

Abstract
Although a crystal structure and a pharmacophore model are available for cytochrome P450 2C8, the role of protein flexibility and specific ligand-protein interactions that govern substrate binding are poorly understood. X-ray crystal structures of P450 2C8 complexed with montelukast (2.8 Å), troglitazone (2.7 Å), felodipine (2.3 Å), and 9-cis-retinoic acid (2.6 Å) were determined to examine ligand-protein interactions for these chemically diverse compounds. Montelukast is a relatively large anionic inhibitor that exhibits a tripartite structure and complements the size and shape of the active-site cavity. The inhibitor troglitazone occupies the upper portion of the active-site cavity, leaving a substantial part of the cavity unoccupied. The smaller neutral felodipine molecule is sequestered with its dichlorophenyl group positioned close to the heme iron, and water molecules fill the distal portion of the cavity. The structure of the 9-cis-retinoic acid complex reveals that two substrate molecules bind simultaneously in the active site of P450 2C8. A second molecule of 9-cis-retinoic acid is located above the proximal molecule and can restrain the position of the latter for more efficient oxygenation. Solution binding studies do not discriminate between cooperative and noncooperative models for multiple substrate binding. The complexes with structurally distinct ligands further demonstrate the conformational adaptability of active site-constituting residues, especially Arg-241, that can reorient in the active-site cavity to stabilize a negatively charged functional group and define two spatially distinct binding sites for anionic moieties of substrates.
Cytochrome P450 2C8 is one of the principal drug-metabolizing P4504 monooxygenases expressed in human liver. It is the predominant hepatic P450 catalyzing the 6α-hydroxylation of paclitaxel (1) and the epoxidation of arachidonic acid (2, 3). Additionally, P450 2C8 contributes extensively to the metabolism of drugs such as pioglitazone, rosiglitazone, troglitazone, amodiaquine, amiodarone, and cerivastatin (4) as well as to the oxidation of retinoic acid (5–8). A screen of 209 commonly used drugs and related compounds identified several potent inhibitors of P450 2C8, including montelukast, candesartan cilexetil, mometasone furoate, clotrimazole, and felodipine (9). In addition, the glucuronide of gemfibrozil is a potent inhibitor of P450 2C8 (10, 11), and this inhibition is thought to underlie a drug-drug interaction between gemfibrozil and cerivastatin that can increase risk of rhabdomyolysis. Interestingly, glucuronides of 17β-estradiol (12) and diclofenac (13) are also P450 2C8 substrates.
A pharmacophore model for substrates of P450 2C8 was proposed by Mansuy and co-workers (14) based on the observation that many substrates are large organic anions at physiologic pH and exhibit sites of oxidation that are located ∼13 Å from the anionic group. Additionally, it was noted that several polar moieties are often present at intermediate distances between the site of hydroxylation and the anionic group. Site-directed mutagenesis further suggested that Arg-241 might play a role in the binding of anionic substrates, but the substrate-free structure of P450 2C8 (Protein Data Bank code 1pq2) indicated that Arg-241 is located outside the active-site cavity (15).
The structure of P450 2C8 was first determined in our laboratory for the enzyme crystallized in the absence of substrates or inhibitors (15). Consistent with the large size of several substrates and inhibitors such as paclitaxel and montelukast, the enzyme exhibits a relatively large substrate-binding cavity compared with the ones evident for structures of most other human P450s (16). Computer-simulated docking indicated that the large active-site cavity is likely to accommodate substrates in several possible binding poses that do not necessarily conform closely to the proposed pharmacophore. Additionally, the docking simulations suggested that anionic groups might be accommodated in a large substrate access channel located between the helix B-C loop and β-sheet 1 with the potential for polar interactions with protein side chains as well as residual water molecules (14). Further computer simulations also indicated that all-trans-retinoic acid might bind in either a proximal site or an alternative distal site near helix B′ that places the retinoid carboxylate close to Arg-241. The latter suggested that conformational changes could allow Arg-241 to neutralize the charge of the retinoid in the distal site.
In this study, we characterize the ligand-protein binding interactions of P450 2C8 with felodipine and three anionic substrates (montelukast, troglitazone, and 9-cis-retinoic acid) by x-ray crystallography. The results indicate that these compounds exhibit single poses in the active site of P450 2C8 and provide information regarding adaptive changes in the structure of the enzyme. In addition, the complex of the enzyme with 9-cis-retinoic acid displays two molecules bound simultaneously in the active-site cavity. The two molecules exhibit two modes for accommodating the anionic substrates in the active site of the enzyme with the proximal substrate molecule positioned for 4-hydroxylation.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—A modified form of P450 2C8 was expressed in Escherichia coli transformed with the pCW2C8dH plasmid and purified as described previously (15), with modification of the buffer used for eluting the protein from the metal ion affinity column to contain 30 mm potassium phosphate (pH 7.4), 300 mm NaCl, 20% glycerol, 10 mm β-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride, 5 mm CYMAL-5 (Anatrace), and 30 mm histidine. The protein was modified to remove the N-terminal membrane targeting sequence and to add a C-terminal histidine tag to facilitate purification as described previously (15).
For comparative substrate binding studies, the pCW2C8mod plasmid described previously for the expression of the membrane-bound enzyme in E. coli (17) was modified to express the enzyme with a C-terminal histidine tag. The C-terminal coding sequence was isolated following digestion of the pCW2C8dH plasmid using the endonucleases SphI and HindIII. The fragment was ligated into the corresponding sites of the pCW2C8mod plasmid, and the construct (pCW2C8modH) was verified by sequence analysis. Membranes were isolated from E. coli strain GC5 (PGC Scientifics, Fredrick, MD) transformed with the pCW2C8modH expression vector, and full-length P450 2C8modH was extracted from preparations of cell membranes using the detergent Nonidet P-40 as described (17). The protein was purified from the detergent extract using the procedures for purification of truncated P450 2C8dH. The wash and elution buffers used for chromatography contained 0.4% (w/v) CHAPS to maintain the solubility of the full-length protein. The concentrations of P450 preparations were estimated by difference spectroscopy of the carbon monoxide complex of the dithionite-reduced enzyme at 450 nm relative to the reduced enzyme in the absence of carbon monoxide using an extinction coefficient of 91 mm-1 cm-1. Alternatively, the concentration of the purified ferric enzyme was estimated from the absorbance at 417 nm using an extinction coefficient of 107 mm-1 cm-1. Spectral data were obtained using a Varian Cary 1E UV-visible spectrophotometer.
Crystallization of P450 2C8 Complexes—Crystals of P450 2C8 complexed with the R-enantiomer of the sodium salt of montelukast (R-(E)-1-(((1-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methylethyl)phenyl)propyl)thio)methyl)cyclopropaneacetic acid; Sequoia Research Products, Pangbourne, UK) (see Fig. 1) were grown in sitting drops obtained by mixing 1 μl of P450/montelukast solution (232 μm P450 2C8, 470 μm R-montelukast, 265 μm sodium palmitate (Sigma), 174 μm C12E8 detergent (Anatrace), 48 mm potassium phosphate (pH 7.4), 17.4 mm HEPES (pH 7.4), 240 mm NaCl, 96 μm dithiothreitol, 480 μm EDTA, 4.8% glycerol, and 9% methanol) with 1.25 μl of precipitant solution (6% polyethylene glycol 3350, 0.1 m HEPES (pH 7.5), 0.1 m Li2SO4, 200 μm R-montelukast, and 4% MeOH). The drop was equilibrated by vapor diffusion at 25 °C with the reservoir solution containing 8% polyethylene glycol 3350, 0.1 m HEPES (pH 7.5), 0.1 m Li2SO4, and 2% ethylene glycol.
FIGURE 1.

Structures of R-felodipine, R-montelukast, 2R,5R-troglitazone, and 9-cis-retinoic acid.
