CPX-5X-1 | A | G | GRTRK | 24a |
| CPX-5X-2 | A | G | GRKRN | 22a |
| CPX-5X-3 | V | G | GATRR | 32a |
| CPX-5X-4 | A | S | GSQSK | 36a |
| Six residue linker library | ||||
| CPX-6X-1 | A | G | GTKRYH | 35a |
| CPX-6X-2 | A | G | GRRHYK | 28a |
| CPX-6X-3 | A | G | GNRRHR | 24a |
| CPX-6X-4 | A | S | GSKQSK | 38a |
| Second generation library | ||||
| CPX-L2-1 | L | S | GSKSRR | 33b |
| CPX-L2-2 | F | S | GRKNSH | 19b |
| CPX-L2-3 | I | S | GTRGSQ | 29b |
| CPX-L2-4 | L | S | GHRSHR | 27b |
| CPX-L2-5 | I | S | GDRKRR | 28b |
| CPX-L2-6 | V | A | GARGRH | 24b |
| CPX-L2-7 | V | S | GTHNSQ | 26b |
| CPX-L2-8 | V | S | GPNKSR | 17b |
| CPX-L2-9 | I | S | GPHNSR | 23b |
| CPX-L2-10 | I | S | HRGYHAQR | 33b |
aFold fluorescence above background after 50 min of expression; bfold fluorescence above background after 25 min of expression.
In an attempt to further enhance peptide display, the amino acid linker that joined the passenger peptide to the N-terminus of the display scaffold was also targeted for mutagenesis and screening for improved variants. A library was created in place of the original linking sequence GGQSGQ by randomizing these six residues. Screening yielded four clones exhibiting a 10-fold improved display when compared with CPX. Sequencing did not reveal a consensus within the target linker region. Instead, clones with enhanced display possessed a non-targeted substitution of either A165V or G166S. Since the randomly selected library members from the initial pool did not possess mutations outside of the intended region, these advantageous substitutions were rare and likely arose from PCR errors. In parallel, a library was generated with random residues at the surface exposed C-terminus of CPX, since native outer membrane proteins (Omps) possess a conserved C-terminal motif thought to aid in Omp membrane insertion or assembly (Bos and Tommassen, 2004). Four clones were isolated from this library that exhibited improved peptide display. Again, these variants did not share consensus in the randomized region, but each carried the spontaneous mutation G166S. These results suggest that the amino acid compositions of the new termini derived from circular permutation have little effect on the rate of assembly and display of CPX, whereas residues A165 and G166 play a key role in proper translocation and insertion of the protein.
A final library was designed to combine the display-enhancing mutations identified within the most improved clones from the first generation libraries. A six residue linker library was chosen to identify optimal linkers to connect the native N- and C-termini, since longer linkers appeared to improve display (Table I). After 50 min of induction, individual library members exhibited higher display levels on average when compared with clones with five amino acid linkers (Table I). The first amino acid of the linker was fixed to glycine since it was highly conserved in the isolated clones. The third and sixth positions were restricted to R/K/S/H/Q/N using the codon MRM, given the increased frequency of these residues at the proposed positions in clones with improved function, and the remaining three positions were fully randomized. Positions A165 and G166, where beneficial substitutions were observed, were also fully randomized. Improved variants were identified using two rounds of MACS followed by two rounds of FACS, sorting clones exhibiting the highest display of the SApep after 30 min of induction. Ten clones were isolated after the final FACS screening (Table I). All variant scaffolds identified possessed a more bulky hydrophobic residue (I/L/V/F) in place of alanine at position 165, a consensus for serine at position 166 and a high frequency of basic residues Arg and Lys within the linking region. The display enhancing substitutions A165V and G166S are located immediately upstream of the native C-terminus of OmpX (Fig. 2).
Fig. 2.
