

Abstract
The efficient syntheses of the ABCD ring system of the originally proposed structure of azaspiracid-1 and the ABCDE ring system of the revised structure of azaspiracid-1 containing the correct stereochemistry at C6, C10, C13, C14, C16, C17, C19, C21, C22, C24 and C25 have been achieved.
Azaspiracid-1 (1) was discovered in 1995 when several individuals became ill after consuming mussels harvested from Killary Harbor in Ireland.1 Yasumoto and co-workers soon concluded a new toxin, azaspiracid-1 (1), was the cause of the outbreak in Ireland (Fig. 1).2 Subsequent to Yasumoto’s original report, several derivatives 2–5 have been isolated from Ireland3 and there is growing evidence of the spread of azaspiracid throughout other regions of Europe.4 A recent report appears to link the presence of azaspiracid to an ubiquitous alga.5 The toxic effects of azaspiracid have been shown to include serious injury to the digestive tracts, liver, pancreas, thymus and spleen in mice.6 The significant effect of this toxin on the European shellfish industry4 and its daunting structure garnered our attention7 as well as the interest of several other laboratories.8
Fig. 1.
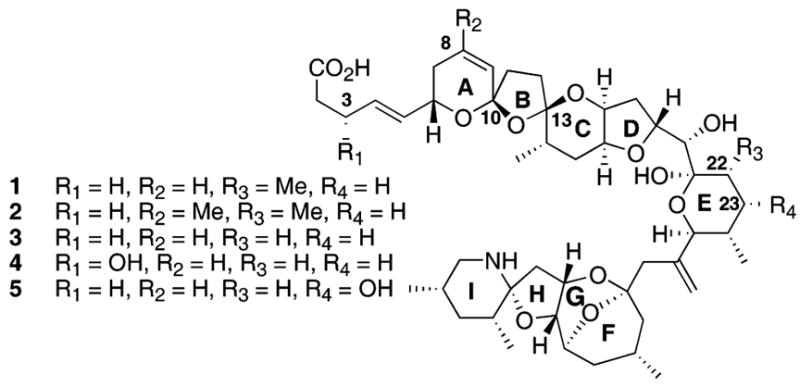
Originally proposed structure assignments for azaspiracid-1 to azaspiracid-5.
One particularly challenging portion of the azaspiracid architecture is the C10, C13 transoidal bisspiroketal moiety. This transoidal stereochemistry in 1 is proposed to exist with the C13 furan oxygen in an equatorial or “non-anomeric” orientation (Fig. 2). Given the fact that no external stabilizing force appears to be present, the non-anomeric orientation at this position has proven to be a demanding structural motif to construct. Our laboratory7d–f as well as the Nicolaou8h and Nishiyama8m laboratories have developed solutions to address this hurdle.
Fig. 2.
