

Abstract
Antigenic peptide loading of major histocompatibility complex class II molecules is enhanced by lysosomal pH and catalyzed by the HLA-DM molecule. The physical mechanism behind the catalytic activity of DM was investigated by using time-resolved fluorescence anisotropy (TRFA) and fluorescence binding studies with the dye 8-anilino-1-naphthalenesulfonic acid (ANS). We demonstrate that the conformations of both HLA-DM and HLA-DR3, irrespective of the composition of bound peptide, are pH sensitive. Both complexes reversibly expose more nonpolar regions upon protonation. Interaction of DM with DR shields these hydrophobic domains from the aqueous environment, leading to stabilization of the DM and DR conformations. At lysosomal pH, the uncovering of additional hydrophobic patches leads to a more extensive DM–DR association. We propose that DM catalyzes class II peptide loading by stabilizing the low-pH conformation of DR, favoring peptide exchange. The DM–DR association involves a larger hydrophobic surface area with DR/class II-associated invariant chain peptides (CLIP) than with stable DR/peptide complexes, explaining the preferred association of DM with the former. The data support a release mechanism of DM from the DM–DR complex through reduction of the interactive surface, upon binding of class II molecules with antigenic peptide or upon neutralization of the DM–DR complex at the cell surface.
Presentation of peptides derived from exogenous proteins predominantly occurs in the context of heterodimeric major histocompatibility complex (MHC) class II molecules (1). Binding of these peptides probably takes place in specialized intracellular vesicles, called MHC class II compartments (MIICs; ref. 2). Class II molecules are directed to the MIICs after dimerization in the endoplasmic reticulum and complexing to the invariant chain (Ii) (3). Before binding of internalized peptides, Ii is proteolytically removed, leaving a nested set of peptides (termed CLIP, for class II-associated invariant chain peptides) in the peptide-binding cleft of the class II dimer (4, 5). Subsequent removal of CLIP allows binding of the peptides that are eventually presented to the immune system.
The process of peptide loading in the MIICs has not been unravelled, but the lysosomal pH value of the MIICs helps to promote peptide loading of class II molecules (6, 7). For instance, low pH increases protease activity, required both for processing of protein antigen (8) and for Ii proteolysis (9). In addition, CLIP removal from class II is facilitated by low pH (10). Finally, pH affects the mechanism of peptide binding itself (11), possibly through protonation of acidic residues involved in peptide complexing in the class II binding cleft (12) or through a conformational change in the class II dimer (13, 14). The HLA-DM molecule is another important component for peptide loading. Cell lines mutated for HLA-DM, or transgenic mice lacking the equivalent murine molecule, accumulate class II/CLIP complexes at the cell surface (15–18). HLA-DM, a nonpolymorphic and unconventional major histocompatibility complex molecule, is predominantly located in the MIICs. It catalyzes the dissociation of CLIP and other peptides from class II molecules and facilitates selection for antigen presentation of peptides that stably bind to class II molecules (19–26). The catalytic action of DM is pH-dependent with an optimal activity between 4.5 and 5.5. Moreover, DM stabilizes class II complexes that are devoid of peptide during peptide exchange (27, 28).
The mechanism by which HLA-DM exerts its function is unknown, but results from a direct association between HLA-DM and HLA-DR, which is most marked at lysosomal pH (29, 30). The mode of interaction between DM and DR has not yet been determined, and this interaction is crucial to understanding the catalytic and chaperoning function of DM. The mechanism by which DM is released, to achieve cell surface expression of DR, and concomitant intracellular retention of DM, is also of relevance. We set out to gain insight into the physical mechanism of DM interaction with class II molecules by investigating the structural states of DM and DR at both neutral and lysosomal pH. We then went on to examine whether the conformations they adopt in these conditions affect the interaction between the two complexes.
MATERIALS AND METHODS
Preparation and Purification of Soluble HLA-DM and HLA-DR.