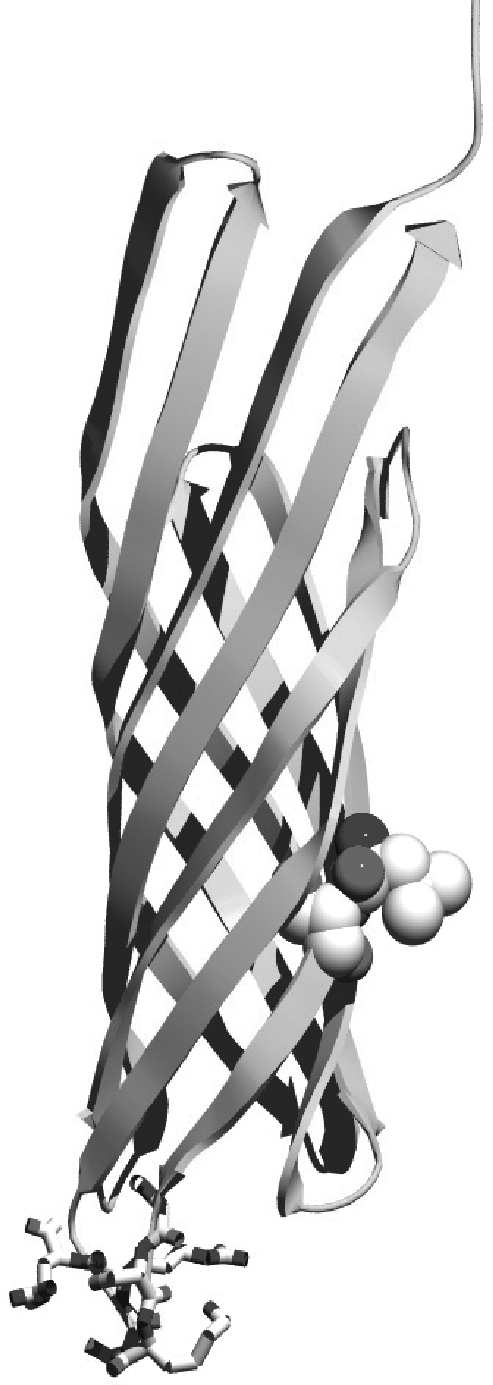
Locations of enhancing mutations within eCPX. Image was created using Molecular Operating Environment using the reported structure of OmpX (Vogt and Schulz, 1999), joining the N- and C-termini with the six residue linker GSKSRR, and creating the new termini within the second extracellular loop. Enhancing substitutions A165L and G166S are shown as space filling residues.
Expression characteristics of optimized CPX
The scaffold variant exhibiting the most improved display characteristics, CPX-L2-1, or enhanced CPX (eCPX), was then compared to parental OmpX and CPX scaffolds. The cell-surface display level of SApep was measured at incremental times after induction of expression for these three scaffolds. This peptide was displayed either at the N-terminus (CPX and eCPX) or as an insertional fusion within the second extracellular loop of OmpX. The level of display was measured before and after 30, 60 and 90 min of induction using flow cytometry (Fig. 1). The display rate of eCPX was substantially improved relative to that of CPX, and even slightly higher than that of OmpX. After only 30 min of expression, the level of display of eCPX-nSApep was 50-fold above background autofluorescence. Introducing A165L and G166S into OmpX resulted in nearly identical display of SApep relative to that obtained with OmpX (data not shown).
To determine whether the enhanced display using the eCPX scaffold was a general effect or specific to the streptavidin binding peptide SApep, several unrelated passenger peptides were fused to the N-terminus of CPX and eCPX and their display levels were compared (Fig. 3). Surprisingly, a disulfide-constrained peptide binding to C-reactive protein (CRPpep) (EWACNDRGFNCQLQR) identified by bacterial display (Bessette et al., 2004) displayed with eCPX yielded nearly 50-fold higher florescence labeling than that for CPX, after only 30 min of expression. In fact, the fluorescence of cells displaying CRPpep from CPX could not be distinguished from background (Fig. 3). Similarly, the T7•tag epitope (T7pep) (MASMTGGQQMG) was displayed more efficiently by eCPX than by CPX. A disulfide-constrained 19-mer peptide binding to vascular endothelial growth factor (VEGF) identified previously using phage display (Fairbrother et al., 1998) was also displayed over 3-fold more efficiently within eCPX. Additionally, an IgG binding mini-protein [a minimized version of the Z-domain from protein A (Braisted and Wells, 1996)] composed of 33-amino acids that form two antiparallel α-helices exhibited a display level roughly 3-fold higher than that from CPX after a 2 h induction period. Finally, P2, a proline-rich peptide (PAPSIDRSTKPPL) known to bind to the C-terminal SH3 domain of Mona (Harkiolaki et al., 2003), was expressed as a C-terminal fusion using both CPX and eCPX. Similar to the improvements of display at the N-terminus, the display level after only 30 min of expression of P2 using eCPX was improved by 9-fold compared to display with CPX. Thus, for all peptides investigated, the eCPX scaffold increased display levels when compared to the parental CPX.
Fig. 3.