Crystals of the P450 2C8 complex with the 2R, 5R-stereoisomer of troglitazone (5R-((4-((6-hydroxy-2,5,7,8-tetramethyl-chroman-2R-yl)methoxy)phenyl)methyl)-1,3-thiazolidine-2,4-dione; kindly provided by Pfizer) (see Fig. 1) were grown in sitting drops obtained by mixing 1.25 μl of P450/substrate/detergent solution with 1.25 μl of reservoir solution. The P450/substrate/detergent solution was prepared by combining 4 μl of 465 μm P450 2C8 in 50 mm potassium phosphate (pH 7.4), 500 mm NaCl, and 20% glycerol; 0.21 μl of 10 mm troglitazone in ethanol; 0.21 μl of 10 mm sodium palmitate in ethanol; and 1 μl of 1 mm C12E8 in water. The drop was equilibrated by vapor diffusion at 25 °C with a reservoir solution consisting of 10% polyethylene glycol 3350, 0.1 m HEPES (pH 7.5), 0.1 m Li2SO4, and 20% glycerol in water.
Crystals of the P450 2C8 complex with felodipine (methyl ethyl 4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate) (Fig. 1) were grown in sitting drops obtained by mixing 1 μl of P450/felodipine solution (194 μm P450 2C8, 556 μm racemic felodipine (Sigma), 265 μm sodium palmitate, 174 μm C12E8, 48 mm potassium phosphate (pH 7.4), 17.4 mm HEPES (pH 7.4), 240 mm NaCl, 96 μm dithiothreitol, 480 μm EDTA, 4.8% glycerol, and 9% methanol) with 1.25 μl of precipitant/felodipine solution (6% polyethylene glycol 3350, 0.1 m HEPES (pH 7.5), 0.1 m Li2SO4, 150 μm racemic felodipine, and 4% MeOH). The drop was equilibrated by vapor diffusion at 25 °C with a reservoir solution containing 80% precipitant solution (6% polyethylene glycol 3350, 0.1 m HEPES (pH 7.5), and 0.1 m Li2SO4) and 20% glycerol.
Crystals of the P450 2C8 complex with 9-cis-retinoic acid (a gift from F. Hoffmann-La Roche Ltd., Basel, Switzerland) were grown in sitting drops obtained by mixing 1 μl of P450/substrate solution (256 μm P450 2C8, 905 μm 9-cis-retinoic acid, 261 μm of sodium palmitate, 174 μm C12E8, 48 mm potassium phosphate (pH 7.4), 17.4 mm HEPES (pH 7.4), 240 mm NaCl, 96 μm dithiothreitol, 480 μm EDTA, 4.8% glycerol, and 13% methanol) with 1.25 μl of precipitant/substrate solution (8% polyethylene glycol 3350, 0.1 m HEPES (pH 7.5), 0.1 m Li2SO4, 400 μm 9-cis-retinoic acid, and 4% MeOH). The drops were equilibrated by vapor diffusion at 25 °C with a reservoir solution containing 8% polyethylene glycol 3350, 0.1 m HEPES (pH 7.5), and 0.1 m Li2SO4.
Data Collection, Processing, and Structure Solution—Crystals were soaked for a few seconds in cryoprotectant solution (10% polyethylene glycol 3350, 80 mm HEPES (pH 7.5), 80 mm Li2SO4, 50 mm NaCl, and 30% ethylene glycol) and flash-frozen in liquid nitrogen prior to data collection. The data used for the determination of the structure of the 2C8-montelukast complex were collected at 100 K using a rotating anode with a CuKα source. The data were integrated and scaled using the program CrystalClear (Molecular Structure Corp., Houston, TX). Data for the other three complexes were collected at 100 K at the Stanford Synchrotron Radiation Laboratory using the beamlines indicated in Table 1, and data from single crystals were integrated and scaled using Mosflm and Scala, respectively (18). Data collection statistics for each data set are given in Table 1.
TABLE 1.
Data collection and processing statistics PDB, Protein Data Bank; SSRL, Stanford Synchrotron Radiation Laboratory.
| Complex | Felodipine | Montelukast | 9-cis-Retinoic Acid | Troglitazone |
|---|---|---|---|---|
| PDB code | 2nnj | 2nni | 2nnh | 2vn0 |
| Space group | I222 | I222 | P21212 | I222 |
| Used wavelength (Å) | 0.98 | 1.5418 | 0.98 | 0.98 |
| Resolution limit (Å)a | 2.28 (2.35–2.28) | 2.80 (2.9–2.8) | 2.60 (2.67–2.60) | 2.70 (2.77–2.70) |
| Cell parameters | a = 74.41, b = 139.22, c = 163.23 Å | a = 74.14, b = 140.52, c = 164.54 Å | a = 139.81, b = 163.62, c = 73.56 Å | a = 74.20, b = 136.09, c = 163.24 Å |
| X-ray source | BL1-5 SSRL | Rotating anode Cu target | BL11-1 SSRL | BL9-1 SSRL |
| Total observations | 281,119 | 98,893 | 357,238 | 181,911 |
| Unique reflectionsa | 38,962 | 21,514 | 51,297 | 23,135 |
| Completeness of all data (%)a | 99.6 (97.6) | 99.6 (97.8) | 97.7 (97.4) | 100.0 (100.0) |
| Redundancya | 7.2 (7.4) | 4.6 (4.4) | 7.0 (7.2) | 7.9 (8.0) |
| Mean I/σa | 8.7 (1.8) | 11.9 (3.7) | 7.7 (1.7) | 7.5 (1.5) |
| Rsym valuea,b | 0.063 (0.381) | 0.079 (0.38) | 0.079 (0.45) | 0.065 (0.52) |
Values in parentheses indicate data in the highest resolution shell
Rsym = ΣhΣj|〈I(h)|〉 – I(h)j|/ΣhΣj|〈I(h)|〉, where I(h) is the intensity of an individual reflection, and |〈I(h)|〉 is the mean intensity of that reflection
Initial phase information was obtained by molecular replacement with the original P450 2C8 structure as the search model (Protein Data Bank code 1pq2). The diffraction data for the 2C8 complexes with montelukast, felodipine, and troglitazone were integrated and scaled in the I222 space group, and a single molecule of 2C8 constitutes the asymmetric unit. Two P450 2C8 molecules were found in the asymmetric unit of the 2C8-retinoic acid complex, which crystallized in the P21212 space group. Structural models were fit to electron density maps using the program O (19) and refined using CNS Version 1.1 (20). The refinement statistics for each model are shown in Table 2.
TABLE 2.
Model refinement statistics PDB, Protein Data Bank.
| Complex | Felodipine | Montelukast | 9-cis-Retinoic Acid | Troglitazone |
|---|---|---|---|---|
| PDB code | 2nnj | 2nni | 2nnh | 2vn0 |
| Resolution range (Å) | 50.0–2.28 | 48.7–2.8 | 50.0–2.60 | 43.88–2.70 |
| Reflections used | 36,898 | 20,432 | 48,714 | 23,134 |
| Reflections used in Rfree set | 1962 | 1082 | 2557 | 1167 |
| Rcryst/Rfreea | 0.24/0.27 | 0.23/0.27 | 0.24/0.29 | 0.22/0.25 |
| Protein atoms (average B-factor; residues 28–490 of native protein) | 3694 (56.0) | 3694 (56.0) | 3694 (46.3)/3694 (49.5) | 3694 (56.2) |
| Heme atoms (average B-factor) | 43 (29.2) | 43 (49.1) | 43 (31.6)/43 (31.0) | 43 (44.8) |
| Ligand atoms (average B-factor) | 25 (45.2) | 41 (71.8) | 22 (50.3)/22 (52.6) | 31 (81.1) |
| 22 (52.2)/22 (44.5) | ||||
| Palmitic acid atoms (average B-factor) | 18 (57.8) | 18 (58.9) | 18 (54.9)/28 (47.5) | 18 (58.6) |
| Solvent atoms (average B-factor) | 179 (51.2) | 11 (36) | 91 (42.8) | 30 (44.8) |
| Sulfate atoms (average B-factor) | 15 (78.9) | 5 (84.5) | 5 (58.9)/5 (81.1) | 0 |
| Ramachandran plot (% residues) | ||||
| Most favored regions | 89.7 | 85.3 | 86.0 | 86.7 |
| Allowed regions | 9.4 | 12.2 | 13.2 | 12.0 |
| Generously allowed regions | 0.5 | 1.9 | 0.7 | 1.0 |
| Disfavored regions | 0.5b,c | 0.6b,d | 0.1e | 0.2b |
| r.m.s. deviationf | ||||
| Bond angle | 1.34° | 1.40° | 2.00° | 1.4° |
| Bond length (Å) | 0.007 | 0.008 | 0.019 | 0.008 |
Rcryst or Rfree = Σ||Fo| – |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively
Gln-184
Leu-476
Ile-38
Arg-A377
Root mean square (r.m.s.) deviations relate to the Engh and Huber parameters
Ligand Binding Studies—The binding of 9-cis-retinoic acid to purified P450 2C8dH and 2C8modH was examined by monitoring the concentration-dependent increase in the high spin form of the enzyme by visible absorption spectroscopy. The sample cuvette (1.000-cm path length) contained 2 or 20 μm P450 in 1 ml of 50 mm potassium phosphate buffer (pH 7.4) containing 500 mm NaCl. The ligand dissolved in methanol was added in 0.5-μl aliquots to the sample cuvette, and spectra were recorded digitally at 0.25-nm intervals from 260 to 700 nm. The final concentration of methanol was ≤2%. Following correction of each spectrum for dilution, difference spectra were computed by subtraction of the spectrum of the enzyme obtained in the absence of the ligand from each of the spectra recorded following the addition of the ligand. The conversion of the enzyme from low spin to high spin was monitored by the increase in absorbance of the enzyme at 646 nm and decrease at 570 nm. The concentration-dependent changes in absorbance were fit with a quadratic form of the binding equation by nonlinear regression using SlideWrite Plus (Advanced Graphics Software, Carlsbad, CA) as described under “Results.” Additional models of multiple equilibrium binding were fit to the data by numerical simulations and nonlinear regression using DynaFit 4.02.089 (BioKin Ltd.) (21).