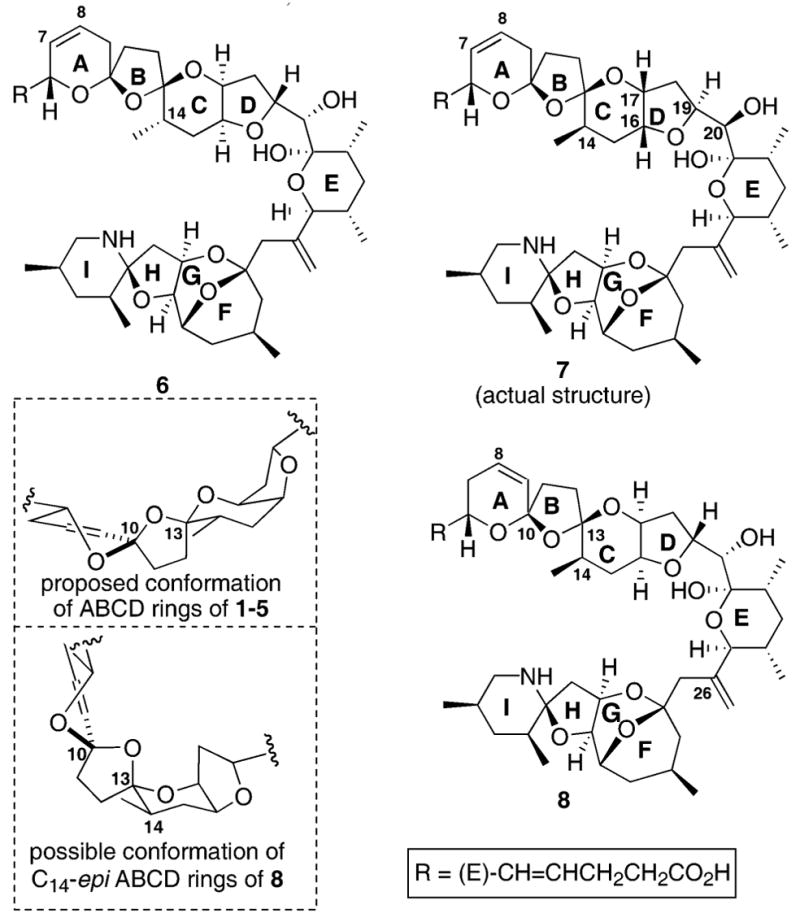
Alternate proposed structures for azaspiracid-1.
Recently, Nicolaou and co-workers revealed, in a series of impressive publications,9 that azaspiracid-1 was actually mis-assigned. They initially proposed an alternate structure 6 involving the relocation of the C8,9 alkene to the C7,8 position and the enantiomer of C28–C47 FGHI ring system (FGHI-ent).9a,b Nicolaou and co-workers asserted that movement of the alkene to the C7,8 position might address their observation of two inseparable compounds (presumably due to the bisspiroketal) versus the one isomer observed by Yasumoto. While the data put forth by Nicolaou to justify his proposed structure 6 was clearly enticing, we were troubled by the complications created by the relocation of the C8,9 olefin.2 It was not apparent to us what stereochemical difference would be relayed to the bisspiroketal by movement of the C8,9 alkene.
It was our belief that the major error(s) in structural assignment of azaspiracid lay in the CD ring system. We were intrigued by the possibility that the actual structure of azaspiracid might instead possess the epimeric C14 stereocenter (e.g. compound 8). This modification would potentially allow the C13 spiroketal to return to its preferred anomeric conformation (Fig. 2). The differences in chemical shifts reported by Nicolaou9a,b at H4–H6 and H8–H9 might be explained by the significant difference in local environment caused by returning the C13 furan oxygen on the C ring to the anomeric conformation. Independent and concurrent to our efforts, the Nicolaou laboratory has revised their original proposal to include the epimeric C14 stereochemistry while establishing the correct structure of azaspiracid-1 (7).9c,d Herein, we describe our synthesis of the ABCD and ABCDE ring systems of azaspiracid. Our overall retrosynthetic strategy for compounds 1 and 8 is shown in Scheme 1.
Scheme 1.
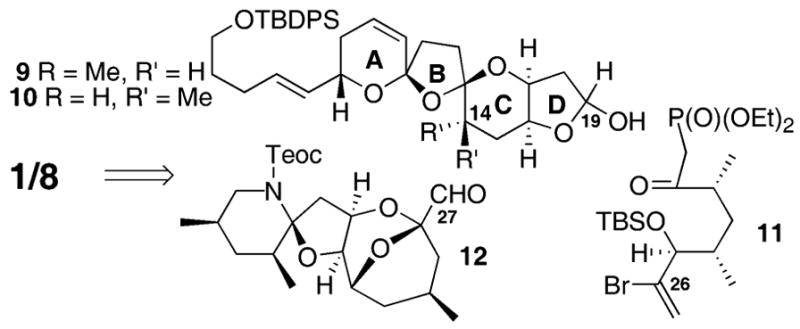
Retrosynthetic plan for targets 1 and 8.