Soluble HLA-DM complexes were generated in a baculovirus expression system using the dual promoter transfer vector pBacp10pol (kindly provided by P. Marrack) (31) to clone the truncated DMA (DMA∗0101) and DMB (DMB∗0101) genes and the BaculoGold vector (PharMingen) to generate recombinant baculoviruses (25). Purification was performed as described (25). Filtration (Amicon; 10,000 molecular weight cut-off) was used to exchange the buffer for phosphate-buffered saline. HLA-DR3 complexes were affinity purified from T2.DR3 cells (kindly provided by P. Cresswell) (32) by affinity chromatography using the HLA-DR-specific antibody L243 (American Type Culture Collection) as described (33). n-Octyl β-d-glucopyranoside (OG; Sigma) was used at 0.5% to solubilize the cell membranes. After elution and filtration (10,000 molecular weight cut-off) to exchange the buffer for phosphate-buffered saline with 0.5% OG, the samples containing HLA-DR3 were subjected to another round of affinity purification to obtain highly pure HLA-DR3 complexes. The purity of DM and DR in the eluted fractions was determined by SDS/PAGE followed by silver staining (34). Only fractions estimated to be more than 90% pure were used in subsequent experiments.
Protein Determinations.
Protein concentrations were assayed on SDS/PAGE using both Coomassie brilliant blue (R-250; GIBCO/BRL) and silver (34) staining techniques with BSA (Sigma), ovalbumin (Sigma), and hen egg white lysozyme (Sigma) as standards.
Preparation of Complexes of HLA-DR and Apolipoprotein B-(2877–2894) peptide [ApoB(2877–2894)].
ApoB(2877–2894) was synthesized as described (33). Purified HLA-DR3 complexes from T2.DR3 cells were incubated with a 20-fold molar excess of ApoB(2877–2894) for 4 days at 37°C in buffer containing 25 mM Na2CO3, 50 mM Tris, 2 mM EDTA, 0.1 mM phenylmethanesulfonyl fluoride, and 0.1% OG adjusted to pH 4.5 by 1 M citric acid. Excess peptide was removed by repeated ultrafiltration with 10,000 molecular weight cut-off, and buffer was exchanged for phosphate-buffered saline containing 0.5% OG. The percentage of DR3/ApoB(2877–2894) complexes was determined by incubation of the complexes in reducing Laemmli sample buffer for 30 min at room temperature, followed by quantitation of the relative amount of SDS-stable DR3αβ complexes on SDS/PAGE.
Time-Resolved Fluorescence Anisotropy (TRFA).
The apparatus used for TRFA measurements has been described (35, 36). Briefly, a coupled Nd–yttrium/aluminum garnet/dye laser system provided polarized light pulses of 300-nm wavelength. Tryptophan fluorescence was detected at 350 nm parallel (i∥) and perpendicular (i⊥) to the excitation polarization by single-photon counting equipment. At time t the fluorescence intensity S(t) and anisotropy R(t) were calculated as:
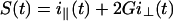 |
and
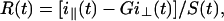 |
with G denoting a correction factor for the polarization dependence of the detection system. Data were analyzed assuming multiexponential decays for the fluorescence intensity
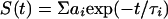 |
and anisotropy
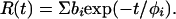 |
The components of the polarized fluorescence
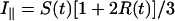 |
and
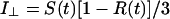 |
were convoluted with the apparatus response function (37) and least-square fits to the experimental data i∥(t) and i⊥(t) were performed. We always started by assuming mono-exponential decays and included more exponentials stepwise until no further improvement of the fit was observed. Visual rating of the residuals and a χ2 test were taken as criteria. Measurements were performed using 200 nM DM in a solution containing 10 mM KPi, 10 mM NaOAc, 150 mM NaCl, and 17 mM OG. To correct for background signals, mock samples lacking the protein were prepared and background data were subtracted from the sample data before data analysis.
The measured relaxation times and their amplitudes may be interpreted within the framework of rotational diffusion. The simplest case is given by a rigid sphere of volume V in an isotropic medium of viscosity η, for which the anisotropy is given by
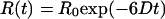 |
with D = kBT/6ηV denoting the coefficient of rotational diffusion (kB being the Boltzmann constant) (38). The rotational diffusion of an entire protein is then described by the diffusion coefficient D = (6φ)−1.