RESULTS
Crystallization of P450 2C8-Ligand Complexes—P450 2C8 was crystallized originally as a dimer with two fatty acid molecules bound at the dimer interface (15). Biochemical characterization indicated that preparations of enzyme contained primarily palmitic acid with some stearic acid. The fatty acids appeared to be acquired from the expression host, and they persisted through the isolation and purification of the enzyme. As preparations of 2C8 exhibited a mixture of dimers and monomers by size exclusion chromatography and contained less than stoichiometric amounts of fatty acid, the effects of adding palmitic acid to the crystallization incubations were examined in the initial phases of this study. It was found that the concentration of P450 2C8 required for crystallization could be significantly reduced in the presence of a molar equivalent of palmitic acid. Additionally, the crystallization conditions used in the original P450 2C8 structure contained 15% ethanol (15), which strongly inhibited ligand binding. These conditions were further optimized to eliminate ethanol from the precipitant solution. The reduction in P450 concentration provided an advantage for the crystallization of ligand complexes because it also reduced concentrations of the poorly soluble ligands that were required to saturate the protein during crystallization. The tendency of the protein to precipitate was also greatly reduced or eliminated during the incubations. The structures of the resulting complexes indicate that the fatty acid-stabilized dimer is compatible with substrate and inhibitor binding.
Structures of P450 2C8 Complexes—P450 2C8 complexed with felodipine, troglitazone, or montelukast crystallized in the I222 space group, and the structures were refined using 2.28-, 2.7-, and 2.8-Å limits of diffraction. In each case, molecular replacement searches using the substrate-free structure (Protein Data Bank code 1pq2) positioned a single P450 2C8 molecule in the asymmetric unit. In contrast, crystals of the complex with 9-cis-retinoic acid exhibited the P21212 space group with a 2.6-Å limit of diffraction, and two P450 2C8 molecules were identified in the asymmetric unit. For each complex, residues 28–490 of the 2C8 polypeptide chains were adjusted and rebuilt, where the numbering corresponds to that of the full-length wild-type protein. Additionally, the heme prosthetic group and a molecule of palmitic acid bound to an external binding site on each chain were defined by the electron density maps. In the 9-cis-retinoic acid complex, the palmitic acid molecules bind in the interface between the two molecules in the asymmetric unit. This interface occurs between the single chain in the asymmetric unit of the crystallized felodipine, troglitazone, and montelukast complexes and a symmetry-related molecule (-x+1,-y,z) in the crystal. As described previously for the 1pq2 structure (15), the interface is formed by the helix F-G portions of the protein. The hydrophobic portion of the fatty acid passes through a turn formed by helices F, F′, G′, and G of one polypeptide chain, and the carboxylate of the fatty acid interacts with the N-terminal end of helix G of the neighboring polypeptide chain in the crystal lattice. The ligands were modeled as discussed in more detail below.
P450 2C8 exhibits two substrate-binding channels that exit the active-site cavity on either side of the helix B-C loop. The two cavities merge and terminate just beyond the heme iron (Fig. 2A), where the reactive iron-oxo intermediate is generated during catalysis. The cavity is bounded additionally by helix I, the helix F-G region, portions of β-sheet 1, the turn in β-sheet 4, and the loop between helix K and β-sheet 1 (15).
FIGURE 2.

Views of the ligand-binding site of P450 2C8 illustrating interactions of R-montelukast (A and B) or 2R,5R-troglitazone (C and D) with the protein. The heme prosthetic group is rendered as a red stick figure, with the central iron atom shown as a sphere. Portions of the secondary structure of the protein are rendered as a cyan ribbon, with side chains shown as stick figures with carbons colored cyan. In some cases, portions of the substrate-free structure (Protein Data Bank code 1pq2) are shown as a gray ribbon, with side chains shown as stick figures with carbons colored gray. The nitrogen, carbon, and oxygen atoms of the backbone are shown in some cases to illustrate hydrogen bonding interactions (black dashed lines). The distances between each ligand and the heme iron are indicated and identified by black dashed lines. Side chains making close contacts (<4 Å) are depicted and labeled if visible. The substrates are depicted as stick figures with carbon atoms colored orange. Other atoms are colored red for oxygen, blue for nitrogen, yellow for sulfur, and green for chlorine. The oxygen atoms of several water molecules that occupy the cavity are rendered as spheres.A gold mesh is used to render 2|Fo| - |Fc|σA-weighted ligand omit maps contoured at 1σ around the ligands. A black mesh is used to depict the solvent-accessible surface of the active-site cavity. The views differ between panels to clearly depict different features of the structures. The transparent solid surface in D illustrates the solvent-accessible surface of the volume that is left unoccupied upon troglitazone binding. The figures were rendered by ray tracing using PyMOL (DeLano Scientific, Palo Alto, CA).
Montelukast was identified as a high affinity inhibitor of P450 2C8 with a Ki of 9 nm by Walsky et al. (9, 22). Montelukast is the largest of the four compounds (Mr = 586) characterized in this study with three branches connected to an asymmetric carbon atom (Fig. 1). The marketed therapeutic compound corresponds to the R-enantiomer, which was used for crystallization of the complex. Montelukast fits the size and shape of the cavity well without major changes in the tertiary structure compared with the substrate-free structure (Protein Data Bank code 1pq2). The positions of the longest and shortest branches are clearly defined by the 2|Fo| - |Fc|σA-weighted electron density map determined for the final model with montelukast omitted (Fig. 2B). The longest and most hydrophobic moiety resides in the substrate access channel that exits on the C-terminal side of helix B′. Its terminal chloroquinoline ring occupies a distal hydrophobic pocket in the active-site cavity that passes over helix I and under helix G (Fig. 2, A and B). This branch exhibits a planar conformation that maintains conjugation of the ethenyl group with flanking aromatic rings and complements the size and shape of the distal part of the cavity well. This portion of montelukast exhibits 32% lower B-factors than the average B-factor observed for montelukast, indicating that it is the most sterically constrained portion of the ligand. The shortest branch is positioned with its terminal carboxylate residing near a cluster of polar residues, Ser-100, Ser-103 and Asn-217, near the N terminus of helix B′. The side chain of Ser-100 and the amide hydrogen of Ser-103 donate hydrogen bonds to the carboxylate moiety of montelukast.
