

Abstract
Motion of integral membrane proteins to the plasma membrane in response to G-protein-coupled receptor signals requires selective cargo recognition motifs that bind adaptor protein 1 and clathrin. Angiotensin II, through the activation of AT1 receptors, promotes the recruitment to the plasma membrane of Na,K-ATPase molecules from intracellular compartments. We present evidence to demonstrate that a tyrosine-based sequence (IVVY-255) present within the Na,K-ATPase α1-subunit is involved in the binding of adaptor protein 1. Mutation of Tyr-255 to a phenylalanine residue in the Na,K-ATPase α1-subunit greatly reduces the angiotensin II-dependent activation of Na,K-ATPase, recruitment of Na,K-ATPase molecules to the plasma membrane, and association of adaptor protein 1 with Na,K-ATPase α1-subunit molecules. To determine protein-protein interaction, we used fluorescence resonance energy transfer between fluorophores attached to the Na,K-ATPase α1-subunit and adaptor protein 1. Although angiotensin II activation of AT1 receptors induces a significant increase in the level of fluorescence resonance energy transfer between the two molecules, this effect was blunted in cells expressing the Tyr-255 mutant. Thus, results from different methods and techniques suggest that the Tyr-255-based sequence within the NKA α1-subunit is the site of adaptor protein 1 binding in response to the G-protein-coupled receptor signals produced by angiotensin II binding to AT1 receptors.
The dopaminergic and renin-angiotensin systems act coordinately to regulate blood pressure, in part, by affecting sodium transport in renal proximal tubules (1-3). The apical Na/H-exchanger and basolateral Na,K-ATPase (NKA)2 are responsible for ∼75% of the renal-filtered sodium reabsorption (4, 5). By maintaining a low intracellular sodium concentration, Na,K-ATPase provides the driving force for renal proximal tubule sodium reabsorption (6). Both the Na/H-exchanger and NKA are regulated by the hormones dopamine and angiotensin II (Ang II). Dopamine activation of type 1 dopamine receptors (D1Rs) induces natriuresis and inhibits renal proximal tubule NKA activity (1, 6-10). The antinatriuretic Ang II, through activation of type 1 angiotensin II receptors (AT1Rs), stimulates sodium reabsorption in renal proximal tubule cells (11-14) and activates NKA (15, 16). We have demonstrated that NKA regulation by dopamine and Ang II is due to a reduced/increased number of NKA molecules at the plasma membrane and not to modulation of the intrinsic NKA activity (16). At the cellular level, Ang II-dependent activation of NKA is mediated by recruitment of NKA molecules from intracellular compartments to the plasma membrane via a clathrin-coated vesicle-dependent mechanism (16, 17). Protein kinase Cβ-dependent phosphorylation of the NKA α1-subunit constitutes the triggering signal for NKA plasma membrane recruitment. Phosphorylation of Ser-11 and Ser-18 in NKA α1 may induce a conformational change in the protein to facilitate the binding of adaptor protein 1 (AP-1). Although we are beginning to better understand the organization of signals initiated by stimulation of AT1Rs and the traffic of NKA molecules from intracellular compartments to the plasma membrane, the mechanism involved in cargo selection remains unclear. The present study was performed to identify the NKA α1 sequence that interacts with AP-1.
EXPERIMENTAL PROCEDURES
Materials—Ouabain, angiotensin II, chelerythrine chloride, and mouse anti-AP-1 antibody were obtained from Sigma. Alexa Fluor 488-tagged anti-mouse and Cy3-tagged anti-rabbit antibodies were purchased from Molecular Probes (Eugene, OR). N-hydroxysulfosuccinimide-biotin was obtained from Pierce. ExactaCruz E antigen detection system was purchased from Santa Cruz Biotechnology. Anti-Na,K-ATPase α1 monoclonal mouse antibody was a generous gift of Dr. Robert W. Mercer (Washington University, St. Louis, MO). Anti-Na,K-ATPase α1 polyclonal rabbit antibody was a generous gift of Dr. Jack H. Kaplan (University of Illinois, Chicago, IL). Other reagents were of the highest quality available.
Cell Culture and Transfection—OK cells stably expressing the wild-type rat Na,K-ATPase α1 or mutant forms of this protein were cultured in Dulbecco's modified Eagle's medium containing 10% calf serum and antibiotics. To prevent the expression of the endogenous NKA α1, cells were maintained all the time in 3 μm ouabain medium (18, 19). Plasmid preparation and site-directed mutagenesis were performed as described previously (20). Stable expression of Na,K-ATPase WT α1 and mutants was performed as described by Pedemonte et al. (18, 19).
Cell Treatment—The optimal concentrations of the reagents and the extension of the treatments used in these experiments were as determined previously (21-23). In some experiments, before and during the treatment with hormones, the cells were incubated with 2 μm chelerythrine chloride for 30 min to inhibit protein kinase C activity.
