

Abstract
Selective resistance to the effects of insulin on glucose metabolism in skeletal muscle and adipose tissue is a key feature of polycystic ovary syndrome (PCOS). The pathogenesis of insulin resistance in skeletal muscle in PCOS involves interaction of environmental factors with intrinsic defects in insulin signaling. We aimed to determine whether 1. intrinsic defects in insulin action/signaling and cytokine secretion were present in adipose cells in PCOS and 2. insulin resistance can be induced in control adipose cells by culture in medium conditioned by insulin-resistant PCOS fibroblasts. Subcutaneous abdominal preadipocytes from obese women with PCOS (n = 7) and age- and BMI-matched controls (n = 5) were cultured for several generations in vitro. Basal and insulin-stimulated glycogen synthesis and basal glucose transport did not differ in the preadipocytes from women with PCOS and controls. Abundance of insulin receptor β-subunit (IRβ), insulin receptor substrate (IRS)-1, IRS-2, p85 subunit of phosphatidylinositol (PI) 3-kinase, and ERK1/2 activation did not differ. Secretion of tumour necrosis factor (TNF)-α and interleukin (IL)-6 did not differ. Insulin action on glycogen synthesis in control preadipocytes was not altered by co-culture with, or growth in media conditioned by PCOS skin fibroblasts with constitutive serine phosphorylation of IRβ (IR ser+), indicating that IR ser+ cells do not secrete an insulin resistance-inducing factor. We conclude that in contrast to skeletal muscle and skin fibroblasts, there is no evidence for intrinsic defects in insulin signaling in the PCOS adipose cell lineage, indicating that insulin resistance in these cells is likely due to factors in the in vivo environment.
1. Introduction
Polycystic ovary syndrome (PCOS) is among the most common endocrine disorders of premenopausal women. It is characterized by hyperandrogenism, disordered gonadotropin secretion, skeletal muscle insulin resistance and an increased risk of type 2 diabetes mellitus (DM) [1–4]. Insulin resistance in PCOS is independent of obesity, but obesity plays an amplifying role [3–5]. Although adipocyte-derived factors are now known to play a key role in modulating insulin action in skeletal muscle [6], the adipose cell lineage in PCOS has not been comprehensively investigated. To date, studies using freshly isolated subcutaneous abdominal adipocytes from women with PCOS have demonstrated obesity-independent post–insulin receptor (IR)-binding resistance to the effects of insulin on glucose transport [7–10]. Additionally, insulin inhibition of lipolysis is impaired in these cells [9, 10], but not in adipocytes isolated from the visceral depot [11].
Recent studies of skeletal muscle in PCOS have provided evidence for the interaction of intrinsic defects in insulin signaling with factors in the in vivo environment in the pathogenesis of insulin resistance in this tissue. Impaired insulin-stimulated glucose uptake in skeletal muscle in vivo of women with PCOS is associated with decreased IRS-1–associated PI3-kinase activation [12]. However, when skeletal muscle cells from women with PCOS are grown in culture, insulin-stimulated glucose uptake is not impaired despite defects in insulin signaling via both insulin receptor substrate (IRS)-1 and IRS-2, indicating that in vivo factors are required for insulin resistance to be manifested [13]. The persistence of these signaling defects in cells cultured for generations outside their in vivo environment suggests that the defects are intrinsic, although whether these changes are genetic or epigenetic remains to be elucidated. Impaired insulin signaling in skeletal muscle in PCOS is selective for glucose metabolic pathways: mitogenic signaling via the mitogen activated protein kinase ERK1/2 is enhanced [14]. When skin fibroblasts from women with PCOS are cultured for multiple generations, these cells also display insulin resistance selective for metabolic pathways [15] due to a distinct molecular mechanism. Insulin receptors (IR) purified from these fibroblasts show constitutive serine phosphorylation (IR ser+) with inhibition of insulin-stimulated tyrosine phosphorylation, caused by a serine kinase extrinsic to the receptor [16, 17].
The potential roles of environmental factors and intrinsic insulin signaling defects in the pathogenesis of insulin resistance have not been evaluated in the adipose cells of women with PCOS. The aim of this study was to determine whether intrinsic defects in insulin signaling were present in PCOS adipose cells by assessing insulin action in cells grown for multiple generations removed from the influences of the in vivo environment. Preadipocytes, the fibroblast-like precursor cells present in the stromal fraction of adipose tissue, differentiate in vivo into mature lipid-laden adipocytes [18]. Although primary cultures of human preadipocytes show a high capacity for differentiation to adipocytes in vitro, this differentiation capacity is lost once preadipocytes have been allowed to divide for only a few generations [19]. We therefore elected to study insulin action in the cultured preadipocytes of women with PCOS and age- and body mass index (BMI)- matched controls. We hypothesized that intrinsic defects would be present in cultured preadipocytes from women with PCOS, similar to our findings in cultured skin fibroblasts [15] and skeletal muscle [13, 14]. Given that proinflammatory cytokines have been implicated in impairment of insulin signaling [20–25], we also measured secretion of tumour necrosis factor (TNF)-α and interleukin (IL)-6 by preadipocyte cultures of women with PCOS and controls. Finally, we carried out experiments using control preadipocyte cultures in order to determine if IR ser+ fibroblasts secreted an insulin-resistance inducing factor.