The third branch terminating with a tertiary alcohol group is positioned close to the heme iron, with carbon 4 of the phenyl ring positioned 4.2 Å from the heme iron (Fig. 2B). The orientation of the tertiary alcohol group is not uniquely defined by the electron density, and it was modeled in the electron density based on the potential for hydrogen bonding of the hydroxyl group with the carbonyl of helix I residue Val-296 (Fig. 2B). Although the electron density surrounding the tertiary alcohol moiety is more complete in maps generated with montelukast included in the model, the connection of this moiety to the central asymmetric carbon is not defined well by the 2|Fo| - |Fc|σA-weighted omit maps compared with the other two branches of montelukast (Fig. 2B). This is likely to reflect reduced steric restraints on the third moiety as evidenced by the reduced number of protein side chains in this region that make close contacts (defined as distances of4Åor less between atom centers) (Fig. 2, A and B). Close contacts with montelukast are much more numerous in the upper and distal portions of the substrate-binding cavity, and the interactions of the protein with the largest subcomponent of montelukast are likely to be major determinants of the binding orientation. Overall, the complementarities of size, shape, hydrophobicity, and polarity between montelukast and the active-site cavity are likely to underlie the Ki of 9 nm exhibited by this compound for the enzyme (9, 22).
Troglitazone (Mr = 442) is smaller than montelukast, displays a more linear shape, and has two chiral carbons (Fig. 1). One asymmetric carbon, C-5, occurs in the terminal thiazolidinedione group and forms the connection with the rest of the molecule. The second asymmetric carbon, C-2, occurs in the chromane ring and connects this group to the glitazone moiety. The 2R,5R-stereoisomer was co-crystallized with P450 2C8 in an orientation that positions the chromane ring near helix I, ∼8 Å above the heme iron, and the thiazolidinedione moiety in the substrate access channel near the C-terminal end of helix B′. The weakly acidic thiazolidinedione group (23) is likely to be ionized at physiologic pH, and the orientation of Arg-241 on helix G changes to position the basic side chain in the substrate access channel, where it forms two charge-stabilized hydrogen bonds: one with a carbonyl oxygen and one with the acidic nitrogen of the thiazolidinedione moiety (Fig. 2C). Asn-204 of helix F donates a hydrogen bond to the other carbonyl oxygen of the thiazolidine ring. The ether oxygen in the middle portion of the troglitazone molecule is hydrogen-bonded to an ordered water molecule that is hydrogen-bonded, in turn, to Ser-103 of helix B′ (Fig. 2, C and D), as well as additional water molecules in the cavity (data not shown).
Racemic troglitazone is oxidized to a quinone-like metabolite by human liver microsomes, which reflects oxidative cleavage of the chromane cyclic ether. The P450 2C8-catalyzed reaction exhibits a Km of 2.7 μm and a kcat of 4.2 min-1 (24). The observed position for the 2R,5R-troglitazone is too distant from the heme iron for direct reaction with the compound I-like intermediate. Interestingly, this position leaves a significant volume (∼500 Å3) unoccupied in the active site, between the inhibitor and the heme prosthetic group. The positioning of the chromane ring is likely to reflect the contribution of hydrogen bonding between its phenol group with the protein rather than steric restraints that limit a closer approach to the heme iron. Two hydrogen bonds involving the phenolic oxygen are evident: one with the carbonyl oxygen of Val-296 on helix I and the other with the amide hydrogen of Glu-300 on helix I (Fig. 2D). Hydroxylation of the 5-methyl group of the chromane ring has also been reported, but has not been characterized for P450 2C8. The active-site cavity of 2C8 is likely to also accommodate other stereoisomers of troglitazone, but the chirality of the asymmetric carbons in the each stereoisomer will require alterations in the positions of the heteroatoms compared with that seen for the 2R,5R-isomer. Attempts to crystallize the 2S,5R-stereoisomer with P450 2C8 did not produce definitive electron density for the chromane group, suggesting that it might be less restrained by hydrogen bonding interactions (data not shown).
Felodipine is a smaller molecule (Mr = 384) that is a high affinity inhibitor of P450 2C8 with a Ki of 90 nm (9). It is more rigid than montelukast or troglitazone and nonionic and nonlinear in shape (Fig. 1). Electron density maps indicate a well defined, single binding position for felodipine in the relatively large active-site cavity (Fig. 3A). Although racemic felodipine was used for co-crystallization with P450 2C8, felodipine was modeled as the single R-enantiomer based on greater electron density defining the ethyl carboxylate for the R-enantiomer. A mixed occupancy by the two stereoisomers might not be evident, as the expected differences arising from a single carbon atom with partial occupancy would be relatively small. The dichlorophenyl ring of felodipine is positioned closest to the heme iron, with carbon 4 of the aromatic ring positioned 4.6 Å from the heme iron (Fig. 3A). Close contacts are evident with protein side chains forming the first and second tiers above the heme surface (Fig. 3, A and B). The substituted dihydropyridine moiety resides in a broader portion of the cavity (Fig. 3), and interactions with the surrounding side chains (data not shown) are less constraining (>4 Å between atom centers). Some deformation of the structure relative to the original substrate-free structure is evident in the vicinity of Ile-476 (Fig. 3B), but overall, the structural backbone is not greatly affected by the binding of felodipine. The remaining cavity volume contains several ordered water molecules that form hydrogen bonds with the protein (Fig. 3A). In contrast, neither the water molecules nor side chains of the protein form hydrogen bonds with the heteroatoms of felodipine, which include the oxygens of the carboxylate ester side chains and the nitrogen of the dihydropyridine ring. Thus, the interactions between the protein and ligand are largely hydrophobic.
FIGURE 3.

Views of the ligand-binding site of P450 2C8 illustrating interactions of (R)-felodipine (A and B) or 9-cis-retinoic acid (C and D) with the protein. The proximal molecule of retinoic acid is designated as RA1, and the distal molecule is labeled as RA2. The atom color code is the same as described in the legend of Fig. 2.
Retinoic acid (Mr = 300) is the smallest of the four compounds. The structure determined for the complex of 9-cis-retinoic acid with P450 2C8 reveals that two molecules of the substrate are simultaneously present in the substrate-binding cavity (Fig. 3, C and D). The proximal molecule is bound with the trimethylcyclohexenyl ring in close proximity to the heme iron (Fig. 3C), with carbon 4 positioned 4.6 and 4.8 Å from the iron for molecules A and B of the asymmetric unit, respectively. The position of carbon 4 is consistent with the observation that 9-cis-retinoic acid is oxidized at this position by P450 2C8 to form the 4S-hydroxy metabolite (7). The carboxylate end of the proximal retinoic acid molecule is positioned close to the backbone of the polypeptide chain near Gly-98. The polypeptide backbone has twisted out relative to the substrate-free structure to accommodate the retinoic acid in this position (Fig. 3C). This change also orients the amide hydrogens of Gly-98 and Ser-100 toward the carboxylate of retinoic acid for hydrogen bonding. The second molecule of 9-cis-retinoic acid binds in the distal portion of the cavity, where the carboxylate of the distal molecule forms a hydrogen bond to Asn-204 as well as a charge-stabilized hydrogen bond with the side chain of Arg-241 (Fig. 3, C and D), which has moved into the active-site cavity as seen for the troglitazone complex. The second molecule of retinoic acid is located in the cavity occupied by the chloroquinoline moiety of montelukast. The retinoid molecules are also able to maintain a planar conformation that allows conjugation of double bonds of the isoprenoid chain. There are extensive contacts between the two molecules of retinoic acid, where they stack next to each other in the more spacious region of the active-site cavity.
Cooperative and Noncooperative Models for Retinoic Acid Binding to P450 2C8—The occupancy of the substrate-binding cavity by two molecules of 9-cis-retinoic acid could reflect the relatively high concentrations of P450 2C8 and 9-cis-retinoic acid that are used for crystallization. To better understand the binding of the substrate to the protein in solution, spectral studies were undertaken to characterize the binding of 9-cis-retinoic acid to both the truncated form of P450 2C8 crystallized in this study and the full-length enzyme (25, 26). The binding of retinoic acid to P450 2C8 increases the high spin character of the heme iron as evidenced by difference spectra displaying a decrease in the absorbance at 417 and 570 nm and an increase at 394 and 646 nm (Fig. 4A). The change in spin state is attributed to the formation of a 5-coordinate iron as a result of diminished binding of water to the open coordination site of the iron in the presence of the substrate (27, 28).