Display of various peptides and mini-proteins using eCPX (shaded) and CPX (white) as measured using flow cytometry. The X-axis indicates the fold fluorescence above background for each protein target in the corresponding fluorescent channel. P2 was labeled with Mona, which is fused to the fluorescent protein YPet. CRPpep and V114 were labeled with biotinylated CRP and VEGF, respectively, then labeled with SA–PE. Mini-Z and T7pep were labeled with Alexa488 -conjugated human IgG and anti-T7•tag monoclonal IgG, respectively. SApep was detected with SA–PE.
Since the increased expression of the scaffold could potentially result in a reduction in cell viability, the viability of E. coli populations displaying two different peptides was measured as a function of the duration of induction of expression. The IgG binding mini-Z helix was selected since it exhibited reduced display in the improved eCPX scaffold. Viability was measured at 30, 60 and 90 min after induction using CFU assays and compared to that for parental strain (MC1061/pBAD33) lacking an expression cassette. Under ordinary display conditions (i.e. induction for less than 60 min), a measurable loss in viability could not be observed. After prolonged induction (90 min), cells expressing the IgG binding minihelix (miniZ) exhibited a 15% loss of viability (data not shown), while cells displaying SApep exhibited viability indistinguishable from that of cells that do not express the scaffold (MC1061/pBAD33).
Biterminal display using eCPX
Two distinct peptides were simultaneously displayed on the structurally adjacent N- and C-termini of eCPX. SApep was fused to the N-terminus, and the P2 peptide fused to the C-terminus (eCPX-nSApep-cP2). Labeling with fluorescent probes SA–PE (red) and YPet-Mona (Nguyen and Daugherty, 2005) (green) enabled independent detection of each peptide using flow cytometry. To determine the ability of eCPX to simultaneously display these two peptides, cells expressing the biterminal display scaffold were labeled with SA–PE only, YPet-Mona only or both probes concurrently. If the peptides bound to their respective receptors independently (i.e. without any steric clashes), there should be no difference between the extent of single color labeling (fluorescence intensity) of the sample labeled with one probe and that labeled with both fluorescent probes simultaneously. However, simultaneous labeling of cells expressing a fusion protein of the form eCPX-nSApep-cP2 with SA–PE and YPet-Mona, or with each probe separately yielded differing extents of labeling, consistent with steric interference between these two large fluorescent probes (290 and 34 kDa, respectively). Specifically, the fluorescence of the cells when labeled with only one probe was always greater than the fluorescence in the corresponding channel of the cells when labeled with both probes simultaneously. In an attempt to reduce steric interference, a long flexible linker of the form (GGGS)5 was inserted between SApep and eCPX, resulting in a total linker length of 26 amino acids causing SApep to be further from the cell surface and thus increasing the distance between the two peptides. Using this long linker, independent labeling of each displayed peptide was improved (Fig. 4). These results indicate that the use of a long, unstructured linker can increase the accessibility of large proteins to peptides simultaneously displayed at both termini of eCPX, without substantially reducing the level of display.
Fig. 4.

Flow cytometric measurement of simultaneous N- and C-terminal display (bi-terminal display). Overlays of two-parameter histograms resulting from clones displaying SApep and P2 on the N- and C-terminus, respectively, with a 6 (A) or a 26 (B) residue linker between SApep and the N-terminus of eCPX. In both (A) and (B) plots, the four distinct populations consist of non-displaying cells mock-labeled with SA–PE and Ypet-Mona (bottom left population), cells that display SApep and P2 labeled with only SA–PE (top left population), with SA–PE and YPet-Mona (top right population), or with only YPet-Mona (bottom right population).
Discussion
The application of bacterial display technology to a broader range of protein engineering applications has been hindered by the absence of robust, validated display scaffolds. We recently reported a circularly permuted outer membrane protein X (CPX) having the unique characteristic that both C- and N-termini of the scaffold become localized on the bacterial cell surface (Rice et al., 2006). This ‘biterminal’ scaffold provides a means for expression normalization to improve affinity screening, analogous to yeast display (Boder and Wittrup, 1997), as well as a means to display dimeric peptides. However, the permuted scaffold exhibited reduced surface localization efficiency, precluding presentation of large peptides or the display of two unique peptides simultaneously from structurally adjacent termini. Here, semi-rational design and directed evolution were used to enhance the surface localization efficiency of CPX to improve the display of diverse peptides. To identify CPX scaffold variants with optimal linker sequences for joining the native C- and N-termini, four separate libraries with three, four, five or six random linker amino acids were screened. Improved scaffold variants revealed a preference for longer linkers of five to six residues, a strict consensus for glycine at the first position of the linker, and a high frequency of basic residues in the remaining positions. Unintended substitutions (A165V, G166S) near the native C-terminus of OmpX greatly increased the display level. These substitutions probably arose from rare errors introduced during PCR which were enriched from the large libraries (109). After directed evolution, the final variant with the most improved display (eCPX) carried substitutions A165L and G166S, with a linker sequence of GSKSRR.