Determination of Rb+ Uptake—Measurements of NKA-mediated transport by Rb+ uptake were performed with attached cells as described previously (18, 19). Cells were transferred to serum-free Dulbecco's modified Eagle's medium containing 50 mm HEPES, pH 7.4, and either 3 μm or 5 mm ouabain (incubation medium). All treatments and determinations were performed at 23 °C. Then, a trace amount of [86Rb]RbCl was added to the cell medium. After 20 min, cells were washed three times with ice-cold saline and dissolved with SDS, and accumulated radioactivity was determined. NKA-mediated Rb+ uptake was calculated from the difference in tracer uptake between samples incubated in 3 μm or 5 mm ouabain. The ouabain-insensitive Rb+ uptake (measured in the presence of 5 mm ouabain) was 25-30% of the total Rb+ uptake measured. In some experiments, cells were treated with hormones before the Rb+ uptake determination. The concentrations used and the time of treatment are described in the respective figures.
Determination of Plasma Membrane Pool of NKA by Biotinylation—For biotinylation experiments, the cells were treated with 1 μm PMA or 1 pm Ang II for 10 min at room temperature, and then the medium was changed to ice-cold 10 mm Tris-HCl, pH 7.5, 2 mm CaCl2, 150 mm NaCl, and 1.5 mg/ml N-hydroxysulfosuccinimide-biotin. After incubation for 1 h at 4°C, the cells were scraped in immunoprecipitation (IP) buffer (20 mm Tris, 2 mm EDTA, 2 mm EGTA, and 30 mm sodium pyrophosphate, pH 7.3) containing a protease inhibitor mixture, frozen in liquid nitrogen, thawed rapidly, probe-sonicated twice in an ice-water bath, and frozen/thawed again. The cell suspension was centrifuged at 14,000 × g at 4 °C for 5 min. The supernatant was separated, and protein concentration was determined. Aliquots containing equal amounts of protein were transferred to clean tubes, and 0.2% Triton X-100 and 0.2% SDS were added. The suspension was incubated at 4 °C for 1 h with anti-α1 antibody and for 2 h with protein A/G-agarose, which had been prewashed three times with phosphate-buffered saline and once with IP buffer containing 0.2% Triton X-100. After separation, the agarose beads were washed four times with IP buffer containing 0.2% Triton X-100 and 0.2% SDS and once with 50 mm Tris-HCl, pH 7.4, and finally resuspended in Laemmli sample buffer. Electrophoresis, Western blot analysis with ExtrAvidin peroxidase conjugate, and densitometric analysis were performed as described previously (16, 21, 24).
Determination of AP-1 Coprecipitation with NKA α1—OK cells stably expressing either the WT or Y255F mutant form of NKA α1 were treated with 1 pm Ang II (or vehicle) for 10 min at room temperature. The cells were then scraped in ice-cold radioimmune precipitation assay buffer followed by the ExactaCruz IP protocol (performed as described by the manufacturer). Cell lysates (containing an equal amount of protein for each treatment) were precleared with Preclearing Matrix E (Santa Cruz Biotechnology) for 30 min at 4 ° C. Preimmune normal mouse serum and control antibody anti-actin C-2 IgG1 (both reagents are from Santa Cruz Biotechnology; catalog nos. sc-45051 and sc-8432, respectively) and NKA α1 IgG1 antibody were incubated with the ExactaCruz IP matrix to form IP complexes. These were washed twice with phosphate-buffered saline and then incubated with cell lysates for 1 h at 4 °C. The IP matrixes were pelleted, washed three times with radioimmune precipitation assay buffer, and finally resuspended in Laemmli sample buffer. After separation by SDS-PAGE, the proteins were transferred to a piece of polyvinylidene difluoride membrane, which was assayed by Western blot analysis with an AP-1 antibody. The protein bands were analyzed by densitometry. The polyvinylidene difluoride membrane was stripped and tested with an anti-NKA α1 antibody to determine precipitated NKA as a control of PAGE loading. The ratio of integral densities of protein bands identified by AP-1 and NKA α1 antibodies in the same membrane was calculated for each sample. Then, the membrane was stripped again and tested with an anti-phosphoserine antibody to determine the level of NKA α1 phosphorylation.
Preparation of Cells for Microscopic Studies—Cells attached to glass coverslips and grown to 90% confluence were incubated for 10 min at room temperature with 1 pm Ang II in serum-free Dulbecco's modified Eagle's medium containing 50 mm HEPES, pH 7.4. The cells were washed twice with phosphate-buffered saline containing 1.2% sucrose (PBSS), fixed with freshly prepared 4% paraformaldehyde in PBSS for 10 min, washed twice with PBSS, incubated with l-lysine/sodium m-periodate for 20 min, washed with PBSS, permeabilized with 0.2% bovine serum albumin and 0.2% Triton X-100 in PBSS for 10 min, and washed again twice with PBSS. The cells were blocked with 5% bovine serum albumin, 1% normal goat serum, and 0.2% Tween 20 for 1 h. Cells were incubated with mouse anti-AP-1 and rabbit anti-NKA α1 antibodies and washed with PBSS. In the dark, cells were incubated with Alexa Fluor 488-tagged anti-mouse and Cy3-tagged anti-rabbit antibodies for 1 h. Then, the coverslips were washed, air-dried, and mounted on glass slides using Gel-Mount containing anti-fading agents (Biomeda, Foster City, CA).