FIGURE 4.

Solution ligand binding studies. A, the progressive effects of 6, 12, 18, 24, 30, 36, 42, 48, 54, and 60 μm 9-cis-retinoic acid on the visible absorption spectra of 20 μm P450 2C8dH. Difference spectra were computed as described under “Experimental Procedures” in Excel and plotted using Slide-Write Plus. Absorbance changes below 400 nm that reflect both changes of the P450 spectrum upon ligand binding and the absorption spectrum of the retinoic acid are not shown. The difference spectra exhibit several isosbestic points as well as positive and negative changes consistent with the conversion of the low spin heme protein to a high spin form. At high concentrations of retinoic acid, the isosbestic points at short wavelengths are lost because of the increased contributions of the absorption spectrum of the retinoid. B, the dependence of changes in the absorbance difference between 646 and 570 nm of truncated P450 2C8 (+) normalized for protein concentration (20 μm) on the total concentration of 9-cis-retinoic acid. The continuous curve is a nonlinear least-squares regression fitting of the quadratic form of the ligand binding equation with n independent binding sites per protein molecule fit to the data shown (Table 3). The predicted values for this data set obtained from alternative models are shown for comparison. The two models are 1) independent binding equilibria for two sites (•), with binding to one site yielding a spectral change, and 2) a sequential binding model for the formation of the singly and doubly occupied complexes, where the single and doubly occupied complexes exhibit distinct differential absorption coefficients (□). The predictions are based on parameters estimated using DynaFit from a global fit to all of the data sets summarized in Table 3.
As 9-cis-retinoic acid exhibits an absorption maximum at 330 nm, the increase in absorption of the P450 2C8 complex at 394 nm could not be used to accurately monitor changes in the spin state. To reduce interference by the spectrum of 9-cis-retinoic acid, the increase in absorbance at 646 nm and the decrease at 570 nm were used to characterize the concentration dependence of retinoic acid binding (Fig. 4A). The isosbestic points flanking the peak at 646 nm and the trough at 570 nm were not significantly disturbed by the spectrum of 9-cis-retinoic acid at the concentrations used in the titrations.
The relatively linear increase in the spectral changes and the sharp transition to a limiting value (Fig. 4B, +) indicated that the concentration of the protein was likely to exceed the dissociation constant governing ligand binding. For this reason, the data points were fit with the quadratic form of the binding equation, where S and P are the total concentration of 9-cis-retinoic acid and P450 2C8, respectively.
Nonlinear least squares minimization was used to estimate values for the maximum difference in the absorbance, the apparent dissociation constant (Kd), and the number of independent 9-cis-retinoic acid-binding sites (n) per P450 based on the assumption that the amount of complex was proportional to the relative change in absorbance.
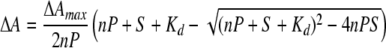 |
(Eq. 1) |
Roughly two independent substrate-binding sites were estimated for 9-cis-retinoic acid per molecule of P450 2C8 at a concentration of either 2 or 20 μm P450 2C8dH (Table 3). The titration at 2 μm P450 2C8 was repeated using the full-length enzyme, yielding similar results (Table 3) and indicating that the truncated and full-length proteins exhibit very similar binding behavior. Additionally, the titration of P450 2C8dH at 20 μm protein was repeated in the presence of 0.8–0.9 mm CHAPS, which dissociates P450 2C8dH dimers and yielded estimates similar to those seen in the absence of the detergent. Titrations at protein concentrations that exceed the apparent Kd favor the estimation of the stoichiometry of binding because the ratio of bound to free 9-cis-retinoic acid is high at each concentration of the ligand until saturation is reached, and the transition to a plateau in the absorbance change is relatively sharp at a 2:1 ratio of 9-cis-retinoic acid to P450 2C8dH, consistent with estimated values of n (Fig. 4B). The estimates for the Kd ranged from 0.24 to 0.64 μm (Table 3), which are consistent with the tight binding seen at 2 and 20 μm P450 2C8. The predicted values for the maximum change in absorbance normalized with respect to the concentration of P450 2C8 ranged from 3.96 to 4.60 mm-1.
TABLE 3.
Characterization of the binding of 9-cis-retinoic acid to P450 2C8 by difference spectroscopy The quadratic form of the ligand binding equation was used to estimate the maximal change in the absorbance between 650 and 590 nm normalized relative to the protein concentration, the dissociation constant (Kd), and the number of independent binding sites (n) by nonlinear least-squares fitting as described under “Results.” P450 2C8dH is the truncated protein crystallized in this study; 2C8modH is the full-length enzyme; and CHAPS is the detergent used to dissociate dimers of 2C8dH. The means ± S.D. obtained for at least three preparations of each protein are shown. The path length of the cuvette was 1.000 cm.
| P450 preparation | ΔAmax/[P450] | Kd | No. binding sites/P450 |
|---|---|---|---|
| mm–1 | μm | ||
| 2C8dH (2 μm) | 4.60 ± 0.30 | 0.24 ± 0.12 | 1.65 ± 0.27 |
| 2C8modH (2 μm) | 4.44 ± 0.35 | 0.41 ± 0.18 | 2.51 ± 0.50 |
| 2C8dH (20 μm) | 3.96 ± 0.09 | 0.64 ± 0.29 | 2.16 ± 0.08 |
| 2C8dH (20 μm) + CHAPS | 4.06 ± 0.31 | 0.55 ± 0.49 | 1.96 ± 0.22 |
| All titrations | 4.30 ± 0.37 | 0.44 ± 0.29 | 2.07 ± 0.45 |
Although this 2:1 stoichiometry is consistent with the presence of two molecules of 9-cis-retinoic acid observed in the structure of the enzyme, the two sites are not structurally equivalent. The proximal 9-cis-retinoic acid-binding site is most likely to affect the presence of water at the sixth coordination site of the heme and produce a direct effect on the spectrum of the enzyme. The binding of 9-cis-retinoic acid in the distal site could affect both the binding of 9-cis-retinoic acid to the proximal site and the magnitude of the spectral change. To examine alternative models with nonequivalent binding constants and differential effects on the spectral change, numerical simulations implemented by the computer program DynaFit (21) were employed to fit the results of the titrations with alternative models that involve complex binding equilibria such as cooperative binding. The program employs a differential evolution search for initial parameters that approximate a solution to differential equations describing the kinetic steady state at equilibrium, conservation relationships between components of the system, and spectral responses that are dependent on the concentrations of specific components of the system. The initial estimates are then refined by nonlinear least-squares minimization to provide final estimates of each parameter. A global fit can be obtained utilizing multiple data sets, and model discrimination analysis provides for consideration of alternative models followed by a statistical comparison of the results. Using this approach, sequential ordered or random binding to two sites and contributions to the observed spectral change from the final doubly occupied model as well as singly occupied complexes were tested. The command file for DynaFit is provided as supplemental material. The models were fit globally to all of the data sets summarized in Table 3. This consisted of 14 independent titrations with a total of 218 data points obtained using several independent preparations of P450 2C8dH and the full-length protein. The program was allowed to vary the concentration of the P450 within 10% of the input value. This compensates for potential differences in the concentration of the P450 that may contribute to the variation in the values for ΔAmax/[P450 (mm)] evident in Table 3. This could arise from both experimental error and the presence of small amounts of P450 that do not respond to ligand binding and that vary in relative amount between independent preparations.
A model for two independent binding sites for the same protein fits the results with the lowest sum of the squares of the residuals. The “two independent binding sites” model was written as two equilibrium reactions using independent descriptors for the two binding sites so that binding to the distal site (Dstl) did not affect the concentration of free protein for the binding to the proximal site (Prxml) and vice versa. Only the complex formed at the proximal site was considered to produce a spectral change.