Two substitutions (A165L, G166S) identified to improve surface localization occur at the native C-terminus of OmpX and have a high degree of similarity to the C-termini of outer membrane proteins that lack periplasmic domains (Table II), even though these Omps share little identity. These results support previous observations that the C-terminal sequence serves an important role in membrane assembly and/or translocation (Struyve et al., 1991). The beneficial eCPX substitutions discovered could potentially enhance the ability of chaperones to recognize and assemble this topological variant. Interestingly, the extended C-terminal motif resulting from directed evolution (−L/VXYRFG) is conserved in OmpA homologs which utilize this motif in their last transmembrane β-strand that precedes a C-terminal periplasmic domain (Table II). Our results suggest that glycine is required to allow the internal, as opposed to terminal, YRF motif to interact with a putative chaperone. The improved linkers also include an increased frequency of charged residues that could serve to increase solubility and decrease aggregation to allow more protein to insert into the membrane.
Table II.
Comparison of bacterial outer membrane proteins’ C-terminal sequences with eCPX
| Omp ID | C-terminal sequencea |
|---|---|
| eCPX | DVGTWILSVGYRFGSKSRRAT… |
| NP_992891 | NIGTWVLGIGYRF |
| YP_624929 | NPIVTFLSVGYRF |
| YP_156535 | NPRTVSLSVGYRF |
| YP_341424 | DPRTVSLTVGYRF |
| ZP_00792472 | TPVTFNLNVGYRF |
| Sequence of last membrane β-strand of OmpA homologsb | |
| EAQ20637 | LVSVGLIYRFGGKTQAPA |
| EAP54071 | YWGLELSYRFGTPVAAAA |
| AAN42586 | LLSLGVSYRFGQGEAAPV |
| NP_992692 | LLSVGVSYRFGQEDAAAP |
| ZP_00826943 | MLSVGVSYRFGQDDVVAP |
aIdentities between eCPX (with substitutions A165L, G166S) and other bacterial outer membrane protein C-termini.
bOmpA homologues with a periplasmic domain following the transmembrane strand exhibit a highly conserved glycine residue following the C-terminal YRF motif of transmembrane proteins.
The evolution of an efficiently localized biterminal display scaffold (eCPX) provides several important new capabilities for peptide engineering. First, eCPX increased the display rate of various unrelated peptide fusions, on either the N- or C-terminus when compared to that achieved using the parental CPX. Increased display improves library screening efficiency using FACS and promotes the recovery of peptides that are otherwise difficult to display. For example, the display of CRPpep using CPX was only marginally detectable after 30 min of expression, but the same peptide displayed using eCPX provided a fluorescent signal 50-fold above background. If a peptide library displayed on CPX was screened for CRP binding peptides, this sequence and others like it may not have been isolated due to the difficulties in presenting this peptide using CPX. Though, it should be noted that excessive display levels, above that required for screening by FACS, should be avoided to minimize avidity interactions with target proteins. In fact, using eCPX, induction periods as short as 10 min can be sufficient for affinity screening of peptide libraries (unpublished observations). Importantly, quantifying the display level during library screening by labeling of a C-terminal peptide such as P2 allows fine affinity discrimination (VanAntwerp and Wittrup, 2000); that is, peptides with a high affinity but low display level can be differentiated from peptides with a high display but moderate affinity. Moreover, efficient biterminal display allows for the potential to generate peptide libraries on each terminus where both peptides can bind to distinct epitopes of a target protein, resulting in increased binding affinity and specificity through avidity. Finally, C-terminal display with eCPX should enable new applications such as screening for preferred PDZ C-terminal binding motifs (Harris and Lim, 2001).
Engineering of the bacterial display scaffold eCPX yielded a circularly permuted transmembrane protein that has both termini presented on the exterior of the cell and inserts into the outer membrane as efficiently as the non-permuted parent. The amino acid sequence used to join the termini played a major role in the proper function of CPX, and two substitutions adjacent to the native C-terminus aided in display of peptides using eCPX. This novel protein scaffold substantially improves the robustness of bacterial surface display via CPX and, when combined with modern cell sorting instrumentation, creates new opportunities for peptide engineering. More generally, similar linker mutagenesis strategies coupled with efficient screening methods should prove useful for optimizing other display scaffolds and circularly permuted proteins.