2. Subjects, materials, and methods
2.1. Subjects
The study was approved by the Institutional Review Board at Brigham and Women’s Hospital and all subjects gave written, informed consent. A total of 7 obese women with PCOS and 5 age-, BMI- and ethnicity-matched control women were studied. Women were aged 21 to 40 years, in good health, and taking no medications known to affect carbohydrate or sex hormone metabolism for at least 1 month prior to the study, except for oral contraceptive agents which were discontinued 3 months before study. Control women had regular menstrual cycles (every 27 – 35 days), no clinical or biochemical evidence of hyperandrogenism and no first degree relatives with type 1 or 2 DM. Women with PCOS had irregular menstrual cycles (≤ 6 menses per year) and elevated total testosterone (T) and/or non-sex-hormone-binding globulin-bound testosterone (uT) levels [1, 5, 7, 16]. Nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency, androgen-secreting neoplasms, and hyperprolactinemia were excluded by appropriate tests in the PCOS women. All subjects underwent a 75 gram oral glucose tolerance test after an overnight fast, with glucose and insulin levels measured basally and 2 hours post-challenge. World Health Organization criteria (WHO) were used to assess glucose tolerance [26]. Fasting glucose levels were also assessed using American Diabetes Association (ADA) criteria [27].
2.2. Harvesting and culture of preadipocytes
Subjects underwent aspiration of subcutaneous abdominal adipose tissue under local anaesthesia after an overnight fast as previously described [7]. The stromal cell fraction was obtained by collagenase digestion and centrifugation using the method of Rodbell [28] and cultured to near confluence in low glucose Dulbecco’s Modified Eagle Medium (DMEM) (Gibco BRL Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Gibco BRL Life Technologies), penicillin 50 IU/ml and streptomycin 50 μg/ml (Cellgro, Mediatech, Herndon VA). All experiments were performed on subcultured cells (passage 2) grown at a density of 3000 cells/ml until near confluence (total population doublings 11 ± 1, mean ± SD), and incubated in Minimum Essential Medium alpha (αMEM; Gibco BRL Life Technologies) supplemented with 0.1% bovine serum albumin (BSA fraction V; Boehringer Mannheim Corp., Indianapolis, IN) for 2 h prior to assays or lysis.
2.3. Glycogen synthesis and glucose transport assays
Glycogen synthesis was determined by measuring glucose incorporation into glycogen in duplicate wells for each condition using the method of Henry et al [29] with the modification that cultures were washed with serum-free medium and then incubated in the same medium ± insulin (human recombinant regular insulin; Novo Nordisk, Princeton, NJ) plus D-[14C-(U)]glucose (NEN Life Science Products, Boston, MA) for 2 h. Lysates from 2 wells per 6-well dish were assayed for protein concentration (Bio-Rad Laboratories, Hercules CA). Glucose uptake in 6 replicate wells for each condition was measured using the method of Ciaraldi et al [30], under conditions where cell membrane glucose transport was rate-limiting. An aliquot of lysate from each well was assayed for protein concentration and the remainder counted using liquid scintillation counting. Specific glucose transport was calculated by subtracting L-[14C]-glucose incorporation from [3H]-deoxy-D-glucose (NEN Life Science Products, Boston, MA) incorporation in each well.
2.4. Western blotting
Confluent preadipocytes were incubated in αMEM + 0.1% BSA for 2 h, followed by incubation in the same medium ± insulin (100 nM) for 10 min, then lysed as reported [16]. Western blotting was carried out as previously described [12], with PCOS and control samples run together on the same gels. Membranes were probed with antibodies to IRβ-subunit (Transduction Laboratories, San Diego, CA), IRS-1, IRS-2 (the gift of Dr M. White, Joslin Diabetes Center, Boston, MA), p85 subunit of PI3-kinase (Upstate Biotechnology, Lake Placid, NY), ERK1/2, or phospho-ERK1/2 (Thr202/Tyr204) (Cell Signaling Technology, Beverly, MA), then incubated with appropriate secondary antibodies (Biorad Laboratories). The products were visualized using enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Buckinghamshire, England) and quantitated using a scanning densitometer (Bio-Rad Laboratories). The same internal standard was run in all immunoblots for each protein and results are expressed as percentage of internal standard.
2.5. Secretion of TNFα and IL-6
Confluent preadipocytes were incubated in αMEM + 0.1% BSA for 12 h, then media replaced with fresh αMEM + 0.1% BSA. After 48 h, the media were collected and aliquots assayed in duplicate for IL-6 and TNFα using ELISA kits according to the manufacturer’s instructions (Quantikine HS; R&D Systems, Minneapolis, MN).
2.6. Conditioned media and co-culture experiments
The PCOS IR ser+ skin fibroblasts used in both conditioned media and co-culture experiments had previously been shown to belong to a sub-population of PCOS fibroblasts with significantly increased baseline IRβ serine phosphorylation with decreased insulin–stimulated tyrosine phosphorylation [16]. Insulin-stimulated glycogen synthesis was measured in control preadipocytes cultured in medium that had been conditioned by prior incubation with PCOS IR ser+ or control skin fibroblast cultures. Fresh growth medium (15% v/v) was added to the conditioned media and media changed twice weekly until near-confluence. In addition, insulin-stimulated glycogen synthesis was measured in control preadipocytes co-cultured with PCOS IR ser+ or control fibroblasts using dual-chamber culture dishes (Becton Dickinson, Franklin Lakes, NJ). Duplicate wells were assayed for each condition.