Optical Setup of Fluorescence Microscopy—To obtain high resolution three-dimensional images of the cells, the fluorescence imaging work station consisted of an Olympus IX-81 inverted fluorescence microscope (Olympus Corp., Tokyo, Japan) equipped with a 60X/100X oil immersion objective lens, cooled Hamamatsu ORCA-ER CCD camera (Hamamatsu Photonics, Hamamatsu-city, Japan), a halogen 100-watt light source, a motorized filter and shutter, and a scan wizard for collection of x/y and z image sequences and wavelength positions over time, all controlled by SimplePCI software (Compix, Cranberry Township, PA). To obtain deconvolved images of cells, at least seven z-stacks of the field with a step of 0.5-1.0 μm were acquired, and deconvolution was performed using Auto-Deblur software (AutoQuant Imaging, Troy, NY). For all intensity measurements, the Dynamic Intensity Analysis module of SimplePCI was used.
Fluorescence Resonance Energy Transfer—For FRET determinations, images were acquired sequentially through three channels using the following filter sets (Chroma Corp., Rockingham, VT): Alexa Fluor 488 (excitation, 470/40 nm; emission, 500/40 nm), Cy3 (excitation, 540/25 nm; emission, 605/55 nm), and Alexa Fluor 488/Cy3 (excitation, 470/40 nm; emission, 605/55 nm). Corrected FRET (FRETC) was calculated on a pixel-by-pixel basis for the entire image using the equation FRETC = FRET - (0.45 × Alexa Fluor 488) - (0.20 × Cy3) (25), where FRET, Alexa Fluor 488, and Cy3 correspond to background-subtracted images of cells stained with Alexa Fluor 488 and Cy3 acquired through the FRET, Alexa Fluor 488, and Cy3 channels, respectively. Values of 0.45 and 0.20 are the fractions that were calculated for the bleed-through of Alexa Fluor 488 and Cy3 fluorescence, respectively, through the FRET filter channel. Calibrations of bleed-through were performed in cells treated with only one primary/secondary set of the antibodies, either anti-AP-1/Alexa Fluor 488 or anti-NKA α1/Cy3. In the cells, treated simultaneously with both sets of antibodies, normalized FRET (FRETN) values were calculated according to the equation FRETN = FRETC/(Alexa Fluor 488 × Cy3)1/2, where FRETC, Alexa Fluor 488, and Cy3 are the mean intensities of FRETC, Alexa Fluor 488, and Cy3 fluorescence, respectively (26).
Pseudocolor FRETN images were obtained using the ISee imaging program from Digital Analysis Technology (Raleigh, NC). Pseudocolor FRETN images are displayed with deep blue (cold) indicating low values and bright red (hot) indicating high values.
Statistical Procedures—Each experiment was repeated at least three times. Comparison between the two experimental groups was determined with the non-paired Student's t test. p < 0.05 was indicated with an asterisk in the figures. For the microscopic images, at least six randomly chosen sets of cells were used in each experimental condition. The figures show representative images for each experiment.
RESULTS
Adaptor proteins such as AP-1 interact with target proteins through binding
to defined protein sequences carrying the NPXY motifs, where N, P,
and Y are asparagine, proline, and tyrosine, and X can be any amino
acid, or the more broadly used
Ypp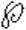 motifs, where Y is
tyrosine, p residues are highly variable but tend to be hydrophilic, and
is a residue with a bulky
hydrophobic chain (27). The
sequence of residues may be in the reverse order. Analysis of the primary
sequence of the NKA subunits revealed the presence of five of these consensus
sequences within the cytosolic loops of α1 at Tyr-50, Tyr-255, Tyr-469,
Tyr-537, and Tyr-679. The small cytosolic loop of the β-subunit has none
of the consensus sequences. One by one, the tyrosine residues in the α1
consensus sequences were mutated to phenylalanine. The mutation introduced the
minimum possible change by eliminating the tyrosine hydroxyl group and keeping
the aromatic characteristic of the residue side chain. Plasmids containing the
rat wild-type NKA α1 (WT α1) and its mutated forms were stably
expressed in OK cells, a cell culture line that is widely used as a model of
renal proximal tubule epithelia
(28-30).
Cells expressing the WT and mutant forms of NKA α1 were selected and
maintained all the time in the presence of 3 μm ouabain medium
to prevent the expression of the endogenous α1-subunit
(19,
31). Transport activity
mediated by the NKA was determined in these cells. The basal ouabain-sensitive
Rb+ uptake in WT α1 cells was 9.0 ± 0.7 nmol/min/mg
protein, and similar values were determined in cells expressing the different
NKA α1 mutants, which indicates that the NKA was expressed at the same
level in the different cell lines. Ang II-dependent stimulation of AT1Rs
(16) increased the
ouabain-sensitive Rb+ uptake to 12.4 ± 0.5 nmol/min/mg
protein (a 38% increase) in cells expressing the wild-type NKA α1
(Fig. 1). Whereas similar
transport levels were observed in most of the NKA α1 mutants, the
hormonal stimulation was significantly reduced in cells expressing the NKA
α1 Y255F mutant (Fig.