Synthesis of the keto phosphonate 11 began from the commercially available Masamune auxiliary (Scheme 2).10 Boron-mediated anti-aldol reaction11 of 2-bromoacrolein with the norephedrine auxiliary produced the anti adduct in good diastereoselectivity. Subsequent protection of the C25 hydroxyl as its TBS ether followed by reduction yielded the alcohol 13. Alteration to the corresponding iodide, Myers alkylation12 and conversion to the keto phosphonate provided 11.
Scheme 2.
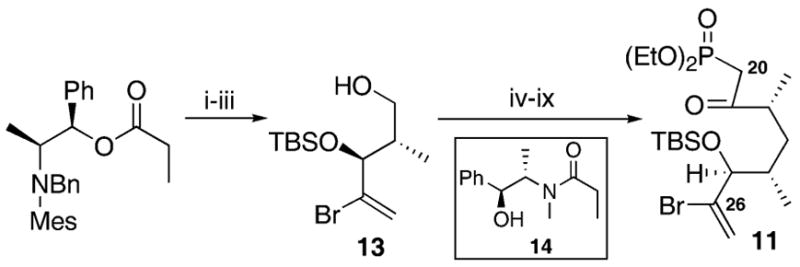
(i) Cyx2BOTf, Et3N, 2-bromoacrolein, Et2O, 92%, 93 : 7 d.r.; (ii) TBSOTf, Et3N, DMAP, CH2Cl2, 62%; (iii) DIBAL-H, CH2Cl2, −78 °C, 86%; (iv) Ph3P, imid., I2, CH2Cl2, 88%; (v) LDA, LiCl, 14, THF, 92%; (vi) LDA, BH3•NH3, THF, 84%; (vii) TPAP, NMO, CH2Cl2, mol. sieves; (viii) Me(O)P(OEt)2, n-BuLi, THF; (ix) PDC, DMF, mol. sieves, 53% over 3 steps.
The synthesis of the northern portion of azaspiracid commenced with previously reported ketone 157e (Scheme 3). Acid-catalyzed bisspirocyclization using PPTS in THF/H2O yielded the two bisspiroketals 16 and 17 in near equal amounts (10 : 9). As observed previously, the unwanted cisoidal bisspiroketal 16 could be recycled to provide the transoidal bisspiroketal 17. These conditions are an improvement on our original CSA, PhMe/t-BuOH conditions7d–f which provided a 5 : 3 ratio (16 : 17). Interestingly, treatment of the ketone 15 with CSA in hexanes led to formation of the C14-epi transoidal compound 1813 as the major product (4.5 : 1 : 6 ratio for 16 : 17 : 18). Unlike the bisspiroketals 16 and 17, the C14-epi compound 18 could not be re-equilibrated to the alternate spiroketals. It appears from this experiment that C14-epi transoidal adduct 18 is the thermodynamic “sink” for the C16-benzyloxy bisspiroketals. Working in parallel, removal of the C16 O-benzyl ether from 17 and 18 using LiDBB14 followed by conversion to the diazoester yielded 19 and C14-epi product 20. Rhodium-catalyzed C–H insertion using traditional catalysts such as Rh2(OAc)4 performed poorly in our hands, yielding none of the desired lactone. Based on recent work by Doyle’s15 and Wee’s16 laboratories using the chiral catalyst Rh2[(4S)-(MPPIM)]4,17 treatment of the diazo ester with 1 mol% of the rhodium catalyst in refluxing dichloromethane yielded the desired lactone in an unoptimized 12% yield for 21. This compound proved to be unstable upon prolonged storage. The transoidal nature of the spiroketal 21 was confirmed by extensive 2D NMR. Unfortunately, the analogous C–H insertion with C14-epi compound 20 appeared to provide only a trace of the presumed product 22. Based on this result, synthesis of a C14 epi-variant of bisspirocyclization precursor 15, which possessed C18 and C19 needed to be constructed.