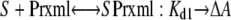 |
(Eq. 2) |
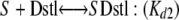 |
The estimated values ± S.E. for the two dissociation constants are Kd1 = 0.75 ± 0.28 and Kd2 = 0.74 ± 0.21 μm with an estimated differential absorption coefficient of 4.19 ± 0.29 mm-1 cm-1 relative to the ligand-free protein. These values are similar to the binding constants and mean value for the normalized ΔAmax obtained using the quadratic form of the binding equation (Table 3). In contrast, the fit obtained with a single binding site exhibited an almost 4-fold higher sum of the squares of the residuals, and the two independent binding sites model was superior based on the corrected Akaike information criteria (29), ΔAICc = 301.
The second best fit was obtained using a sequential binding model with distinct equilibrium constants for the binding of the first and second molecules of 9-cis-retinoic acid (S) to P450 2C8 (P) and distinct differential absorption coefficients for the PS and SPS complexes relative to the ligand-free protein.
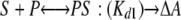 |
(Eq. 3) |
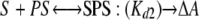 |
The estimated values ± S.E. for the dissociation constants are Kd1 = 0.17 ± 0.31 μm and Kd2 = 3.2 ± 1.6 μm, which is consistent with negative cooperativity for binding of the second molecule. The estimates for the apparent differential absorption coefficients were 2.11 ± 0.08 and 4.38 ± 0.3 mm-1 cm-1 for the singly and doubly occupied complexes, respectively, relative to the ligand-free protein. This result also suggests that binding of the second molecule enhances the change in the spin state of the enzyme elicited by the binding of the first molecule by ∼2-fold. Compared with the “two independent sites” model, the sequential binding model exhibited a 1.75-fold larger sum of the squares of the residuals and a ΔAICc value of 124. Moreover, the S.E. values for the estimates of the dissociation constants are larger than those determined for the model with two independent binding sites. The predicted results for these two best models are shown for the specific data set displayed in Fig. 4B, using the parameters based on the global fit of all data sets. It should be noted that each of these models can fit individual data sets very well, but the variation in the individual parameters is greater than that obtained for the global fit. As the sensitivity of the method was limited, these titrations could not be repeated using 10-fold lower concentrations of the protein, where the ratio of bound to free substrate would be lower, and better discrimination between the models might be evident. In summary, the results of the solution binding studies are consistent with two molecules of retinoic acid binding to P450 2C8, but they do not distinguish clearly between cooperative and noncooperative models for binding.
DISCUSSION
The large active-site cavity of P450 2C8 exhibits a trifurcated architecture that approximates a T or Y shape with branches of differing lengths, widths, and chemical properties. The bottom branch provides access to the heme iron, the catalytic center of the protein, and the other two terminate in solvent and substrate access channels that exit the buried active-site cavity on either side of helix B′. The cavity is rather large at the junction of the three branches. The structures of the four complexes presented in this study sample diverse aspects of ligand binding to the P450 2C8 active-site cavity. Overall, only small localized deformations of the protein backbone were noted, and the predominant interactions between the ligands and the protein are hydrophobic. Our results indicate that the dimer of 2C8, which is stabilized by the binding of two palmitic acid molecules at the dimer interface, is compatible with substrate and inhibitor binding. The formation of the dimer is not required for retinoic acid binding because CHAPS at concentrations that dissociate the dimer into monomers did not significantly affect the binding parameters for 9-cis-retinoic acid.
Montelukast is a relatively large molecule with a tripartite structure that fits into the three branches of the cavity. The relative size of each branch of the cavity dictates the orientation of the largest subcomponent of montelukast, whereas polarity is likely to determine the orientation of the two smaller subcomponents of montelukast, positioning the carboxylate moiety in the solvent access channel and the hydrophobic moiety near the heme iron. The more linear troglitazone molecule fills the upper part of the active-site cavity. Its thiazolidinedione moiety is stabilized by the Arg-241 and Asn-204 side chains in the substrate access channel near the C-terminal end of helix B′, whereas the chromane ring system is bound 8 Å above the heme iron in a position favoring hydrogen bonding with helix I residues. Binding of this inhibitor further enlarges the cavity at the branching point and leaves a significant residual active-site volume close to the heme iron unoccupied. The dichlorophenyl moiety of the rather compact and rigid felodipine molecule occupies the branch of the cavity near the heme iron with the substituted dihydropyridine ring fitting against the surface of the cavity at the branching point. The opposite surface of the dihydropyridine ring exhibits fewer close contacts with protein side chains, and water molecules are evident in the remaining portions of the cavity. The distal portion of the cavity contains a number of polar amino acid side chains and exposed peptide backbone hydrogen bond donors and acceptors for stabilization of water molecules in the distal portion of the cavity. In contrast, the binding of water in the apolar portion of the cavity is likely to be less stable and thus favors binding of the hydrophobic substrate in the proximal portion of the cavity near the heme iron. In contrast to a single molecule of montelukast, troglitazone, or felodipine, two molecules of 9-cis-retinoic acid bind to P450 2C8 and together occupy the three branches of the cavity and largely fill the available space. A significant interaction between the two molecules of retinoic acid is evident, where the two molecules both reside in the most spacious portion of the cavity near the helix B-B′ loop and helix F′. The interaction between the two molecules of retinoic acid restrains the dynamics of each molecule in the active site.
The binding of the proximal molecule of 9-cis-retinoic acid in the lower portion of the cavity positions the retinoic acid for oxidation at the observed site (carbon 4) by the iron-bound reactive oxygen intermediate. The benzyl ring of montelukast is also positioned close to the iron, suggesting that it could also be oxidized by the enzyme. Montelukast is a potent and relatively selective competitive inhibitor of P450 2C8 in vitro (9, 22), but montelukast does not appear to alter the clearance of pioglitazone or repaglinide in vivo, which is largely mediated by P450 2C8 (30, 31). This is likely to reflect the pharmacokinetic properties of montelukast that limit the in vivo concentration of montelukast available for P450 2C8 binding. The benzylic side chain of montelukast is known to be oxidized in vivo (32) and in vitro (33), but to the best of our knowledge, the oxidation of montelukast by P450 2C8 has not been reported. Felodipine is also reported to be a potent competitive inhibitor of P450 2C8 (9). The dichlorophenyl ring is positioned appropriately in 2C8 for potential oxidation with carbon 4 residing 4.6 Å from the heme iron, but we are not aware of evidence for the oxidation of the compound by P450 2C8. Felodipine is largely converted to the pyridine analog by oxidative dehydrogenation of the dihydropyridine ring, which is predominantly catalyzed by P450 3A4 (34, 35). In contrast, troglitazone is not positioned for oxidation, but is known to be oxidized by P450 2C8 (24). However, rotation of the chromane ring into a position for oxidation to occur is not sterically hindered.
The carboxylate moieties of montelukast and retinoic acid are likely to be negatively charged at physiologic pH. The carboxylate group of montelukast is located near the helix B-B′ loop and helix F′ in an open solvent channel, where it exhibits hydrogen bonds with the side chain of Ser-100 and the backbone amide hydrogen of Ser-103. The carboxylate of the proximal retinoic acid is similarly positioned to accept hydrogen bonds from the amide hydrogens of Gly-98 and Ser-100. A deformation of the backbone in the helix B-B′ loop increases the available space and reorients the amide hydrogens toward the substrate carboxylate for hydrogen bonding. The solvation of this portion of the activesite cavity is also likely to facilitate the binding of the carboxylate moieties of both substrates to these neutral sites. It is also possible that a counterion such as Na+ or K+ could be present in the solvent channel and could not be detected by x-ray diffraction. The binding interactions seen for these anionic compounds are similar in nature to the binding of fatty acids to P450 102A1, where the carboxylate occupies the analogous site without assistance of a positively charged protein side chain (36).