1A). We have previously demonstrated that 8-OH-DPAT, a
serotonin 1A receptor agonist, and the phorbol ester PMA, an activator of
protein kinase C, stimulate the same signaling pathway as Ang II to induce the
plasma membrane recruitment of NKA molecules
(32). Consistent with this
observation, the substitution of Tyr-255 for phenylalanine in NKA α1
impairs the stimulation of NKA activity by either 8-OH-DPAT or PMA
(Fig. 1B).
motifs, where Y is
tyrosine, p residues are highly variable but tend to be hydrophilic, and
is a residue with a bulky
hydrophobic chain (27). The
sequence of residues may be in the reverse order. Analysis of the primary
sequence of the NKA subunits revealed the presence of five of these consensus
sequences within the cytosolic loops of α1 at Tyr-50, Tyr-255, Tyr-469,
Tyr-537, and Tyr-679. The small cytosolic loop of the β-subunit has none
of the consensus sequences. One by one, the tyrosine residues in the α1
consensus sequences were mutated to phenylalanine. The mutation introduced the
minimum possible change by eliminating the tyrosine hydroxyl group and keeping
the aromatic characteristic of the residue side chain. Plasmids containing the
rat wild-type NKA α1 (WT α1) and its mutated forms were stably
expressed in OK cells, a cell culture line that is widely used as a model of
renal proximal tubule epithelia
(28-30).
Cells expressing the WT and mutant forms of NKA α1 were selected and
maintained all the time in the presence of 3 μm ouabain medium
to prevent the expression of the endogenous α1-subunit
(19,
31). Transport activity
mediated by the NKA was determined in these cells. The basal ouabain-sensitive
Rb+ uptake in WT α1 cells was 9.0 ± 0.7 nmol/min/mg
protein, and similar values were determined in cells expressing the different
NKA α1 mutants, which indicates that the NKA was expressed at the same
level in the different cell lines. Ang II-dependent stimulation of AT1Rs
(16) increased the
ouabain-sensitive Rb+ uptake to 12.4 ± 0.5 nmol/min/mg
protein (a 38% increase) in cells expressing the wild-type NKA α1
(Fig. 1). Whereas similar
transport levels were observed in most of the NKA α1 mutants, the
hormonal stimulation was significantly reduced in cells expressing the NKA
α1 Y255F mutant (Fig.
1A). We have previously demonstrated that 8-OH-DPAT, a
serotonin 1A receptor agonist, and the phorbol ester PMA, an activator of
protein kinase C, stimulate the same signaling pathway as Ang II to induce the
plasma membrane recruitment of NKA molecules
(32). Consistent with this
observation, the substitution of Tyr-255 for phenylalanine in NKA α1
impairs the stimulation of NKA activity by either 8-OH-DPAT or PMA
(Fig. 1B).
FIGURE 1.

Importance of Tyr-255 for the Ang II-dependent stimulation of NKA activity. A, the effect of Tyr-255 mutation on the Ang II stimulation of NKA-mediated Rb+ uptake. OK cells expressing WT or mutant forms of NKA α1 were treated with 1 pm Ang II for 10 min before the Rb+ uptake assay. The percentage of Rb+ uptake increase for each experimental condition was calculated with respect to a control that was not treated with Ang II. *, p < 0.05 with respect to OK WT α1 cells. B, the effect of the Y255F mutation on the stimulation of NKA-mediated Rb+ uptake by different activator/agonists. OK cells expressing the WT or Y255F mutant form of NKA α1 were treated for 10 min with 1 μm PMA, 3 μm 8-OH-DPAT, or 1 pm Ang II as described under “Experimental Procedures” before the Rb+ uptake assay. The percentage of increase for each experimental condition was calculated with respect to a non-treated control. *, p < 0.05 with respect to the control. C, the effect of the Y255F mutation on the Ang II-induced plasma membrane recruitment of NKA molecules. Cells were treated with either PMA or Ang II, and then the abundance of NKA molecules at the plasma membrane was determined by biotinylation as described under “Experimental Procedures.” Representative Western blots are shown in the upper panel. Quantitation of the Western blots is presented in the lower panel as the percentage of increase of NKA abundance at the plasma membrane. *, p < 0.05 with respect to the same treatment of OK cells expressing WT α1. D, the effect of the Y255F mutation on NKA-AP-1 coprecipitation. OK cells expressing either the WT or Y255F mutant form of NKA α1 were treated with 1 pm Ang II for 10 min and then dissolved with radioimmune precipitation assay buffer. Cell lysates were precleared with Preclearing Matrix E and incubated with ExactaCruz IP matrix conjugated to preimmune normal mouse serum (Preim), anti-actin C-2 (IgG1), and NKA α1 antibodies for 1 h. The matrix was separated by centrifugation and treated with Laemmli buffer. The precipitated proteins were separated by PAGE and transferred to polyvinylidene difluoride membrane, and Western blot analysis was performed with an anti-AP-1 antibody. Representative Western blots for AP-1 (AP1) and precipitated NKA α1 are shown in the upper panel. Quantitation data of precipitated AP-1 to NKA ratios are presented in the bar graph (lower panel) as a percentage of change of the Ang II-induced coprecipitation AP-1/NKA ratio with respect to a non-treated control. *, p < 0.05 with respect to the same treatment of OK cells expressing WT α1. E, Ang II-mediated phosphorylation of NKA α1. OK cells were treated with 1 pm Ang II for 10 min, NKA α1 was immunoprecipitated, and Western blot analysis was performed with an anti-phosphoserine antibody. Representative Western blots for NKA α1 phosphorylation (phosph) and precipitated NKA α1 are shown in the upper panel. Quantitation data of phosphorylated NKA α1 to precipitated NKA ratios are presented in the bar graph (lower panel) as a percentage of change of the Ang II-induced NKA α1 phosphorylation/NKA ratio with respect to a non-treated control.