Scheme 3.
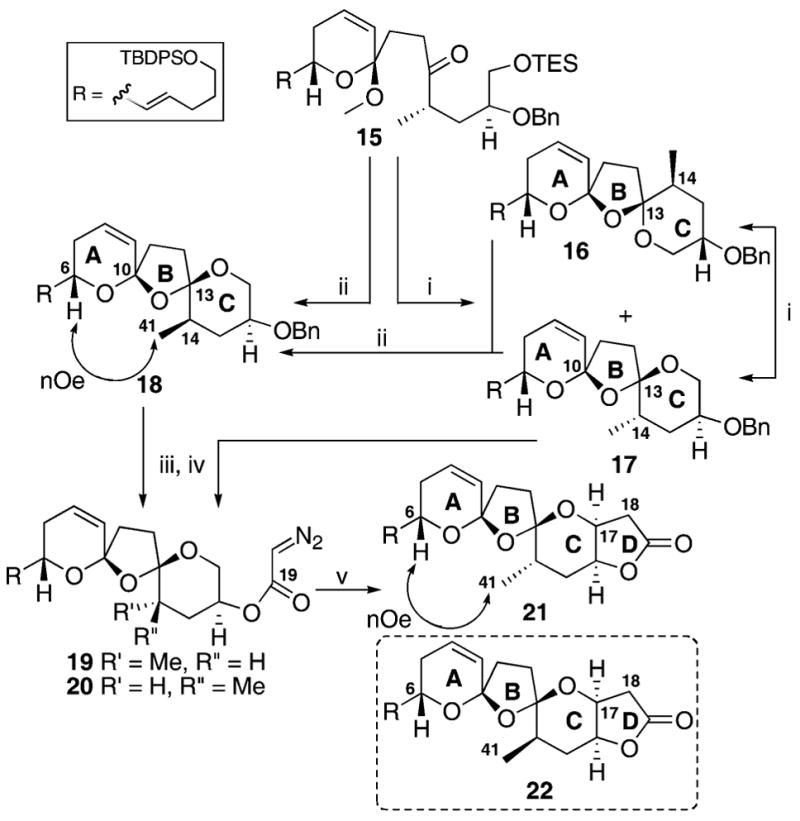
(i) PPTS, THF, H2O, 40% 16, 36% 17; (ii) CSA, hexanes, 42% 18, 32% 16, 7% 17; (iii) LiDBB, THF, −78 °C, 10 min; (iv) ClCOCH=N-NHTs, N,N-dimethylaniline, Et3N, CH2Cl2, 60% 19 over 2 steps, 65% 20 over 2 steps; (v) Rh2(4S)-(MPPIM)4, CH2Cl2, reflux, 12% 21.
Synthesis of the required aldehyde began with the readily available Evans alkylated product 24 (Scheme 4). We initially hypothesized that Sharpless dihydroxylation with AD mix β should provide the desired stereochemical combination at C16,17. This stereochemical result would have been opposite of what would be predicted by the accepted Sharpless model;18 however, we expected preferential π-stacking of the 1° O-benzyl ether7a with the AD mix ligands would reverse the selectivity. Subsequent functional group manipulation, in accord with our prior work,7e,f gave aldehyde 26. After careful inspection of lactone 25 using Mosher ester analysis,19 we discovered that our π-stacking hypothesis had proven to be in error. As the exact structure of azaspiracid-1 was still unknown at this point, we chose to pursue the bisspiroketalization with the aldehyde 26.
Scheme 4.