2.7. Statistical analysis
Data were analyzed using Student’s t-test for comparison between (unpaired) and within (paired) groups. Data were log transformed as necessary to achieve homogeneity of variance. Data are presented as mean ± SEM with significance at P < 0.05.
3. Results
3.1. Subject characteristics (Table 1)
Table 1.
Subject characteristics.
| Control | PCOS | |
|---|---|---|
| Number of subjects | 5 | 7 |
| Age (years) | 30 ± 3 | 29 ± 2 |
| BMI (kg/m2) | 37.5 ± 2.9 | 38.8 ± 1 |
| Total testosterone (nmol/l) | 0.8 ± 0.1 | 2.6 ± 0.3*** |
| Unbound testosterone (nmol/l) | 0.2 ± 0.1 | 0.9 ± 0.1*** |
| DHEAS (μmol/l) | 4.8 ± 0.8 | 5.8 ± 0.8 |
| Glucose (mmol/l) fasting | 4.4 ± 0.2 | 5.2 ± 0.2* |
| Glucose (mmol/l) 2 h post 75g glucose | 5.9 ± 0.3 | 8.1 ± 0.8 |
| Insulin (pmol/l) fasting | 100 ± 21 | 186 ± 43 |
| Insulin (pmol/l) 2 h post 75g glucose | 387 ± 29 | 1134 ± 316** |
Data are mean ± SEM.
P < 0.05,
P < 0.01,
P < 0.0001, unpaired t-test.
By design, the PCOS and control subjects were comparable for age and BMI. Compared with control women, women with PCOS had significant elevations of serum total and biologically available testosterone. No subject had abnormal fasting glucose levels but two PCOS subjects had impaired glucose tolerance and one PCOS subject fulfilled criteria for DM by WHO criteria. Control subjects had normal fasting and post-glucose load plasma glucose levels. Mean fasting glucose was significantly higher in the PCOS subjects (P < 0.05) and also tended to be higher at 2 h post - glucose load (P = 0.06). Plasma insulin 2 h post - 75 g glucose load was significantly elevated in women with PCOS compared with controls (P < 0.01), consistent with the presence of insulin resistance.
3.2. Glucose transport and glycogen synthesis
Basal glucose transport did not differ in cultured preadipocytes from women with PCOS and controls (Figure 1a). There was no stimulation of glucose transport by insulin (data not shown), in keeping with the absence of GLUT4 transporter in preadipocytes [31]. However, glucose incorporation into glycogen was stimulated by a mean of 2.4 fold (range 1.5 – 3.5) in both groups, probably through activation of glycogen synthase [32]. Glucose incorporation into glycogen did not differ in cultures from women with PCOS and controls at baseline or after insulin stimulation (Figure 1b).
Figure 1.
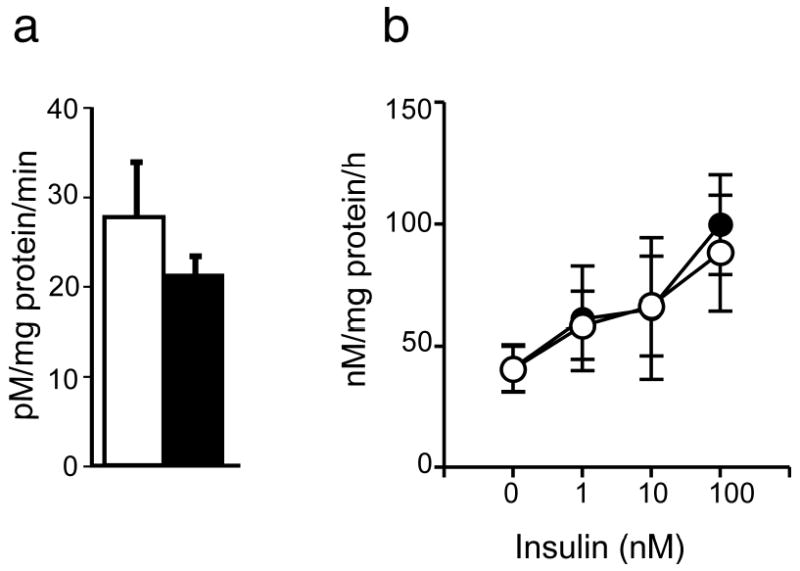
Basal glucose uptake and insulin-stimulated glycogen synthesis in cultured preadipocytes of obese PCOS women and age- and BMI-matched controls. Glucose uptake (n = 7 PCOS, n = 5 controls) was assessed at baseline (a). Glycogen synthesis (n = 6 PCOS, n = 5 controls) was measured at baseline and following incubation with insulin (1 – 100 nmol/l) (b). Filled circles represent PCOS and open circles represent controls. Filled bars represent PCOS and open bars represent controls. Data are presented as mean ± SEM.