This binding mode contrasts with the charge-stabilized interactions displayed for the binding of the negatively charged thiazolidinedione group of troglitazone and for the binding of the carboxylate moiety of the distal retinoic acid molecule. The binding of the proximal retinoic acid in the lower portion of the cavity close to the heme leaves sufficient residual space for a second molecule of retinoic acid to bind in the remaining portion of the substrate-binding cavity. The carboxylate moiety of the distal retinoic acid molecule is positioned to accept hydrogen bonds from both Arg-241 and Asn-204. The Arg-241 side chain moves from a position outside the cavity, as seen in other structures determined for P450 2C8, to the inside of the cavity, where it can provide a strong, charge-stabilized hydrogen bond with the carboxylate of the retinoic acid. A similar reorientation of Arg-241 is evident in the troglitazone complex, where both Arg-241 and Asn-204 interact with the weakly acidic thiazolidinedione ring. This result indicates that Arg-241 can play a role in substrate binding, as suggested by the mutagenesis studies of Melet et al. (14). The latter studies indicated that the substitution of Arg-241 with alanine had differential effects on the apparent Km values for substrate oxidation, ranging from little or no effect for paclitaxel to a 4-fold increase for fluvastatin. The mutation also increased the IC50 for inhibition by troglitazone by 6-fold. The flexibility of fluvastatin may allow the carboxylate side chain of fluvastatin to interact with Arg-241 when the molecule is positioned for oxidation. This flexibility could also allow fluvastatin to reposition so that, in the R241A mutant, the negatively charged side chain is positioned much like the carboxylate of montelukast. The carboxylate function of fluvastatin would then reside near the solvent channel close to the N-terminal end of helix B′ in a position predicted by the docking studies with the original 2C8 structure (14). This suggests that the stronger, charge-stabilized hydrogen bonding to Arg-241 might contribute to a lower apparent Km for the reactions catalyzed by the wild-type enzyme. On the other hand, the effect of the R241A substitution was less pronounced for all-trans-retinoic acid (3-fold) (14). The isoprenoid chain of the retinoic acid is relatively rigid because of the extensive conjugation of the unsaturated bonds. Thus, the cyclohexenyl ring cannot easily be positioned near the heme iron for oxidation with the isoprenoid chain occupying the distal cavity. It is possible that two molecules of all-trans-retinoic acid also bind to P450 2C8 and that the R241A mutation reduces the binding of the distal molecule and only indirectly affects the binding of the proximal substrate molecule. Together, these data illustrate the versatility of P450 2C8 in the binding of high affinity anionic ligands that could not be foreseen with the proposed pharmacophore model (14). The P450 2C8 complex structures demonstrate that at least two different binding modes should be considered in the evaluation of structural determinants of substrate or inhibitor binding.
The spectral binding studies reported here are also consistent with the binding of two molecules of 9-cis-retinoic acid either to the truncated construct crystallized for structure determination or to the full-length enzyme in solution. The estimated binding constants are low relative to the concentrations of the protein, leading to a well defined change in the slope of the binding curve at a 2:1 ratio of ligand to protein and a consistently better fit to the results by equilibrium binding models that involve two binding sites. The results can be fit well either by a model with two independent binding sites or by sequential binding models with evidence for cooperativity. The relatively high ratios of bound to free ligand for concentrations of the protein that are amenable to spectral analysis are likely to preclude significant discrimination between these alternative models and accurate estimates of the binding constants. The Km for the 4-hydroxylation of 9-cis-retinoic acid catalyzed by microsomes prepared from lymphoblastoid cells expressing P450 2C8 is reported to be 7 μm (7), which is greater than the estimates for the Kd values from the spectral binding experiments. Significantly different Km values (1.4 and 50 μm) for the 4-hydroxylation of all-trans-retinoic acid by P450 2C8 (6, 8) have also been reported, and these discrepancies may reflect the limited solubility of the retinoic acid in aqueous solutions as well as the potential for partitioning of retinoic acid in membranes present in these assays.
The simultaneous binding of two molecules in the active site of P450 3A4 is thought to underlie the non-Michaelis-Menten kinetics exhibited by the enzyme with a number of substrates. The effect of simultaneous occupancy can lead to heterotropic and homotropic activation of oxidation and absent or reduced competitive inhibition (37). Binding studies support a mechanism of sequential binding to P450 3A4 in which the first molecule binds to a high affinity site that does not elicit a shift in the spin state of the heme, and subsequent binding of a second molecule increases the high spin character of the enzyme (38–40). In contrast, our results indicate that the first molecule of retinoic acid that binds to P450 2C8 in a sequential binding model elicits a significant change in the high spin character of the enzyme. A structure of P450 3A4 was published that includes two molecules of the inhibitor ketoconazole bound in the active-site cavity (41). Similarly, a structure of P450 158A2 from Streptomyces coelicolor A3(2) exhibits two molecules of flaviolin bound in the substrate-binding cavity (42), which may facilitate the polymerization of the two molecules. Moreover, the bacterial enzyme P450 eryF exhibits homotropic cooperativity for the binding of androstenedione or 9-aminoanthracene, and structures of the enzyme indicate that two molecules of each ligand bind in the relatively large active site of the enzyme (43). The active-site cavities of P450 3A4, 158A2, and eryF are relatively large and open so that the ligands can stack on each other. In the case of P450 2C8, the two retinoic acid molecules overlap in a portion of the cavity but bind to more clearly defined, discrete sites in the substrate-binding cavity because of the branched shape of the cavity. This may reduce the cooperativity of the binding to P450 2C8 relative to P450 3A4. On the other hand, recent reports of substrate inhibition for oxidation of pioglitazone by P450 2C8 (44) could reflect the binding of a second substrate molecule to the distal site with a negative effect on the oxidation of the substrate bound to the proximal site. The structure of the troglitazone complex suggests that binding of pioglitazone in a similar manner would leave sufficient additional space for a second molecule to bind.
In conclusion, P450 2C8 complexes with montelukast, troglitazone, and 9-cis-retinoic acid illustrate two different binding modes for anionic functional groups, with full, partial, or double occupancy of the active-site cavity, respectively. The P450 2C8-felodipine complex structure shows the compact hydrophobic inhibitor to be sequestered close to the heme and several ordered water molecules in the distal portion of the active-site cavity. Overall, the large, trifurcated, active-site cavity is capable of binding structurally divergent substrates without extensive conformational changes. Ligand-induced protein conformational changes are limited to specific interactions, with the most pronounced effects occurring in the helix B-B′ region and the turn in β-sheet 4. A pronounced side chain movement is seen in complexes with troglitazone and retinoic acid, where Arg-241 reorients to stabilize the negatively charged functional groups. The large size of the P450 2C8 active site and the great diversity of preferred substrates complicate the use of a general pharmacophore model. The different ligand-binding modes exhibited by the experimental structures determined in this study sample many aspects of P450 2C8 ligand recognition and should facilitate the generation of more realistic pharmacophore models.
Supplementary Material
Acknowledgments
We thank Daniel Mansuy, Caroline Lee, Mike Wester, Hans Parge, Robert Love, and Ben Burke for helpful comments during this study and the preparation of the manuscript and Qiping Zhao for technical support.