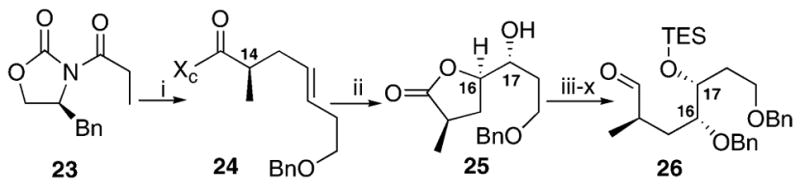
(i) NaHMDS, ICH2CH=CHCH2CH2OBn, THF, 92%; (ii) AD mix β*, NaHCO3, t-BuOH, H2O, r.t., 4 : 1 d.r., 55%; (iii) TIPSOTf, imid. DMF, 88%; (iv) LiBH4, MeOH, THF, 99%; (v) PivCl, DMAP, Et3N, CH2Cl2, 82%; (vi) BnBr, NaH, DMF; (vii) TBAF, THF, 24% over 2 steps; (viii) TESCl, DMAP, Et3N, CH2Cl2, 99%; (ix) LiBH4, MeOH, THF, 89%; (x) TPAP, NMO, CH2Cl2, mol. sieves, 95%.
Our standard Julia coupling approach7c,f with the previously prepared sulfone 277a followed by TPAP oxidation gave the keto sulfone 28 (Scheme 5). Na/Hg amalgam reduction and treatment with PPTS, THF/H2O yielded the expected transoidal product 29 as the sole bisspiroketal. The formation of a single transoidal bisspiroketal coupled with the observed H6-H41 NOE (also found in azaspiracid) led us to suspect that compound 29 possessed the correct stereochemistry present in the natural product. Finally, conversion of the lactone 30 was accomplished through LiDBB debenzylation and TPAP oxidation. Similar NOE correlations were again observed confirming the transoidal nature of both compounds.
Scheme 5.
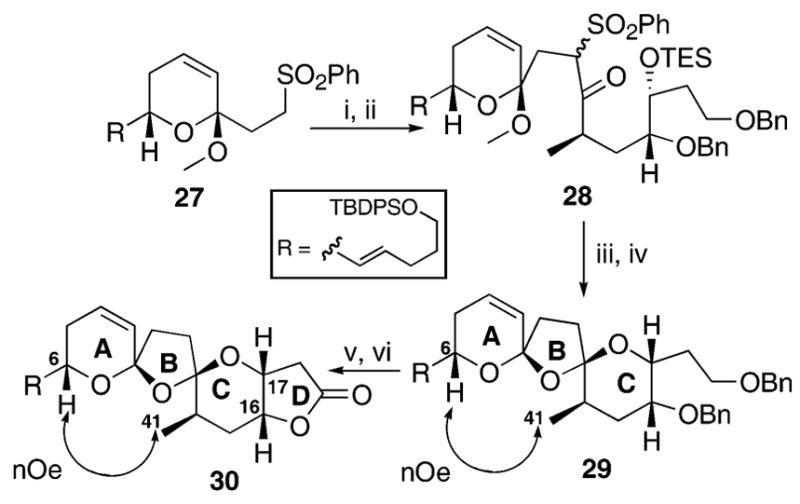
(i) 27, LDA, THF, then 26, −78 °C; (ii) TPAP, NMO, CH2Cl2, mol. sieves, 66% yield over 2 steps; (iii) Na/Hg, Na2HPO4, THF, H2O, 82%; (iv) PPTS, THF, H2O, 50%; (v) LiDBB, THF, −78 °C; (vi) TPAP, NMO, CH2Cl2, mol. sieves, 73% over 2 steps.
With the synthesis of the lactone 30 complete, conversion to the ABCDE ring system was undertaken (Scheme 6). Reduction with DIBAL-H provided the lactol as a mixture of undetermined epimers. Subsequent Wadsworth–Emmons olefination with in situ cyclization20 gave the coupled material 32 in reasonable (45%) yield. Finally, TBAF removal of the protecting groups and oxidation at C1 gave the ABCDE ring system 34.