Materials and methods
Bacterial strains, reagents and plasmids
All experiments were performed with E.coli strain MC1061 (F- araD139 Δ(ara-leu)7696 galE15 galK16 Δ(lac)X74 rpsL (StrR) hsdR2 (rK− mK+) mcrA mcrB1) (Casadaban and Cohen, 1980). All plasmid constructs utilize pBAD33 (Cmr) (Guzman et al., 1995), with the promoter araBAD operon and the p15A origin of replication (low-copy number). KOD Hot Start DNA Polymerase (Novagen) was used for PCRs. Primers were from Operon, restriction enzymes from New England BioLabs, SA–PE from Molecular Probes and streptavidin-coated magnetic microbeads (MyOne™ Streptavidin T1) were from Invitrogen. Qiagen mini-preps and gel extraction kits were used for DNA preparation. Ni-NTA agarose for protein purification was from Qiagen, and B-PER II bacterial protein extraction reagent was from Pierce Biotechnology.
Vector and library construction
Construction of circularly permutated OmpX (CPX) was described previously (Rice et al., 2006). To monitor the display level, a streptavidin binding peptide was fused to the N-terminus of CPX as described in earlier work; this plasmid was termed pB33CPX-nSApep. To generate the libraries that joined the original N- and C-termini of OmpX with three to six random residues, primers PD1237–1240 (see Supplementary data available at PEDS Online) were used (respectively) as the reverse primer and PD179 as the forward primer, with pB33CPX-nSApep as the template. Primers are listed in the supplementary material. The random positions were encoded using NNK codons allowing for all amino acids and the amber stop codon. The product of the PCR reaction was gel purified then used as a forward primer for the next reaction, using PD180 as a reverse primer, and again with pB33CPX-nSApep as the template. The product was then gel purified, digested with SfiI and gel purified again. The digested insert was ligated into the similarly digested vector pB33CPX-nSApep. Ligation products were desalted and electroporated into electro-competent MC1061 yielding 7.5 × 107, 7.5 × 107, 1.5 × 108, 5.0 × 108 transformants, respectively. To create the second generation library (CPX-directed), primers PD1282 and PD179 were used to randomize positions A165 and G166 using pB33CPX-nSApep as the template. The product was then used as a template for PCR with PD1281 and PD179, adding the second generation library residues. The primer encoded for G at the first position of the linker and used a restricted codon of MRM to encode for residues R, K, S, H, Q or N at positions 3 and 6 of the linker; the remaining positions used NNK. The product of the previous PCR reaction was then used as a forward primer for the next reaction, using PD180 as a reverse primer and pB33CPX-nSApep as the template. The product was then digested and ligated into the similarly digested vector pB33CPX-nSApep. Ligation products were desalted and electroporated into electro-competent MC1061 yielding 1.0 × 109 transformants.
The various binding peptides were fused to the N-terminus of CPX and eCPX using a linker of GGQSGQS. PCR was used with pB33CPX-nSApep as the template, PD180 as the reverse primer and with forward primers PD1192/PD1193 for the CRP binding peptide (EWACNDRGF.NCQLQR) or PD961/PD962 for the VEGF binding peptide (VEPNCDIHVMWEWECFERL). The products were digested with SfiI and ligated into similarly digested vector and electroporated into MC1061. Primers PD1130–1133 were used in an assembly PCR to create the forward primer for the mini-Z-domain (FNMQQQRRFYEALHDPNLNEEQRNAKIKSIRDD). This primer with PD180 and template pB33CPX-nSApep was used in PCR. The product was digested with SfiI and ligated into similarly digested vector and electroporated into MC1061. The CPX-T7pep (MASMTGGQQMG) was created using overlap extension PCR with the products from the PCR reaction with primers PD179/PD705 and PD180/PD706. The products were digested with SfiI and ligated into similarly digested vector and electroporated into MC1061. To transfer these peptides to eCPX, the vectors containing the peptide CPX fusion was digested with PstI and KpnI, the smaller fragment was gel extracted and ligated to the similarly digested vector of eCPX, transferring the displayed peptide to the eCPX plasmid. To insert the P2 peptide (PAPSIDRSTKPPL) at the C-terminus of CPX and eCPX, PCR was used with PD179 as the forward primer and primers PD950/PD951 as the reverse primers with pB33CPX-nSApep at template. The primers also encoded a linker of GGQSGQ preceding the P2 peptide. The products were digested with SfiI and ligated into similarly digested vector and electroporated into MC1061. The gene is eCPX-nSApep-cP2. The streptavidin binding peptide was removed using KpnI and HindIII and ligation with a similarly cut insert that contained no N-terminally fused peptide, creating CPX-cP2.