3.3. Abundance of proximal insulin signaling proteins
The baseline abundance of IRβ (Figure 2a), IRS-1 (Figure 2b), p85 subunit of PI3-kinase (Figure 2c) and IRS-2 (Figure 2d) did not differ in preadipocytes from women with PCOS and controls.
Figure 2.
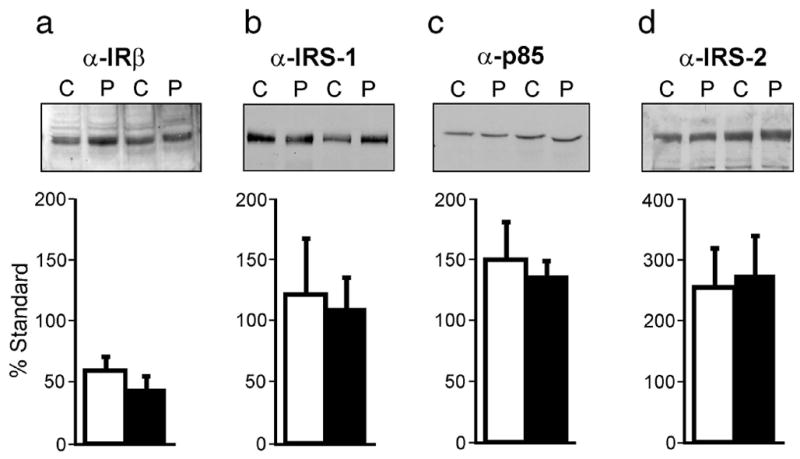
Abundance of proximal insulin signaling proteins in cultured preadipocytes from obese women with PCOS and age- and BMI-matched controls. Abundance of IRβ (a), IRS-1 (b), p85 subunit of PI3-kinase (c) and IRS-2 (d) (n = 6 PCOS, n = 5 controls) determined by immunoblot analysis of preadipocytes from women with PCOS (P) and controls (C). Basal lysates (50 μg) were separated by SDS-PAGE (7.5% gels) and representative immunoblots are shown. Filled bars represent PCOS and open bars represent controls. Data are presented as mean ± SEM.
3.4. ERK1/2 expresssion and activation
Phospho-ERK1/2 abundance did not differ at baseline or after insulin stimulation in preadipocytes from women with PCOS and controls (Figure 3a). Stimulation of ERK1/2 phosphorylation with insulin, mean 1.3 fold, did not reach significance compared with baseline in either group. The abundance of total ERK1/2 protein did not differ in PCOS and controls (Figure 3b).
Figure 3.
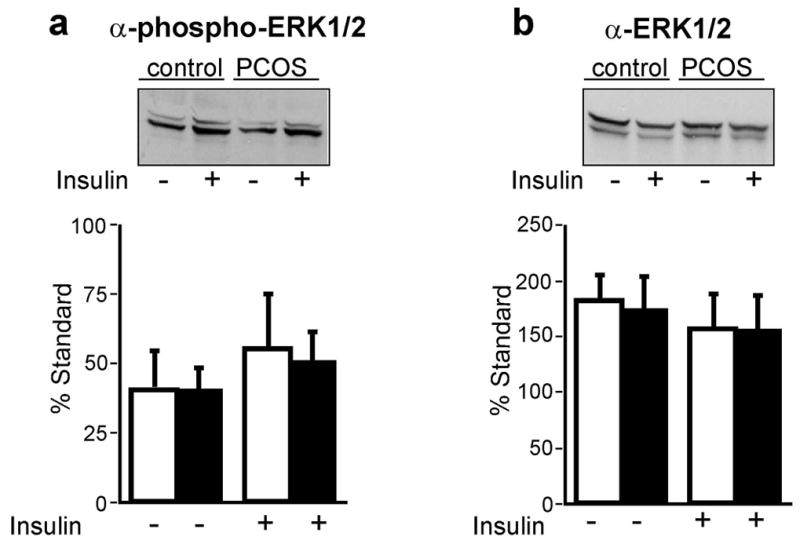
Phospho- and total- ERK1/2 in cultured preadipocytes from obese women with PCOS and age- and BMI-matched controls. Cultured preadipocytes (n = 6 PCOS, n = 5 controls) were incubated with (+) or without (-) insulin (100 nmol/l) for 10 min and lysates (50 μg) separated by SDS-PAGE (7.5% gels). Blots were probed with anti-phospho-ERK1/2, (a) then stripped and reprobed with anti-total ERK1/2 (b). Representative immunoblots are shown. Filled bars represent PCOS and open bars represent controls. Data are presented as mean ± SEM.
3.5. Secretion of TNFα and IL-6
Media concentrations of TNFα and IL-6 did not differ significantly in cultured preadipocytes from women with PCOS and controls, whether expressed as pg/ml/mg protein (TNFα PCOS 3.4 ± 0.6, control 2.7 ± 0.4; IL-6 PCOS 3595 ± 340, control 3098 ± 533) (Figure 4a, 4b) or pg/ml/million cells (TNFα PCOS 1.7 ± 0.4, control 1.1 ± 0.2; IL-6 PCOS 891 ± 285, control 1068 ± 262). A sample of ~29 PCOS and 10 controls would have been needed for the observed differences in TNFα secretion (pg/ml/million cells) to achieve statistical significance at P < 0.05.