Scheme 6.
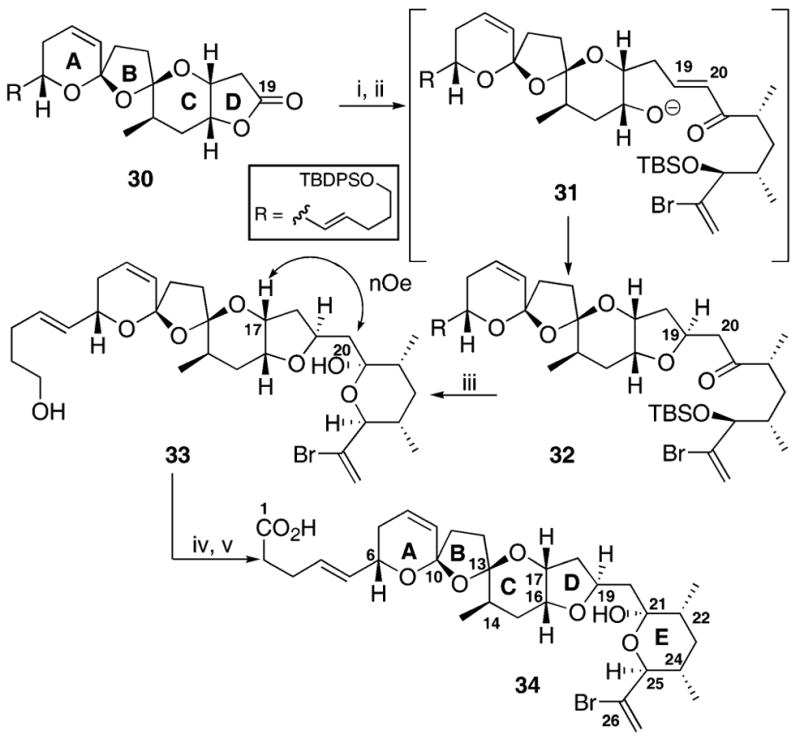
(i) DIBAL-H, CH2Cl2, −78 °C, 78%; (ii) 11, KHMDS, THF, −78 °C to r.t., 45%; (iii) TBAF, THF, 57%; (iv) TPAP, NMO, CH2Cl2, mol. sieves, 68%; (v) NaClO2, t-BuOH, H2O, 2-methyl-2-butene, 60%.
Comparison of the NMR spectra of synthetic 33 and 34 and azaspiracid-1 in identical solvents (CD3OD + 0.5% CD3CO2D) revealed some intriguing results. Large sections of the synthetic materials 33 and 34 were in good agreement with published data for azaspiracid-1.2 Major points of divergence proved to be the H8,9 alkene position and H6 (1H NMR: 33 H6 = 4.36, 34 H6 = 4.35, azaspiracid-1 H6 = 4.81; 33 H8 = 5.94, 34 H8 = 5.95, azaspiracid-1 H8 = 5.76). Given the results presented, we concluded that the relocation of the alkene to the C7,8 position (e.g. compound 7) was necessary for the actual structure of azaspiracid-1. Subsequent to this conclusion, Professor Nicolaou independently reported the confirmation of this assignment.9d
In summary, efficient approaches to the originally proposed ABCD ring system (17 steps from oxazolidinone ent-23) and the revised ABCDE ring system (21 steps from oxazolidinone 23) of azaspiracid are presented. It is important to note that acid 34 contains the correct stereochemistry at C6, C10, C13, C14, C16, C17, C19, C21, C22, C24 and C25 necessary for the actual structure of azaspiracid-1 (7).9d Key transformations include the Wadsworth–Emmons coupling to form the C19,20 linkage and bisspiroketalization of the ketone 28 to provide a single transoidal bisspiroketal 29. Further progress toward the total synthesis of the actual structure of azaspiracid-1 (7) will be reported in due course.