To insert an extended linker of (GGGS)5 between the streptavidin binding peptide and eCPX-cP2, PCR was used with forward primer PD179 and reverse primers PD1429/PD1430/PD31 with pB33eCPX-nSApep-cP2 as template. The product was then gel extracted and used as the forward primer with PD180 as the reverse primer and pB33eCPX-nSApep-cP2 as template. The product was gel extracted and digested with SfiI and ligated to similarly digested vector. The portion after the OmpX signal sequence was now GQSGQ (encoding a SfiI site), AECHPQGPPCIEGRK (the streptavidin binding peptide), (GGGS)5 (the additional linker) and GGQSGQ (original linker) followed by the S54 of eCPX with the P2 peptide on the C-terminus; the gene construct is termed eCPX-nSApep-linker-cP2.
Magnetic selection and screening by FACS
Magnetic selections were performed for the first round of selection using the libraries CPX-5x, CPX-6x and CPX-directed. An overnight culture of cells corresponding to 5× the library diversity was inoculated to LB medium containing 34 µg/ml chloramphenicol (Cm) for a final cell concentration of 0.05 OD600, or 100 µl of overnight cultures into 5 ml LB Cm, which ever was greater. The cultures were then grown at 37°C to 0.5 OD600 with shaking (250 rpm), at which time the culture was moved to room temperature (22°C) to equilibrate and then induced with l-arabinose to a final concentration of 0.04% (w/v). The cells were induced by shaking (250 rpm) for 50 min, at room temperature. A volume of cells corresponding to 5× the library diversity was concentrated by centrifugation (3000 g, 4°C, 5 min) and resuspended in cold PBS to 10–30 OD600. Dynal MyOne SA beads were added to a ratio of approximately one bead per four cells. Magnetic separation was used to wash the beads four times with a volume of LB equivalent to the volume used in the initial labeling, and the beads plus bound cells were finally resuspended in LB with Cm and 0.2% glucose (w/v) for overnight growth.
For flow cytometric sorting, 50 µl of overnight cultures of the libraries were inoculated to 5 ml LB Cm. Cells were induced as described in the previous paragraph, in future rounds of sorting the induction time was decreased to 30 min. Ten microliter of cells were labeled with 100 µl of 100 nM SA–PE in PBS on ice for 45 min, pelleted by centrifugation and the supernatant was removed. Cells were resuspended in ice-cold PBS at approximately 107 cells/ml and immediately analyzed and sorted using a FACSAria cytometer with 488 nm excitation. Between 1 and 5% of the most labeled cells were collected and amplified for further rounds of analysis and/or sorting by growing overnight in LB medium containing glucose and Cm. A subset of the sort was plated directly on agar for isolation of single clones. Typically 4–10 selected clones were assayed for antigen binding by flow cytometry, and the identity of each peptide insert was determined by DNA sequencing.
Determination of peptide display levels
To compare the display level of CPX and eCPX as a function of induction time, CPX and eCPX, cells were subcultured 1:50 from overnight stocks into 5 ml LB Cm and grown for 2 h with shaking (250 rpm) at 37°C. The cells were then moved to room temperature (22°C) to equilibrate and induced with 0.04% (w/v) l-arabinose still shaking at 250 rpm. Samples (5 µl) were taken prior to induction, 30, 60 and 90 min after induction then added to 50 µl of 100 nM SA–PE in PBS and incubated on ice for 45 min. The cells were then centrifuged (3000 g, 5 min), supernatant removed and resuspended in 500 µl ice cold PBS. Cells were immediately analyzed with a FACSAria using 488 nm excitation and fluorescence data collected at 576 nm.