The atomic coordinates and structure factors (codes 2nnh, 2nni, 2nnj, and 2vn0) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant GM031001 (to E. F. J.). This work was also supported by Pfizer Global Research and Development and the Sam and Rose Stein Charitable Trust. Portions of this work were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the United States Department of Energy, Office of Basic Energy Sciences. The Stanford Synchrotron Radiation Laboratory Structural Molecular Biology Program is supported by the United States Department of Energy, Office of Biological and Environmental Research, by the National Center for Research Resources Biomedical Technology Program, and by NIGMS, National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental material.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: P450, a generic term for a cytochrome P450 enzyme (individual P450s are identified using a number-letter-number format based on amino acid sequence relatedness); CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
References
- 1.Rahman, A., Korzekwa, K. R., Grogan, J., Gonzalez, F. J., and Harris, J. W. (1994) Cancer Res. 54 5543-5546 [PubMed] [Google Scholar]
- 2.Daikh, B. E., Lasker, J. M., Raucy, J. L., and Koop, D. R. (1994) J. Pharmacol. Exp. Ther. 271 1427-1433 [PubMed] [Google Scholar]
- 3.Zeldin, D. C., DuBois, R. N., Falck, J. R., and Capdevila, J. H. (1995) Arch. Biochem. Biophys. 322 76-86 [DOI] [PubMed] [Google Scholar]
- 4.Totah, R. A., and Rettie, A. E. (2005) Clin. Pharmacol. Ther. 77 341-352 [DOI] [PubMed] [Google Scholar]
- 5.Leo, M. A., Lasker, J. M., Raucy, J. L., Kim, C.-I., Black, M., and Lieber, C. S. (1989) Arch. Biochem. Biophys. 269 305-312 [DOI] [PubMed] [Google Scholar]
- 6.McSorley, L. C., and Daly, A. K. (2000) Biochem. Pharmacol. 60 517-526 [DOI] [PubMed] [Google Scholar]
- 7.Marill, J., Capron, C. C., Idres, N., and Chabot, G. G. (2002) Biochem. Pharmacol. 63 933-943 [DOI] [PubMed] [Google Scholar]
- 8.Marill, J., Cresteil, T., Lanotte, M., and Chabot, G. G. (2000) Mol. Pharmacol. 58 1341-1348 [DOI] [PubMed] [Google Scholar]
- 9.Walsky, R. L., Gaman, E. A., and Obach, R. S. (2005) J. Clin. Pharmacol. 45 68-78 [DOI] [PubMed] [Google Scholar]
- 10.Shitara, Y., Hirano, M., Sato, H., and Sugiyama, Y. (2004) J. Pharmacol. Exp. Ther. 311 228-236 [DOI] [PubMed] [Google Scholar]
- 11.Ogilvie, B. W., Zhang, D., Li, W., Rodrigues, A. D., Gipson, A. E., Holsapple, J., Toren, P., and Parkinson, A. (2006) Drug Metab. Dispos. 34 191-197 [DOI] [PubMed] [Google Scholar]
- 12.Delaforge, M., Pruvost, A., Perrin, L., and Andre, F. (2005) Drug Metab. Dispos. 33 466-473 [DOI] [PubMed] [Google Scholar]
- 13.Kumar, S., Samuel, K., Subramanian, R., Braun, M. P., Stearns, R. A., Chiu, S. H., Evans, D. C., and Baillie, T. A. (2002) J. Pharmacol. Exp. Ther. 303 969-978 [DOI] [PubMed] [Google Scholar]
- 14.Melet, A., Marques-Soares, C., Schoch, G. A., Macherey, A. C., Jaouen, M., Dansette, P. M., Sari, M. A., Johnson, E. F., and Mansuy, D. (2004) Biochemistry 43 15379-15392 [DOI] [PubMed] [Google Scholar]
- 15.Schoch, G. A., Yano, J. K., Wester, M. R., Griffin, K. J., Stout, C. D., and Johnson, E. F. (2004) J. Biol. Chem. 279 9497-9503 [DOI] [PubMed] [Google Scholar]
- 16.Johnson, E. F., and Stout, C. D. (2005) Biochem. Biophys. Res. Commun. 338 331-336 [DOI] [PubMed] [Google Scholar]
- 17.Richardson, T. H., Jung, F., Griffin, K. J., Wester, M., Raucy, J. L., Kemper, B., Bornheim, L. M., Hassett, C., Omiecinski, C. J., and Johnson, E. F. (1995) Arch. Biochem. Biophys. 323 87-96 [DOI] [PubMed] [Google Scholar]
- 18.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 19.Jones, T. A., and Kjeldgaard, M. (1997) Methods Enzymol. 277 173-208 [DOI] [PubMed] [Google Scholar]
- 20.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 21.Kuzmic, P. (1996) Anal. Biochem. 237 260-273 [DOI] [PubMed] [Google Scholar]
- 22.Walsky, R. L., Obach, R. S., Gaman, E. A., Gleesen, J. P., and Proctor, W. R. (2005) Drug Metab. Dispos. 33 413-418 [DOI] [PubMed] [Google Scholar]
- 23.Giaginis, C., Theocharis, S., and Tsantili-Kakoulidou, A. (2007) J. Chromatogr. B 857 181-187 [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki, H., Shibata, A., Suzuki, M., Nakajima, M., Shimada, N., Guengerich, F. P., and Yokoi, T. (1999) Drug Metab. Dispos. 27 1260-1266 [PubMed] [Google Scholar]
- 25.Schenkman, J. B., Remmer, H., and Estabrook, R. W. (1967) Mol. Pharmacol. 3 113-123 [PubMed] [Google Scholar]
- 26.Jefcoate, C. R. (1978) Methods Enzymol. 52 258-279 [DOI] [PubMed] [Google Scholar]
- 27.Stern, J. O., Peisach, J., Blumberg, W. E., Lu, A. Y., and Levin, W. (1973) Arch. Biochem. Biophys. 156 404-413 [DOI] [PubMed] [Google Scholar]
- 28.Tsai, R., Yu, C. A., Gunsalus, I. C., Peisach, J., Blumberg, W., Orme-Johnson, W. H., and Beinert, H. (1970) Proc. Natl. Acad. Sci. U. S. A. 66 1157-1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hurvich, C. M., and Tsai, C. L. (1995) Biometrics 51 1077-1084 [PubMed] [Google Scholar]
- 30.Jaakkola, T., Backman, J. T., Neuvonen, M., Niemi, M., and Neuvonen, P. J. (2006) Eur. J. Clin. Pharmacol. 62 503-509 [DOI] [PubMed] [Google Scholar]
- 31.Kajosaari, L. I., Niemi, M., Backman, J. T., and Neuvonen, P. J. (2006) Clin. Pharmacol. Ther. 79 231-242 [DOI] [PubMed] [Google Scholar]
- 32.Balani, S. K., Xu, X., Pratha, V., Koss, M. A., Amin, R. D., Dufresne, C., Miller, R. R., Arison, B. H., Doss, G. A., Chiba, M., Freeman, A., Holland, S. D., Schwartz, J. I., Lasseter, K. C., Gertz, B. J., Isenberg, J. I., Rogers, J. D., Lin, J. H., and Baillie, T. A. (1997) Drug Metab. Dispos. 25 1282-1287 [PubMed] [Google Scholar]
- 33.Chiba, M., Xu, X., Nishime, J. A., Balani, S. K., and Lin, J. H. (1997) Drug Metab. Dispos. 25 1022-1031 [PubMed] [Google Scholar]
- 34.Baarnhielm, C., Dahlback, H., and Skanberg, I. (1986) Acta Pharmacol. Toxicol. 59 113-122 [DOI] [PubMed] [Google Scholar]
- 35.Eriksson, U. G., Lundahl, J., Baarnhielm, C., and Regardh, C. G. (1991) Drug Metab. Dispos. 19 889-894 [PubMed] [Google Scholar]
- 36.Li, H. Y., and Poulos, T. L. (1997) Nat. Struct. Biol. 4 140-146 [DOI] [PubMed] [Google Scholar]
- 37.Korzekwa, K. R., Krishnamachary, N., Shou, M., Ogai, A., Parise, R. A., Rettie, A. E., Gonzalez, F. J., and Tracy, T. S. (1998) Biochemistry 37 4137-4147 [DOI] [PubMed] [Google Scholar]
- 38.Lampe, J. N., and Atkins, W. M. (2006) Biochemistry 45 12204-12215 [DOI] [PubMed] [Google Scholar]
- 39.Roberts, A. G., Campbell, A. P., and Atkins, W. M. (2005) Biochemistry 44 1353-1366 [DOI] [PubMed] [Google Scholar]
- 40.Denisov, I. G., Baas, B. J., Grinkova, Y. V., and Sligar, S. G. (2007) J. Biol. Chem. 282 7066-7076 [DOI] [PubMed] [Google Scholar]
- 41.Ekroos, M., and Sjogren, T. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 13682-13687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao, B., Guengerich, F. P., Bellamine, A., Lamb, D. C., Izumikawa, M., Lei, L., Podust, L. M., Sundaramoorthy, M., Kalaitzis, J. A., Manmohan, R. L., Kelly, S. L., Moore, B. S., Stec, D., Voehler, M., Falck, J. R., Shimada, T., and Waterman, M. R. (2005) J. Biol. Chem. 280 11599-11607 [DOI] [PubMed] [Google Scholar]
- 43.Cupp-Vickery, J., Anderson, R., and Hatziris, Z. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 3050-3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tornio, A., Niemi, M., Neuvonen, P. J., and Backman, J. T. (2008) Drug Metab. Dispos. 36 73-80 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.