Financial support was provided by the National Institutes of Health (GM63723). We thank Professor Michael Doyle (University of Maryland) for his generous gift of the rhodium catalysts. The authors would also like to thank Professor Max Dienzer (OSU) and Dr. Jeff Morre′ (OSU) for mass spectral data, Roger Kohnert (OSU) for NMR assistance, Professor David A. Horne (OSU), Dr. Daniel P. Furkert (OSU) and Dr. Roger Hanselmann (Rib-X Pharmaceuticals) for their helpful discussions.
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: Complete experimental procedures and 1H and 13C spectra are provided for all new compounds. See http://www.rsc.org/suppdata/cc/b4/b410092a/
Dedicated to Professor Li-Xin Dai on the occasion of his 80th birthday.
Notes and references
- 1.MacMahon T, Silke J. Harmful Algae News. 1996;14:2. [Google Scholar]
- 2.Satake M, Ofuji K, Naoki H, James KJ, Furey A, McMahon T, Silke J, Yasumoto T. J Am Chem Soc. 1998;120:9967–68. [Google Scholar]
- 3.Furey A, Moroney C, Magdalena AB, Fidalgo Saez MJ, Lehane M, James KJ. Environ Sci Technol. 2003;37:3078–84. doi: 10.1021/es020246z. [DOI] [PubMed] [Google Scholar]
- 4.James KJ, Furey A, Lehane M, Ramstad H, Aune T, Hovgaard P, Morris S, Higman W, Satake M, Yasumoto T. Toxicon. 2002;40:909–15. doi: 10.1016/s0041-0101(02)00082-x. [DOI] [PubMed] [Google Scholar]
- 5.James KJ, Moroney C, Roden C, Satake M, Yasumoto T, Lehane M, Furey A. Toxicon. 2003;41:145–51. doi: 10.1016/s0041-0101(02)00244-1. [DOI] [PubMed] [Google Scholar]
- 6.Ito E, Satake M, Ofuji K, Kurita N, McMahon T, James K, Yasumoto T. Toxicon. 2000;38:917–30. doi: 10.1016/s0041-0101(99)00203-2. [DOI] [PubMed] [Google Scholar]
- 7.(a) Carter RG, Weldon DJ. Org Lett. 2000;2:3913–16. doi: 10.1021/ol006674w. [DOI] [PubMed] [Google Scholar]; (b) Carter RG, Weldon DJ, Bourland TC. 221st National ACS Meeting; San Diego, ORG-479. [Google Scholar]; (c) Carter RG, Graves DE. Tetrahedron Lett. 2001;42:6035–39. [Google Scholar]; (d) Carter RG, Bourland TC, Graves DE. Org Lett. 2002;4:2177–79. doi: 10.1021/ol026033w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Carter RG, Graves DE, Gronemeyer MA, Tschumper GS. Org Lett. 2002;4:2181–4. doi: 10.1021/ol026034o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Carter RG, Bourland TC, Zhou X-T, Gronemeyer MA. Tetrahedron. 2003;59:8963–74. [Google Scholar]
- 8.(a) Dounay AB, Forsyth CJ. Org Lett. 2001;3:975–78. [PubMed] [Google Scholar]; (b) Aiguade J, Hao J, Forsyth CJ. Org Lett. 2001;3:979–82. [PubMed] [Google Scholar]; (c) Aiguade J, Hao J, Forsyth CJ. Tetrahedron Lett. 2001;42:817–20. [Google Scholar]; (d) Hao J, Aiguade J, Forsyth CJ. Tetrahedron Lett. 2001;42:821–24. [Google Scholar]; (e) Nicolaou KC, Pihko PM, Diedrichs N, Zou N, Bernal F. Angew Chem Int Ed. 2001;40:1262–65. [PubMed] [Google Scholar]; (f) Buszek KR. 221st National ACS Meeting; San Diego, ORGN-570. [Google