To determine the effect of the α1 Y255F mutation on the Ang II-induced plasma membrane recruitment of NKA molecules, we measured the plasma membrane abundance of NKA molecules in cells that have been treated or not treated with Ang II. For this, we used the technique of plasma membrane protein biotinylation, which has been validated in several of our previous publications (16, 21, 24). After treatment of the cells with either PMA or Ang II, the cells were rapidly transferred to a 4 °C bath, the temperature at which the plasma membrane proteins were labeled with N-hydroxysulfosuccinimide-biotin. The low temperature stops the hormonal reaction and prevents the intracellular trafficking of NKA molecules. Changes in NKA activity in response to AT1Rs activation are paralleled by increases in abundance of newly recruited NKA units at the plasma membrane (Fig. 1C). On the contrary, in cells expressing the α1 Y255F mutant, stimulation of AT1Rs failed to increase NKA abundance at the plasma membrane. Basal amounts of NKA molecules at the plasma membrane (controls, Fig. 1C) determined in cells expressing the NKA WT α1 and the α1 Y255F mutant were not significantly different, indicating that NKA was expressed at the same level in both cell lines.
We further evaluated whether the IVVY-255 sequence represents the AP-1 binding site by looking at its interaction with NKA using co-IP and FRET techniques. As determined by co-IP assays, stimulation of AT1Rs increases the association of NKA and AP-1 molecules in cells expressing the wild-type α1, whereas this effect was greatly reduced in cells expressing the α1 Y255F mutant (Fig. 1D). Besides being used for IP, aliquots from the cell lysates were separated by PAGE and analyzed by Western blotting with an anti-NKA α1 antibody (data not shown). The same amount of NKA α1 was determined in the cell lysates corresponding to both cell types and all treatment conditions. Thus, the differences presented in the figure are not due to different amounts of NKA available for IP. Furthermore, as shown in Fig. 1D, similar amounts of NKA α1 were immunoprecipitated from cells expressing WT α1 and the Y255F mutant. In cells expressing the NKA α1 Y255F mutant, the antibody recognition for AP-1 is not significantly different from the background level observed for the IP controls performed with preimmune serum or anti-actin control antibody (the same IgG type as the anti-NKA α1 antibody). Results presented in Fig. 1D suggest that treatment of the cells with Ang II specifically increases the association of AP-1 with NKA molecules and that this association is prevented in cells expressing the NKA α1 Y255F mutant. These results are consistent with those presented in Figs. 1 and 2.
FIGURE 2.

Interaction between NKA α1 and AP-1 determined by FRET. A, images of OK cells expressing NKA WT α 1 and treated with Ang II (1 pm, 10 min), the protein kinase C inhibitor chelerythrine chloride (2 μm, 30 min), or the D1R agonist fenoldopam (1 μm, 10 min). Left panels show bright-field images of representative cells for each treatment. Fluorescence images (right panels) were acquired through the Cy3 channel, the Alexa Fluor 488 channel, and the FRET channel. Cells were treated and images were acquired as described under “Experimental Procedures.” Cont, control; BF, brightfield. B, levels of normalized FRET. FRETN was calculated as described under “Experimental Procedures.” *, p < 0.05 with respect to non-treated cells. Cnt, control; A, angiotensin II; C+A, chelerythrine + angiotensin II; F, fenoldopam.
We have previously demonstrated that stimulation of AT1Rs induces the phosphorylation of Ser-11 and Ser-18 of NKA α1 and that mutation of these amino acids prevents the Ang II-dependent regulation of NKA activity (16, 17). Consistent with this, Fig. 1E shows that the Ang II-dependent increased coprecipitation of NKA and AP-1 is accompanied by an increased phosphorylation of NKA α1. The absence of difference in phosphorylation level between cells expressing WT and mutant Y255F α1 shows that the process of AP-1-mediated recruitment of NKA α1 lies downstream of its phosphorylation.
Further support for the interaction between NKA and AP-1 molecules was obtained using FRET between fluorophores attached to the two proteins. Because it can only occur at a very close distance, FRET is the best technique to measure protein-protein interaction. We have previously optimized the use of this technique (33). After treatment with Ang II and/or other reagents, the cells were exposed to polyclonal rabbit and monoclonal mouse antibodies against NKA α1 and AP-1, respectively. Subsequently, the cells were treated with Cy3-tagged anti-rabbit and Alexa Fluor 488-tagged anti-mouse secondary antibodies. Cy3 and Alexa Fluor 488 are an excellent donor-acceptor pair for FRET determinations (34-36). Results depicted in Fig. 2A, vertical panels, demonstrate bright-field images (first panel), as well as images of the cells obtained in the Cy3 channel (second panel), Alexa Fluor 488 channel (third panel), and FRET channel (fourth panel). For determinations of normalized FRET, images of cells labeled with only one of the fluorophores were also acquired (data not shown). FRETN, which is independent of the fluorophore concentrations (37), was determined through pixel-by-pixel intensity recalculation, and it is presented in Fig. 2 as pseudocolor images. In non-treated cells, NKA α1 is mostly localized in the plasma membrane region, and AP-1 is in cytosolic regions surrounding the nucleus. We have previously demonstrated that the intracellular compartments containing NKA molecules are close to the plasma membrane and that it is difficult to distinguish between NKA molecules that are at the plasma membrane and in intracellular compartments (38). In cells treated with Ang II, it can be observed that AP-1 molecules have been recruited to the plasma membrane region where the NKA-containing intracellular compartments are located, and consequently the level of FRETN between the fluorophores attached to NKA and AP-1 has been significantly increased (arrows on Fig. 2A). In cells in which the Ang II-dependent phosphorylation of NKA α1 has been impaired by pretreatment with the protein kinase C inhibitor chelerythrine chloride, the increased level of FRETN was greatly reduced. As a control, some of the cells were treated with the D1R agonist fenoldopam, which, contrary to Ang II, induces the endocytosis of NKA molecules (39, 40), and no increased level of FRETN was observed (Fig. 2B).