In the case of a prolate ellipsoidal body fluctuating in a strong
ordering potential, a description that holds well for side-chain
movements in a protein, the diffusion coefficient for wobbling motions
of the long axis is given by D⊥ =
〈θ2〉/2φ with the mean-square amplitude
〈θ2〉 = 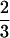 (1 −
〈P2〉) and the order parameter
〈P2〉 =
[b2/(b1 +
b2)]1/2 (39). The orientational
fluctuations of a protein, therefore, can be described in terms of
diffusional coefficients D and (root-mean-square) angular
amplitudes θ.
(1 −
〈P2〉) and the order parameter
〈P2〉 =
[b2/(b1 +
b2)]1/2 (39). The orientational
fluctuations of a protein, therefore, can be described in terms of
diffusional coefficients D and (root-mean-square) angular
amplitudes θ.
The statement that the observed DM diffusion coefficient Dsphere agrees with the presence of DM dimers (see Results and Discussion) is supported by the following calculations. For a given protein mass, its dry volume V = Mr/ρ⋅NA may be estimated by using a mass density of ρ = 1.35 ± 0.05 (g/cm3) and the Avogadro number NA. Its radius follows as r = (3V/4π)1/3. A reasonable average hydration layer of thickness 0.5 ± 0.1 nm has to be added to the radius to yield the hydrodynamic radius R′. For the soluble DMαβ heterodimer with a molar mass (including glycosyls) of 61.7 kDa, the dry volume equals V = 75.9 nm3, and the hydrated volume V′ = 128 ± 12 nm3. This results, for a sphere of this volume, in a rotational diffusion coefficient of D = 5.4 ± 0.5 μs−1. For comparison, the smallest aggregation unit, a dimer of dimers, would have a double molar mass and hydrated volume of 232 nm3, which leads to an expected diffusion coefficient of D = 2.9 μs−1.
Fluorescence Spectroscopy.
Fluorescence binding studies of 8-anilino-1-naphthalenesulfonic acid (ANS) were performed with a luminescence spectrometer (Perkin–Elmer, model LS50B; Figs. 1 and 3 represent data collected in Tübingen, and Figs. 2 and 4, St. Louis), operated at slit widths of 10 nm for both excitation and emission. Measurements were carried out at 22°C in thermostatically controlled cell holders containing 1-cm quartz cuvettes. Excitation was set at 350 nm, with recording of the emission spectrum from 400 to 600 nm. Samples contained 50 nM DM or hen egg white lysozyme (stock solution containing 0.5% OG) or 20–40 nM DR3 complexes and 20 μM ANS in a buffer consisting of 10 mM KPi, 10 mM NaOAc, and 150 mM NaCl. OG was added at 17 mM to keep the DR3 complexes (containing transmembrane regions) in solution, while avoiding formation of micelles by using concentrations below the critical micelle concentration (CMC). For sake of comparison, OG was included in the other experiments as well. Previously, detergent was shown not to affect the outcome of ANS binding experiments to murine class II complexes (14), thus permitting the use of OG in these experiments. Moreover, although OG is known to induce CLIP dissocation at low pH, it does not interfere with the catalytic action of DM (10, 23). Spectra were recorded 2 min after pH adjustment using concentrated NaOH and HCl, and pH values were measured after fluorescence recording. Values are reported as relative fluorescence, being a measure of fluorescence intensity in arbitrary units (fluorescence intensity/reference intensity).
Figure 1.

ANS binding studies show a conformational change of HLA-DM and HLA-DR upon pH variation. Binding of ANS to 50 nM HLA-DM (A), 50 nM hen egg white lysozyme (B), or 20 nM HLA-DR/CLIP (C) was monitored after excitation at 350 nm by recording of the fluorescence emission spectra (400–600 nm) in the presence or absence of protein 2 min after addition of the protein or after adjustment of the pH.
Figure 3.

Interaction between HLA-DM and HLA-DR/CLIP involves conformation-sensitive hydrophobic domains of the protein(s). The difference in ANS fluorescence (arbitrary units) compared with the initial fluorescence value of the sample before DM addition is depicted as a function of added HLA-DM at pH 5.0 (A) or pH 7.3 (B). Solid lines represent titration of HLA-DM into a solution containing 50 nM HLA-DR3 complexed to CLIP and 10 μM ANS. Dashed lines represent titration of HLA-DM into a solution containing 50 nM hen egg white lysozyme. All data shown are derived from a representative set of values of four to five individual experiments.