Figure 4.
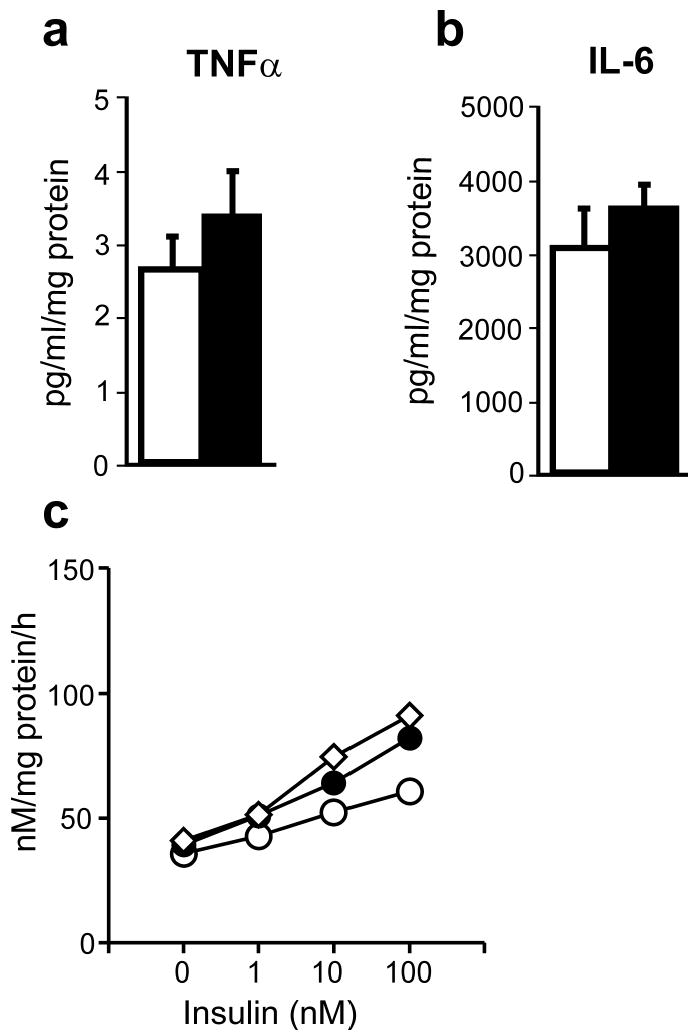
Secretion of cytokines by cultured preadipocytes from obese women with PCOS and age- and BMI-matched controls. Serum-free media from confluent preadipocytes (n = 7 PCOS, n = 5 controls) were collected after 48 h incubation and assayed for TNFα (a) and IL-6 (b) using ELISA. Filled bars represent PCOS and open bars represent controls. Results are expressed as pg/ml of media, and adjusted for protein content of each dish. Insulin-stimulated glycogen synthesis was measured in control preadipocytes cultured with media that had been conditioned by control cultured skin fibroblasts (open circles), by PCOS fibroblasts with constitutive increased serine phosphorylation of IRβ (IR ser+) (filled circles) or non-conditioned media (open diamonds) (c). Data are presented as mean ± SEM.
3.6. Media conditioning and co-culture with PCOS IR ser+ fibroblasts
Basal and insulin-stimulated glycogen synthesis in control preadipocytes were not significantly changed by culturing in media conditioned by PCOS IR ser+ skin fibroblasts (Figure 4c). Similarly, co-culture with PCOS IR ser+ skin fibroblasts did not alter glycogen synthesis in control preadipocytes (data not shown).
4. Discussion
The present study was carried out in order to investigate whether the impairment in insulin action observed in vivo in adipocytes from women with PCOS results from intrinsic or acquired defects. While it was not possible to assess glucose transport [31], insulin-stimulated glycogen synthesis was not impaired in cultured preadipocytes from women with PCOS, in contrast with previous studies of cultured PCOS skin fibroblasts [15]. The abundance of key proteins of the proximal insulin signaling pathway did not differ in PCOS and controls. These findings, in cells grown for many generations outside their in vivo environment, suggest that the defects in glucose metabolism observed in adipose cells in vivo of women with PCOS are acquired secondary to the hormonal/metabolic milieu rather than due to intrinsic defects. Ideally, the current experiments would have utilized preadipocytes cultured for several generations and then terminally differentiated in vitro to produce mature adipocytes, similar to the approach used in our previous studies of skeletal muscle in PCOS [13, 14]. This approach was not feasible as preadipocytes which have been allowed to divide for several generations lose their capacity to differentiate [19]. While it is theoretically possible that intrinsic defects in insulin action in the adipocyte lineage may be manifested only in fully-differentiated adipose cells, studies of the other major insulin-target tissue, skeletal muscle, have shown defects to be present in undifferentiated cells (myoblasts) as well as in mature terminally-differentiated cells (myotubes) [33 – 36].