Determinations of FRETN were also performed in cells expressing the Y255F and Y537F mutants of α1. Ang II stimulation of AT1Rs induced an increased level of FRETN between the fluorophores attached to NKA α1 and AP-1 in cells expressing the Y537F mutant, but not in cells expressing the Y255F mutant (Fig. 2B).
DISCUSSION
Interaction of a clathrin-dependent translocated protein with AP-1 is a key event in the recruitment of target proteins to the plasma membrane (41). In this report, we present evidence that the sequence IVVY-255 within the NKA α1 is the site of binding of AP-1 in response to AT1R stimulation. In support of this conclusion, we present results obtained by different and independent methods and techniques. The reduced Ang II-dependent stimulation of NKA activity was not due to a reduced expression of the α1 Y255F mutant. That the level of expression of NKA α1 is the same in cells expressing the wild-type α1 and the α1 Y255F mutant is illustrated by the display, under basal conditions, of the same level of NKA activity, NKA molecule abundance at the plasma membrane, and fluorescence.
Besides binding to Tyr-based sequences, AP-1 may also interact with dileucine motifs with the sequence z2-4xLL, where z is a negatively charged residue and x a polar residue (42). The NKA α1 cytosolic loops have two of these consensus sequences, EPKHLL-499 and LLLPDE-559. We have previously demonstrated that PMA, which activates the NKA activity through the same signaling pathway as Ang II (16), stimulates the NKA containing the WT α 1 at the same level as NKA molecules containing the mutants L499A and L554A (20). Therefore, so far, the NKA α1 IVVY-255 sequence is the only site of AP-1 binding that is relevant to the Ang II-dependent regulation of NKA activity.
Cells expressing the WT and mutant forms of NKA α1 were selected and maintained all the time in 3 μm ouabain medium, which prevents the expression of the endogenous α1-subunit (19, 31). Thus, endogenous non-mutated α1 should not have interfered with determinations performed in cells expressing the various mutants of α1. We have previously demonstrated that, as part of the mechanism of endocytosis of NKA molecules induced by stimulation of D1Rs, AP-2 interacts with the 537-YLEL sequence within NKA α1 (20). We determined that the mutation Y537F has no effect on Ang II-dependent stimulation of NKA activity and in the NKA α1/AP-1 interaction (as determined by FRET). Furthermore, contrary to the effect of Ang II, stimulation of D1Rs (which produced inhibition of NKA activity) did not increase the interaction of NKA α1 with AP-1. These results, as well as the observation that preventing the Ang II-induced phosphorylation of NKA α1 impairs the NKA α1/AP-1 interaction, support the conclusion that the NKA α1/AP-1 interaction is a very specific response to the activation of AT1Rs. Ang II-dependent plasma membrane recruitment of NKA molecules requires the protein kinase Cβ-mediated phosphorylation of NKA α1 (16). The fact that the Ang II-dependent increment of FRETN was impaired when the cells were treated with the protein kinase C inhibitor chelerythrine chloride is evidence of the causal link between the specific phosphorylation of α1 and the NKA/AP-1 interaction. The results of these experiments strongly suggest that the Ang II-dependent increment on FRETN is not due to some unknown artifact, such as membrane ruffling or constitutive protein delivery to the plasma membrane. It should be noted that the effect of Ang II is an acute effect produced in a few minutes. Thus, the effects observed are not due to the delivery of newly synthesized proteins to the plasma membrane.
AP-1, through its μ-subunit, interacts with the cargo Tyr-based sequences (43, 44). Although AP-1 containing the μA isoform has been implicated in the bidirectional traffic between the trans-Golgi network and endosomes, AP-1 containing the μB isoform seems to be selectively expressed in polarized epithelial cells and to be responsible for delivery of proteins to the basolateral membrane domain (43, 44). Although we have not studied it, it is likely that the μB isoform is expressed in OK, which is derived from opossum kidney proximal tubule epithelial cells. Because the regulation of NKA by Ang II occurs in a short term fashion and does not require protein synthesis (and thereby no traffic through the trans-Golgi network), it seems likely that AP-1 containing the μB isoform would be responsible for the results described in this report.