To compare the display of various peptides using CPX and eCPX, cultures were started using 50 µl of overnight culture in 5 ml LB Cm. Cultures expressing the mini-Z domain, SApep, P2 and T7pep were grown until an OD600 of 0.4 and moved to room temperature (22°) to equilibrate and then induced with l-arabinose to a final concentration of 0.04% (w/v) for 30 min or 2 h for the mini-Z domain. Cultures expressing the CRPpep and V114 peptides were induced at 37°C at an OD600 of 0.4 for 30 min. Induced cells (5 µl) were added to 50 µl of PBS containing the respective antigens at the following concentrations: 50 nM YPet-Mona, 65 nM biotinylated VEGF, 100 nM biotinylated CRP, 300 nM Alexa labeled human IgG, 100 nM SA–PE and 6.7 nM anti-T7•tag monoclonal IgG. Samples were labeled on ice for 45 min. Biotinylated samples were spun down at 3000 g for 5 min and the supernatant removed. Cells were resuspended in 50 µl of PBS with 10 nM SA–PE and put on ice for 45 min. Before cytometric analysis, samples were spun down at 3000 g for 5 min, supernatants removed and 500 µl of PBS added to resuspend the cells. Samples were excited at 488 nm; fluorescence data were collected at 576 nm for SA–PE labeled samples and 530 nm for Alexa labeled samples and YPet conjugated samples.
For the dual labeling experiments, cells were subcultured 1:50 from overnight stocks into 5 ml LB Cm and grown for 2 h shaking (250 rpm) at 37°C. The cells were then induced with 0.04% (w/v) l-arabinose. Cells expressing eCPX-nSApep-cP2 were expressed for 25 min at 37°C, and cells expressing eCPX-nSApep-linker-cP2 were expressed for 45 min at 37°C. Cells (5 µl) were labeled with 50 µl of PBS with 100 nM SA–PE only, 40 nM Ypet-Mona only or with both probes simultaneously. The negative controls were non-displaying cells labeled with SA–PE and Ypet-Mona. The cells were incubated at room temperature for 45 min, centrifuged at 3000 g for 5 min and supernatant removed. The cells were left on ice before resuspension with 500 µl ice cold PBS and analyzed using cytometry with 488 nm excitation and measuring emission at 576 and 530 nm.
Supplementary data
Funding
National Science Foundation CAREER award (BES-0449399) to P.S.D., U.S. Army Research Office—Institute for Collaborative Biotechnologies (DAAD19-03-D-0004).
Supplementary Material
Acknowledgements
We thank Sophia Kenrick and Karen Dane for critically reading the manuscript.
Footnotes
Edited by Andreas Pluekthun
References
- Baird G.S., Zacharias D.A., Tsien R.Y. Proc. Natl Acad. Sci. USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S., Theile S., Heppeler N., Michalczyk A., Wentzel A., Wilhelm S., Jaeger K.E., Kolmar H. FEBS Lett. 2005;579:1177–1182. doi: 10.1016/j.febslet.2004.12.087. [DOI] [PubMed] [Google Scholar]
- Bessette P.H., Rice J.J., Daugherty P.S. Protein Eng. Des. Sel. 2004;17:731–739. doi: 10.1093/protein/gzh084. [DOI] [PubMed] [Google Scholar]
- Boder E.T., Wittrup K.D. Nat. Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- Bos M.P., Tommassen J. Curr. Opin. Microbiol. 2004;7:610–616. doi: 10.1016/j.mib.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Boulware K.T., Daugherty P.S. Proc. Natl Acad. Sci. USA. 2006;103:7583–7588. doi: 10.1073/pnas.0511108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braisted A.C., Wells J.A. Proc. Natl Acad. Sci. USA. 1996;93:5688–5692. doi: 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bupp K., Roth M.J. Mol. Ther. 2002;5:329–335. doi: 10.1006/mthe.2002.0546. [DOI] [PubMed] [Google Scholar]
- Casadaban M.J., Cohen S.N. J. Mol. Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- Charbit A., Boulain J.C., Ryter A., Hofnung M. EMBO J. 1986;5:3029–3037. doi: 10.1002/j.1460-2075.1986.tb04602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann A., Walter K., Wentzel A., Kratzner R., Kolmar H. Protein Eng. 1999;12:797–806. doi: 10.1093/protein/12.9.797. [DOI] [PubMed] [Google Scholar]
- Dane K.Y., Chan L.A., Rice J.J., Daugherty P.S. J. Immunol. Methods. 2006;309:120–129. doi: 10.1016/j.jim.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Daugherty P.S., Olsen M.J., Iverson B.L., Georgiou G. Protein Eng. 1999;12:613–621. doi: 10.1093/protein/12.7.613. [DOI] [PubMed] [Google Scholar]