In contrast to the findings in preadipocytes, our studies in skeletal muscle have shown that both intrinsic defects in insulin signaling and factors in the in vivo environment contribute to the development of insulin resistance in this tissue in women with PCOS. In PCOS, both in vivo and in cultured skeletal muscle cells, IRS-1 - associated PI3-kinase activity was reduced [12, 13]. However, in contrast to skeletal muscle in vivo, insulin-stimulated glucose uptake was not impaired in cultured skeletal muscle of women with PCOS, associated with a compensatory increase in IRS-1 protein abundance [13]. Several additional defects were present in the cultured skeletal muscle cells in PCOS, consistent with intrinsic defects. These included defects in signaling via IRS-2, abnormal electrophoretic mobility of IRS-2 suggestive of partial degradation, and increased non-insulin-mediated glucose transport associated with increased GLUT1 protein abundance [13]. In addition, we have also reported enhanced ERK1/2 phosphorylation in skeletal muscle in vivo and in vitro of women with PCOS, an abnormality which may contribute to insulin resistance through phosphorylation of IRS-1 at the critical Ser312 site [14]. While the molecular basis for these abnormalities in skeletal muscle in PCOS has not yet been determined, it is clear that they represent tissue-specific defects as they are not present in preadipocytes in PCOS.
Several adipose cell products are candidate factors for the induction of insulin resistance in skeletal muscle. These include free fatty acids (FFA), which are thought to inhibit insulin signaling by activating protein kinase C isoforms that serine phosphorylate IRS-1 [37]. TNFα also inhibits insulin signaling through serine phosphorylation of IRS-1 [20]. Other proinflammatory cytokines have been implicated in the pathogenesis of insulin resistance, including IL-6 [25]. In women with PCOS, circulating TNFα levels have not consistently been found to be elevated [38, 39] although mononuclear cells from obese women with PCOS show enhanced TNFα release in response to hyperglycemia compared with controls [40]. Likewise, circulating IL-6 levels do not differ in women with PCOS and age- and BMI-matched controls [41, 42]. Adipose cell products may also act locally by paracrine/autocrine mechanisms to induce insulin resistance. Adipose tissue expression of TNF and IL-6 is higher in insulin-resistant subjects [23] and adipose tissue IL-6 content correlates with impairment of insulin action in adipocytes in obese subjects with and without DM [24]. In the current study, there were no differences in production of TNFα or IL-6 by preadipocytes in vitro in PCOS compared with controls. However, the study was underpowered to detect a significant difference in TNFα production.
We also investigated the possibility that cultured PCOS skin fibroblasts with constitutive increase in serine phosphorylation of IR (IR ser+) could secrete an insulin-resistance-inducing factor. Cultured human skin fibroblasts have the capacity to synthesize several growth factors and cytokines [43] including IL-6 [44]. However, neither growth in IR ser+ conditioned media or co-culture with IR ser+ fibroblasts altered insulin responsiveness in control preadipocytes, indicating that factors secreted by PCOS skin fibroblasts which act via a paracrine mechanism are not implicated in the selective insulin resistance of these cells [15].
In conclusion, the present studies have provided evidence that impaired adipocyte insulin responsiveness in vivo in PCOS is acquired due to factors in the in vivo environment rather than due to intrinsic defects in insulin signaling. The abnormalities we observed in cultured skeletal muscle in PCOS including elevated non-insulin-mediated glucose uptake and enhanced activation of ERK1/2 were not present in preadipocytes. Additionally, preadipocytes from women with PCOS did not demonstrate resistance to insulin’s effects on glycogen synthesis as found in cultured skin fibroblasts, indicating important tissue-specific differences in this syndrome.
Acknowledgments
The authors thank Anna Lee and Nordine Benhaga for their expert technical assistance, Dr M. White (Joslin Diabetes Center, Boston, MA) for the generous gift of IRS-1 and IRS-2 antibodies, and the staff of the Brigham and Women’s Hospital General Clinical Research Center for assistance with the clinical studies. This work was supported by NIH grants R01 DK40605 (AD), U54 HD34449 (AD) and RR 02635 (BWH GCRC), as well as a grant from the American Diabetes Association (AD). AC was supported by fellowships from the Royal Australasian College of Physicians and the American Association of University Women.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dunaif A. Insulin resistance and the polycystic ovary syndrome; mechanism and implications for pathogenesis. Endocr Rev. 1997;18:774–800. doi: 10.1210/edrv.18.6.0318. [DOI] [PubMed] [Google Scholar]