Figure 2.

Binding of ANS to HLA-DR complexed to CLIP or ApoB(2877–2894) shows similar conformational changes of the complexes upon pH variation. Solid lines show binding of ANS to 40 nM DR3/CLIP complexes at pH 7.3 and 5.0 and dashed lines represent binding of ANS to 40 nM DR3/ApoB(2877–2894) complexes at the same pH values. The fluorescence emission spectra (415–600 nm) after excitation at 350 nm were recorded 2 min after addition of the class II molecules or after adjustment of the pH values.
Figure 4.

Interaction of HLA-DM with HLA-DR3/CLIP involves more hydrophobic regions than does interaction with HLA-DR3/ApoB(2877–2894). The difference in ANS fluorescence (arbitrary units) compared with the initial fluorescence value of the sample before DM addition is shown as a function of added HLA-DM. DM was titrated into a solution containing 50 nM DR3/CLIP complexes (filled bars) or 50 nM DR3/ApoB(2877–2894) (empty bars) and 20 μM ANS. The data shown are derived from a representative experiment that was repeated three times.
For ANS fluorescence studies measuring protein–protein interactions, DM or hen egg white lysozyme was titrated into a solution containing 50 nM DR3 or lysozyme and 10–20 μM ANS in 100 mM NaOAc buffer with 17 mM OG at either pH 7.3 or pH 5.0. After stabilization of the signal upon addition of DM, measurements were carried out near the emission maximum of 480 nm. pH values were checked for stability after measurements. Values are reported as the difference in relative fluorescence compared with the initial fluorescence value of the sample containing only 50 nM DR complexes and no DM. Aggregation of the proteins in the samples was checked by monitoring the steady-state anisotropy of the samples at 350 nm with excitation set at 300 nm.
RESULTS AND DISCUSSION
To perform structural studies on HLA-DM, soluble DM was produced as a heterodimeric complex in a baculovirus expression system. The recombinant DM is catalytically active on peptide loading of HLA-DR3 (25). To monitor the conformation of DM at various pH values we performed TRFA experiments on DM. TRFA studies provide a measure of the internal flexibility of proteins and their state of aggregation by determining the hydrodynamic radius (40) and are thus useful to monitor conformational changes. Because measurements are performed on samples in solution, TRFA experiments avoid problems such as interference of solid supports on the conformational status of the molecule. We used the intrinsic fluorescence of the 11 tryptophan residues in the soluble DM to further avoid any influence by an artificially inserted label. Table 1 summarizes the TRFA measurements on DM at neutral and acidic pH. At pH 7.3, the value of 5.3 ± 0.7 μs−1 of the diffusion coefficient Dsphere is consistent with the presence of a DM complex, with a hydrodynamic radius maximally corresponding to a single DM dimer (as explained in Materials and Methods). Thus, DM does not aggregate under the conditions used. There were no detectable conformational changes upon protonation of DM. The aggregation behavior of DM stayed the same, as expressed in a constant diffusion coefficient Dsphere and therefore a constant hydrodynamic radius of the heterodimer. Moreover, the internal dynamics remained similar, as represented by a constant amplitude θrms and diffusion coefficient D⊥ of faster orientational motions of parts of the protein, most presumably side-chain fluctuations of the tryptophan. Thus, no structural transition of DM is observed upon lowering of the pH to values like those in the lysosome. This points to a stable, compact conformation of the DM heterodimer with a tertiary and quaternary structure that is not prone to gross alterations in its physiological pH range. Therefore, any pH-induced structural alterations in DM must be subtle, without affecting the overall fluorescence of its tryptophan residues.
Table 1.