Scholar]; (g) Forsyth CJ, Hao J, Aiguade J. Angew Chem Int Ed. 2001;40:3663–67. doi: 10.1002/1521-3773(20011001)40:19<3663::aid-anie3663>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (h) Nicolaou KC, Qian W, Bernal F, Uesaka N, Pihko PM, Hinrichs J. Angew Chem Int Ed. 2001;40:4068–71. [PubMed] [Google Scholar]; (i) Sasaki M, Iwamuro Y, Nemoto J, Oikawa M. Tetrahedron Lett. 2003;44:6199–201. [Google Scholar]; (j) Buszek KR, Gibson TS, Reinhardt BC, Sunde JR. 226th ACS National Meeting; New York, ORGN-179. [Google Scholar]; (k) Ishikawa Y, Nishiyama S. Tetrahedron Lett. 2004;45:351–54. [Google Scholar]; (l) Ishikawa Y, Nishiyama S. Heterocycles. 2004;63:539–65. [Google Scholar]; (m) Ishikawa Y, Nishiyama S. Heterocycles. 2004;63:885–93. [Google Scholar]
- 9.(a) Nicolaou KC, Li Y, Uesaka N, Koftis TV, Vyskocil S, Ling T, Govindasamy M, Qian W, Bernal F, Chen DY-K. Angew Chem Int Ed. 2003;42:3643–48. doi: 10.1002/anie.200351825. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Chen DY-K, Li Y, Qian W, Ling T, Vyskocil S, Koftis TV, Govindasamy M, Uesaka N. Angew Chem Int Ed. 2003;42:3649–53. doi: 10.1002/anie.200351826. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Vyskocil S, Koftis TV, Yamada YMA, Ling T, Chen DY-K, Tang W, Petrovic G, Frederick MO, Satake YM. Angew Chem Int Ed. 2004;43:4312–4318. doi: 10.1002/anie.200460695. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Koftis TV, Vyskocil S, Petrovic G, Ling T, Yamada YMA, Tang W, Frederick MO. Angew Chem Int Ed. 2004;43:4318–4324. doi: 10.1002/anie.200460696. [DOI] [PubMed] [Google Scholar]
- 10.Abiko A, Liu J-F, Masamune S. J Am Chem Soc. 1997;119:2586–87. [Google Scholar]
- 11.Inoue T, Liu J-F, Buskke DC, Abiko A. J Org Chem. 2002;67:5250–56. doi: 10.1021/jo0257896. [DOI] [PubMed] [Google Scholar]
- 12.Myers AG, McKinstry L. J Org Chem. 1996;61:2428–40. [Google Scholar]
- 13.Dounay and Forsyth have reported isolation of an additional bisspiroketal product from selected conditions. Their compound was assigned to be the C14-epi product bearing a cisoidal bisspiroketal. See ref. 8a and A. B. Dounay, Ph.D. 2001, University of Minnesota, pp. 160–166.
- 14.Freeman PK, Hutchinson LL. J Org Chem. 1980;45:1924–30. [Google Scholar]
- 15.Doyle MP, Catino AJ. Tetrahedron: Asymm. 2003;14:925–28. [Google Scholar]
- 16.Wee AGH. J Org Chem. 2001;66:8513–17. doi: 10.1021/jo010753j. [DOI] [PubMed] [Google Scholar]
- 17.Doyle MP, Zhou Q-L, Raab CE, Roos GHP, Simonsen SH, Lynch V. Inorg Chem. 1996;35:6064–73. [Google Scholar]
- 18.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483–547. [Google Scholar]
- 19.See ESI for full account of Mosher model. I. Ohtani T, Kusumi Y, Kashman Kakisawa H. J Am Chem Soc. 1991;113:4092–96.
- 20.Boger DL, Ichikawa S, Zhong W. J Am Chem Soc. 2001;123:4161–67. doi: 10.1021/ja010195q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.