- 2.Legro RS, Driscoll D, Strauss JF, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci USA. 1998;95:14956–14960. doi: 10.1073/pnas.95.25.14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Legro RS, Kunselman A, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84:165–169. doi: 10.1210/jcem.84.1.5393. [DOI] [PubMed] [Google Scholar]
- 4.Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care. 1999;22:141–146. doi: 10.2337/diacare.22.1.141. [DOI] [PubMed] [Google Scholar]
- 5.Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165–1174. doi: 10.2337/diab.38.9.1165. [DOI] [PubMed] [Google Scholar]
- 6.Abel ED, Peroni O, Kim JK, Kim Y-B, Boss O, Hadro E, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 7.Dunaif A, Segal KR, Shelley DR, Green G, Dobrjansky A, Licholai T. Evidence for distinctive and intrinsic defects in insulin action in polycystic ovary syndrome. Diabetes. 1992;41:1257–1266. doi: 10.2337/diab.41.10.1257. [DOI] [PubMed] [Google Scholar]
- 8.Ciaraldi TP, el-Roeiy A, Madar Z, Reichart D, Olefsky JM, Yen SS. Cellular mechanisms of insulin resistance in polycystic ovarian syndrome. J Clin Endocrinol Metab. 1992;75:577–583. doi: 10.1210/jcem.75.2.1322430. [DOI] [PubMed] [Google Scholar]
- 9.Marsden PJ, Murdoch A, Taylor R. Severe impairment of insulin action in adipocytes from amenorrheic subjects with polycystic ovary syndrome. Metabolism. 1994;43:1536–1542. doi: 10.1016/0026-0495(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 10.Ciaraldi TP, Morales AJ, Hickman MG, Odom-Ford R, Olefsky JM, Yen SSC. Cellular insulin resistance in adipocytes from obese polycystic ovary syndrome subjects involves adenosine modulation of insulin sensitivity. J Clin Endocrinol Metab. 1997;82:1421–1425. doi: 10.1210/jcem.82.5.3961. [DOI] [PubMed] [Google Scholar]
- 11.Ek I, Arner P, Ryden M, Holm C, Thorne A, Hoffstedt J, et al. A unique defect in the regulation of visceral fat cell lipolysis in the polycystic ovary syndrome as an early link to insulin resistance. Diabetes. 2002;51:484–492. doi: 10.2337/diabetes.51.2.484. [DOI] [PubMed] [Google Scholar]
- 12.Dunaif A, Wu X, Lee A, Diamanti-Kandarakis E. Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS) Am J Physiol Endocrinol Metab. 2001;281:E392–E399. doi: 10.1152/ajpendo.2001.281.2.E392. [DOI] [PubMed] [Google Scholar]
- 13.Corbould A, Kim Y-B, Youngren JF, Pender C, Kahn BB, Lee A, et al. Insulin resistance in the skeletal muscle of women with polycystic ovary syndrome involves both intrinsic and acquired defects in insulin signaling. Am J Physiol Endocrinol Metab. 2005;288:1047–1054. doi: 10.1152/ajpendo.00361.2004. [DOI] [PubMed] [Google Scholar]
- 14.Corbould A, Zhao H, Mirzoeva S, Aird F, Dunaif A. Enhanced mitogenic signaling in skeletal muscle of women with polycystic ovary syndrome. Diabetes. 2006;55:751–759. doi: 10.2337/diabetes.55.03.06.db05-0453. [DOI] [PubMed] [Google Scholar]
- 15.Book CB, Dunaif A. Selective insulin resistance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1999;84:3110–3116. doi: 10.1210/jcem.84.9.6010. [DOI] [PubMed] [Google Scholar]
- 16.Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in polycystic ovarian syndrome. J Clin Invest. 1995;96:801–810. doi: 10.1172/JCI118126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Youngren JF, Dunaif A, Goldfine ID, Maddux BA, Zhang BB, et al. Decreased insulin receptor (IR) autophosphorylation in fibroblasts from patients with PCOS: effects of serine kinase inhibitors and IR activators. J Clin Endocrinol Metab. 2002;87:4088–4093. doi: 10.1210/jc.2002-020363. [DOI] [PubMed] [Google Scholar]
- 18.Ailhaud G, Grimaldi P, Négrel R. Cellular and molecular aspects of adipose tissue development. Annu Rev Nutr. 1992;12:207–233. doi: 10.1146/annurev.nu.12.070192.001231. [DOI] [PubMed] [Google Scholar]
- 19.Wabitsch M, Brenner RE, Melzner I, Braun M, Möller P, Heinze E, et al. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int J Obes. 2001;25:8–15. doi: 10.1038/sj.ijo.0801520. [DOI] [PubMed] [Google Scholar]
- 20.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1 – mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 21.Miles PD, Romeo OM, Higo K, Cohen A, Rafaat K, Olefsky JM. TNF-alpha-induced insulin resistance in vivo and its prevention by troglitazone. Diabetes. 1997;46:1678–1683. doi: 10.2337/diab.46.11.1678. [DOI] [PubMed] [Google Scholar]
- 22.Cheung AT, Ree D, Kolls JK, Fuselier J, Coy DH, Bryer-Ash M. An in vivo model for elucidation of the mechanism of tumor necrosis factor-alpha (TNF-alpha)-induced insulin resistance: evidence for differential regulation of insulin signaling by TNF-alpha. Endocrinology. 1998;139:4928–4935. doi: 10.1210/endo.139.12.6336. [DOI] [PubMed] [Google Scholar]
- 23.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumour necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2001;280:E745–751. doi: 10.1152/ajpendo.2001.280.5.E745. [DOI] [PubMed] [Google Scholar]
- 24.Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, et al. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab. 2002;87:2084–2089. doi: 10.1210/jcem.87.5.8450. [DOI] [PubMed] [Google Scholar]