Clathrin vesicle-dependent traffic of membrane proteins requires the specific interaction of the protein cargo with clathrin adaptors (42). Such adaptors are organ-specific and have a particular distribution within the cell cytoplasm. Whereas, induced by D1Rs activation, AP-2 binds to the NKA α1 Tyr-537-based sequence during plasma membrane endocytosis (20), in this report we present evidence that Ang II-induced clathrin-dependent traffic of NKA molecules requires the binding of AP-1 to another sequence located at Tyr-255 within the NKA α1. These observations unequivocally demonstrate the existence of two specific clathrin adaptor-binding sequences that provide selectivity regarding whether the NKA molecules would traffic from or to the plasma membrane in response to G-protein-coupled receptor signals.
Acknowledgments
We thank Dr. Enrique Torre (Emory University) for advice on the fluorescence determinations.
This work was supported, in whole or in part, by National Institutes of Health Grant DK62195 (to C. H. P.). This work was also supported by American Heart Association, Texas Affiliate, Grant 0455110Y (to C. H. P.) and Grants 32X-10860 and 32P-14879 from the Swedish Research Council and a grant from the Swedish Heart and Lung Foundation and the Swedish Foundation for Kidney Research (to A. M. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NKA, Na,K-ATPase; Ang II, angiotensin II; AP-1, adaptor protein 1; AT1R, type 1 angiotensin II receptor; D1R, type 1 dopamine receptor; IP, immunoprecipitation; FRET, fluorescence resonance energy transfer; FRETN, normalized FRET; FRETC, corrected FRET; WT, wild-type; PMA, phorbol 12-myristate 13-acetate.
References
- 1.Aperia, A. C. (2000) Annu Rev. Physiol. 62 621-647 [DOI] [PubMed] [Google Scholar]
- 2.Leong, P. K., Devillez, A., Sandberg, M. B., Yang, L. E., Yip, D. K., Klein, J. B., and McDonough, A. A. (2006) Am. J. Physiol. 290 F854-F863 [DOI] [PubMed] [Google Scholar]
- 3.Zeng, C., Eisner, G. M., Felder, R. A., and Jose, P. A. (2005) Curr. Med. Chem. Cardiovasc. Hematol. Agents 3 69-77 [DOI] [PubMed] [Google Scholar]
- 4.Jose, P. A., Eisner, G. M., and Felder, R. A. (1998) Pharmacol. Ther. 80 149-182 [DOI] [PubMed] [Google Scholar]
- 5.Moe, O. W. (1999) J. Am. Soc. Nephrol. 10 2412-2425 [DOI] [PubMed] [Google Scholar]
- 6.Féraille, E., and Doucet, A. (2001) Physiol. Rev. 81 345-418 [DOI] [PubMed] [Google Scholar]
- 7.Chen, C. J., and Lokhandwala, M. F. (1992) Pharmacol. Toxicol. 70 11-17 [DOI] [PubMed] [Google Scholar]
- 8.Hussain, T., and Lokhandwala, M. F. (1998) Hypertension 32 187-197 [DOI] [PubMed] [Google Scholar]
- 9.Kansra, V., Chen, C., and Lokhandwala, M. F. (1995) Eur. J. Pharmacol. 289 391-394 [DOI] [PubMed] [Google Scholar]
- 10.Zhang, A., Devarajan, P., Dorfman, A. L., and Morrow, J. S. (1998) J. Biol. Chem. 273 18681-18684 [DOI] [PubMed] [Google Scholar]
- 11.Cheng, H. F., Becker, B. N., and Harris, R. C. (1996) J. Clin. Investig. 97 2745-2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inagami, T., Kambayashi, Y., Ichiki, T., Tsuzuki, S., Eguchi, S., and Yamakawa, T. (1999) Clin. Exp. Pharmacol. Physiol. 26 544-549 [DOI] [PubMed] [Google Scholar]
- 13.Navar, L. G., Harrison-Bernard, L. M., Imig, J. D., Wang, C. T., Cervenka, L., and Mitchell, K. D. (1999) J. Am. Soc. Nephrol. 10 Suppl. 12, S266-S272 [PubMed] [Google Scholar]
- 14.Navar, L. G., Lewis, L., Hymel, A., Braam, B., and Mitchell, K. D. (1994) J. Am. Soc. Nephrol. 5 1153-1158 [DOI] [PubMed] [Google Scholar]
- 15.Aperia, A., Holtback, U., Syren, M. L., Svensson, L. B., Fryckstedt, J., and Greengard, P. (1994) FASEB J. 8 436-439 [DOI] [PubMed] [Google Scholar]
- 16.Efendiev, R., Budu, C. E., Cinelli, A. R., Bertorello, A. M., and Pedemonte, C. H. (2003) J. Biol. Chem. 278 28719-28726 [DOI] [PubMed] [Google Scholar]
- 17.Pedemonte, C. H., Efendiev, R., and Bertorello, A. M. (2005) Semin. Nephrol. 25 322-327 [DOI] [PubMed] [Google Scholar]