- Daugherty P.S., Chen G., Iverson B.L., Georgiou G. Proc. Natl Acad. Sci. USA. 2000;97:2029–2034. doi: 10.1073/pnas.030527597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother W.J., Christinger H.W., Cochran A.G., Fuh G., Keenan C.J., Quan C., Shriver S.K., Tom J.Y., Wells J.A., Cunningham B.C. Biochemistry. 1998;37:17754–17764. doi: 10.1021/bi981931e. [DOI] [PubMed] [Google Scholar]
- Francisco J.A., Earhart C.F., Georgiou G. Proc. Natl Acad. Sci. USA. 1992;89:2713–2717. doi: 10.1073/pnas.89.7.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel L.B., Cass R.T., Milligan D.L., Young D.C., Arze R., Johnson C.R. Biochemistry. 1995;34:15430–15435. doi: 10.1021/bi00047a006. [DOI] [PubMed] [Google Scholar]
- Graf R., Schachman H.K. Proc. Natl Acad. Sci. USA. 1996;93:11591–11596. doi: 10.1073/pnas.93.21.11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntas G., Mansell T.J., Kim J.R., Ostermeier M. Proc. Natl Acad. Sci. USA. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman L.M., Belin D., Carson M.J., Beckwith J. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes J., Pluckthun A. Proc. Natl Acad. Sci. USA. 1997;94:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkiolaki M., Lewitzky M., Gilbert R.J., Jones E.Y., Bourette R.P., Mouchiroud G., Sondermann H., Moarefi I., Feller S.M. EMBO J. 2003;22:2571–2582. doi: 10.1093/emboj/cdg258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris B.Z., Lim W.A. J. Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- Heinemann U., Hahn M. Prog. Biophys. Mol. Biol. 1995;64:121–143. doi: 10.1016/0079-6107(95)00013-5. [DOI] [PubMed] [Google Scholar]
- Jung H.C., Lebeault J.M., Pan J.G. Nat. Biotechnol. 1998;16:576–580. doi: 10.1038/nbt0698-576. [DOI] [PubMed] [Google Scholar]
- Lindqvist Y., Schneider G. Curr. Opin. Struct. Biol. 1997;7:422–427. doi: 10.1016/s0959-440x(97)80061-9. [DOI] [PubMed] [Google Scholar]
- Lu Z., Murray K.S., Van Cleave V., LaVallie E.R., Stahl M.L., McCoy J.M. Biotechnology (NY) 1995;13:366–372. doi: 10.1038/nbt0495-366. [DOI] [PubMed] [Google Scholar]
- Mattheakis L.C., Bhatt R.R., Dower W.J. Proc. Natl Acad. Sci. USA. 1994;91:9022–9026. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer J., Jose J., Meyer T.F. J. Bacteriol. 1997;179:794–804. doi: 10.1128/jb.179.3.794-804.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller O.J., Kaul F., Weitzman M.D., Pasqualini R., Arap W., Kleinschmidt J.A., Trepel M. Nat. Biotechnol. 2003;3:3. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- Nakajima H., Shimbara N., Shimonishi Y., Mimori T., Niwa S., Saya H. Gene. 2000;260:121–131. doi: 10.1016/s0378-1119(00)00461-3. [DOI] [PubMed] [Google Scholar]
- Nguyen A.W., Daugherty P.S. Nat. Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- Rice J.J., Schohn A., Bessette P.H., Boulware K.T., Daugherty P.S. Protein Sci. 2006;15:825–836. doi: 10.1110/ps.051897806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G.P. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Struyve M., Moons M., Tommassen J. J. Mol. Biol. 1991;218:141–148. doi: 10.1016/0022-2836(91)90880-f. [DOI] [PubMed] [Google Scholar]
- Taschner S., Meinke A., von Gabain A., Boyd A.P. Biochem. J. 2002;367:393–402. doi: 10.1042/BJ20020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanAntwerp J.J., Wittrup K.D. Biotechnol. Prog. 2000;16:31–37. doi: 10.1021/bp990133s. [DOI] [PubMed] [Google Scholar]
- Vogt J., Schulz G.E. Structure. 1999;7:1301–1309. doi: 10.1016/s0969-2126(00)80063-5. [DOI] [PubMed] [Google Scholar]
- Wilson D.S., Keefe A.D., Szostak J.W. Proc. Natl Acad. Sci. USA. 2001;98:3750–3755. doi: 10.1073/pnas.061028198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrup K.D. Curr. Opin. Biotechnol. 2001;12:395–399. doi: 10.1016/s0958-1669(00)00233-0. [DOI] [PubMed] [Google Scholar]
- Yonezawa M., Doi N., Kawahashi Y., Higashinakagawa T., Yanagawa H. Nucleic Acids Res. 2003;31:e118. doi: 10.1093/nar/gng119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.