- 25.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2001;51:3391–3399. doi: 10.2337/diabetes.51.12.3391. [DOI] [PubMed] [Google Scholar]
- 26.Modan M, Harris MI, Halkin H. Evaluation of WHO and NDDG criteria for impaired glucose tolerance. Results from two national samples Diabetes. 1989;38:1630–1635. doi: 10.2337/diab.38.12.1630. [DOI] [PubMed] [Google Scholar]
- 27.American Diabetes Association. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care. 1997;20:1183–1198. doi: 10.2337/diacare.20.7.1183. [DOI] [PubMed] [Google Scholar]
- 28.Rodbell NMJ. Metabolism of isolated fat cells. J Biol Chem. 1964;239:375–380. [PubMed] [Google Scholar]
- 29.Henry RR, Abrams L, Nikoulina S, Ciaraldi TP. Insulin action and glucose metabolism in non-diabetic control and NIDDM subjects. Comparison using human skeletal muscle cell cultures. Diabetes. 1995;44:936–946. doi: 10.2337/diab.44.8.936. [DOI] [PubMed] [Google Scholar]
- 30.Ciaraldi TP, Abrams L, Nikoulina S, Mudaliar S, Henry RR. Glucose transport in cultured human skeletal muscle cells. Regulation by insulin and glucose in nondiabetic and non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 1995;96:2820–2827. doi: 10.1172/JCI118352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hauner H, Rohrig K, Spelleken M, Lui LS, Eckel J. Development of insulin-responsive glucose uptake and GLUT4 expression in differentiating human adipocyte precursor cells. Int J Obes. 1998;22:448–453. doi: 10.1038/sj.ijo.0800606. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava AK, Pandey SK. Potential mechanism(s) involved in the regulation of glycogen synthesis by insulin. Mol Cell Biochem. 1998;182:135–141. [PubMed] [Google Scholar]
- 33.Jackson S, Bagstaff SM, Lynn S, Yeaman SJ, Turnbull DM, Walker M. Decreased insulin responsiveness of glucose uptake in cultured skeletal muscle cells from insulin-resistant non-diabetic relatives of type 2 diabetic families. Diabetes. 2000;49:1169–1177. doi: 10.2337/diabetes.49.7.1169. [DOI] [PubMed] [Google Scholar]
- 34.Krützfeldt J, Kausch C, Volk A, Klein HH, Rett K, Häring H-U, et al. Insulin signaling and action in cultured skeletal muscle cells from lean healthy humans with high and low insulin sensitivity. Diabetes. 2000;49:992–998. doi: 10.2337/diabetes.49.6.992. [DOI] [PubMed] [Google Scholar]
- 35.Thompson DB, Prately R, Ossowski V. Human primary myoblast cell cultures from non-diabetic insulin resistant subjects retain defects in insulin action. J Clin Invest. 1996;98:2346–2350. doi: 10.1172/JCI119046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Youngren JF, Goldfine ID, Pratley RE. Insulin receptor autophosphorylation in cultured myoblasts correlates to glucose disposal in Pima Indians. Am J Physiol Endocrinol Metab. 1999;276:E990–E994. doi: 10.1152/ajpendo.1999.276.5.E990. [DOI] [PubMed] [Google Scholar]
- 37.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C θ and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 38.Naz RK, Thurston D, Santoro N. Circulating tumor necrosis factor (TNF)-α in normally cycling women and patients with premature ovarian failure and polycystic ovaries. Am J Reprod Immunol. 1995;34:170–175. doi: 10.1111/j.1600-0897.1995.tb00934.x. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez F, Thusu K, Abdel-Rahman E, Prabhala A, Tomani M, Dandona P. Elevated serum levels of tumor necrosis factor alpha in normal-weight women with polycystic ovary syndrome. Metabolism. 1999;48:437–441. doi: 10.1016/s0026-0495(99)90100-2. [DOI] [PubMed] [Google Scholar]
- 40.González F, Rote NS, Minium J, Kirwan JP. In vitro evidence that hyperglycemia stimulates tumor necrosis factor-α release in obese women with polycystic ovary syndrome. J Endocrinol. 2006;188:521–529. doi: 10.1677/joe.1.06579. [DOI] [PubMed] [Google Scholar]
- 41.Escobar-Morreale HF, Villuendas G, Botella-Carretero JI, Sancho J, Millan JL. Obesity, and not insulin resistance, is the major determinant of serum inflammatory cardiovascular risk markers in pre-menopausal women. Diabetologia. 2003;46:625–633. doi: 10.1007/s00125-003-1090-z. [DOI] [PubMed] [Google Scholar]
- 42.Mohlig M, Spranger J, Osterhoff M, Ristow M, Pfeiffer AF, Schill T, et al. The polycystic ovary syndrome per se is not associated with increased chronic inflammation. Eur J Endocrinol. 2004;150:525–532. doi: 10.1530/eje.0.1500525. [DOI] [PubMed] [Google Scholar]
- 43.Kessler-Becker D, Krieg T, Eckes B. Expression of pro-inflammatory markers by human dermal fibroblasts in a three-dimensional culture model is mediated by an autocrine interleukin-1 loop. Biochem J. 2004;379:351–358. doi: 10.1042/BJ20031371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feghali CA, Bost KL, Boulware DW, Levy LS. Human recombinant interleukin-4 induces proliferation and interleukin-6 production by cultured human skin fibroblasts. Clin Immunol Immunopathol. 1992;63:182–187. doi: 10.1016/0090-1229(92)90011-c. [DOI] [PubMed] [Google Scholar]