- 18.Pedemonte, C. H., Pressley, T. A., Cinelli, A. R., and Lokhandwala, M. F. (1997) Mol. Pharmacol. 52 88-97 [DOI] [PubMed] [Google Scholar]
- 19.Pedemonte, C. H., Pressley, T. A., Lokhandwala, M. F., and Cinelli, A. R. (1997) J. Membr. Biol. 155 219-227 [DOI] [PubMed] [Google Scholar]
- 20.Done, S. C., Leibiger, I. B., Efendiev, R., Katz, A. I., Leibiger, B., Berggren, P. O., Pedemonte, C. H., and Bertorello, A. M. (2002) J. Biol. Chem. 277 17108-17111 [DOI] [PubMed] [Google Scholar]
- 21.Efendiev, R., Bertorello, A. M., Pressley, T. A., Rousselot, M., Feraille, E., and Pedemonte, C. H. (2000) Biochemistry 39 9884-9892 [DOI] [PubMed] [Google Scholar]
- 22.Efendiev, R., Bertorello, A. M., Zandomeni, R., Cinelli, A. R., and Pedemonte, C. H. (2002) J. Biol. Chem. 277 11489-11496 [DOI] [PubMed] [Google Scholar]
- 23.Efendiev, R., and Pedemonte, C. H. (2006) J. Am. Soc. Nephrol. 17 31-38 [DOI] [PubMed] [Google Scholar]
- 24.Efendiev, R., Yudowski, G. A., Zwiller, J., Leibiger, B., Katz, A. I., Berggren, P. O., Pedemonte, C. H., Leibiger, I. B., and Bertorello, A. M. (2002) J. Biol. Chem. 277 44108-44114 [DOI] [PubMed] [Google Scholar]
- 25.Gordon, G. W., Berry, G., Liang, X. H., Levine, B., and Herman, B. (1998) Biophys. J. 74 2702-2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia, Z., and Liu, Y. (2001) Biophys. J. 81 2395-2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonifacino, J. S., and Dell'Angelica, E. C. (1999) J. Cell Biol. 145 923-926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guimaraes, J., Vieira-Coelho, M., Serrao, M., and Soares-da-Silva, P. (1997) Int. J. Biochem. Cell Biol. 29 681-688 [DOI] [PubMed] [Google Scholar]
- 29.Malstrom, K., Stange, G., and Murer, H. (1987) Biochim. Biophys. Acta 902 269-277 [DOI] [PubMed] [Google Scholar]
- 30.Nash, S. R., Godinot, N., and Caron, M. G. (1993) Mol. Pharmacol. 44 918-925 [PubMed] [Google Scholar]
- 31.Lane, L. K., Feldmann, J. M., Flarsheim, C. E., and Rybczynski, C. L. (1993) J. Biol. Chem. 268 17930-17934 [PubMed] [Google Scholar]
- 32.Budu, C. E., Efendiev, R., Bertorello, A. M., and Pedemonte, C. H. (2002) Br. J. Pharmacol. 137 1380-1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Efendiev, R., Cinelli, A. R., Leibiger, I. B., Bertorello, A. M., and Pedemonte, C. H. (2006) FEBS Lett. 580 5067-5070 [DOI] [PubMed] [Google Scholar]
- 34.Achnine, L., Blancaflor, E. B., Rasmussen, S., and Dixon, R. A. (2004) Plant Cell 16 3098-3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gidwani, A., Brown, H. A., Holowka, D., and Baird, B. (2003) J. Cell Sci. 116 3177-3187 [DOI] [PubMed] [Google Scholar]
- 36.Park, J. S., Gamboni-Robertson, F., He, Q., Svetkauskaite, D., Kim, J. Y., Strassheim, D., Sohn, J. W., Yamada, S., Maruyama, I., Banerjee, A., Ishizaka, A., and Abraham, E. (2006) Am. J. Physiol. 290 C917-C924 [DOI] [PubMed] [Google Scholar]
- 37.Xia, Z., Zhou, Q., Lin, J., and Liu, Y. (2001) J. Biol. Chem. 276 1766-1771 [DOI] [PubMed] [Google Scholar]
- 38.Efendiev, R., Das-Panja, K., Cinelli, A. R., Bertorello, A. M., and Pedemonte, C. H. (2007) Br. J. Pharmacol. 151 1006-1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hussain, T., and Lokhandwala, M. F. (1997) Clin. Exp. Hypertens. 19 131-140 [DOI] [PubMed] [Google Scholar]
- 40.Hussain, T., and Lokhandwala, M. F. (1997) Am. J. Physiol. 272 F339-F346 [DOI] [PubMed] [Google Scholar]
- 41.Ohno, H., Stewart, J., Fournier, M.-C., Bosshart, H., Rhee, I., Miyatake, S., Saito, T., Gallusser, A., Kirchhausen, T., and Bonifacino, J. S. (1995) Science 269 1872-1875 [DOI] [PubMed] [Google Scholar]
- 42.Kirchhausen, T. (1999) Annu. Rev. Cell Dev. Biol. 15 705-732 [DOI] [PubMed] [Google Scholar]
- 43.Folsch, H., Ohno, H., Bonifacino, J. S., and Mellman, I. (1999) Cell 99 189-198 [DOI] [PubMed] [Google Scholar]
- 44.Ohno, H., Tomemori, T., Nakatsu, F., Okazaki, Y., Aguilar, R. C., Foelsch, H., Mellman, I., Saito, T., Shirasawa, T., and Bonifacino, J. S. (1999) FEBS Lett. 449 215-220 [DOI] [PubMed] [Google Scholar]