TRFA measurements on DM in solution show that pH variation does not result in gross alterations of the DM conformation
| pH | D⊥ μs−1 | θrms, ° | Dsphere, μs−1 |
|---|---|---|---|
| 7.3 | 22.5 ± 2.6 | 20.7 ± 0.7 | 5.3 ± 0.7 |
| 5.0 | 18.2 ± 2.4 | 19.9 ± 0.7 | 4.6 ± 0.7 |
The diffusion coefficient D⊥ and the amplitude θrms are parameters calculated from the TRFA measurements to monitor the internal dynamics of the DM complex. The diffusion coefficient Dsphere was calculated as a parameter expressing the aggregation behavior of DM. All measurements were performed in duplicate.
To detect subtle changes in the DM complex, we used the environment-sensitive probe ANS, whose fluorescence quantum yield depends on the polarity of the solution. The dye binds to hydrophobic patches of proteins, causing ANS to emit fluorescence at higher magnitude and shorter wavelength (41). Changes in protein conformation leading to exposure of previously buried nonpolar regions can thus be visualized. At pH 7.3, addition of DM to an ANS solution resulted in a small, stable increase in the fluorescence emission spectrum (Fig. 1A). Autofluorescence of DM alone was not significant (data not shown). Upon acidification of the sample to pH 5.0, the fluorescence intensity markedly increased and the maximal emission wavelength (λmax) shifted from 510 nm to 500 nm. The eventual fluorescence signals were stable over time and protein aggregation was not detected. The spectral changes show that DM displays some ANS binding sites at neutral pH and undergoes a structural change when going to lysosomal-like pH either by exposing internal nonpolar regions or by increasing hydrophobicity of the ANS-bound regions at acidic pH through titration of exposed functional groups between pH 7 and pH 5. ANS binding to a control protein, hen egg white lysozyme, essentially did not show any changes in the emission spectra between pH 7.3 and pH 5.0 (Fig. 1B). Retitration of the pH to 7.3 quickly reverted the fluorescence emission spectra to their original values, implying that the structural transitions of DM between neutral and lysosomal-like pH are fully reversible (Fig. 1A). Both the ANS and TRFA data are consistent with the idea that protonation induces reversible exposure of localized parts of the DM heterodimer, while leaving the overall structure relatively unchanged.
To evaluate if human class II molecules show a similar behavior upon pH variation, as observed for various rodent class II molecules (13, 14), ANS binding experiments were performed on HLA-DR3 isolated from T2.DR3 cells, in which class II molecules are almost exclusively associated with CLIP (6). Addition of DR3/CLIP to an ANS solution at pH 7.3 had a more prominent effect on the fluorescence emission spectrum of ANS than did addition of DM (Fig. 1C). Lowering of the pH to 5.0 resulted in a much stronger increase of fluorescence than with DM and an additional shifting of λmax from 485 nm to 475 nm. Thus, DR3/CLIP molecules expose more nonpolar regions than DM at neutral pH, and they undergo a far greater pH-induced structural transition. As with DM, the conformational changes were fully reversible upon retitration of the pH to original values (Fig. 1C). Because CLIP bound to DR3 readily dissociates under low-pH conditions in the presence of OG (5, 10, 27), it is possible that during the ANS binding experiments part of the class II molecules were in in the process of becoming “empty” through CLIP release. This possibility, however, did not affect the ANS binding experiments at acidic pH, since the fluoresence emission spectra were stable over time. Moreover, retitration of the pH to neutral values reverted the fluoresence spectra to original values.
To investigate if the stability of peptide binding to the class II binding groove influenced the pH sensitivity of the complex, comparative studies were performed on DR3 dimers complexed to ApoB(2877–2894), a stably binding antigenic peptide (33). CLIP bound to the isolated DR3 was exchanged for ApoB(2877–2894) in vitro. The efficiency of this exchange was assessed by measuring the formation of SDS-stable DR3αβ complexes; DR3αβ/CLIP complexes dissociate into single chains upon incubation in SDS prior to SDS/gel electrophoresis, in contrast to DR3αβ/ApoB(2877–2894) (6). The amount of DR3αβ/ApoB(2877–2894) complexes was estimated to be over 85% (data not shown). ANS binding studies demonstrated that pH titration led to conformational alterations in the DR3αβ/ApoB(2877–2894) complexes that were indistinguishable from those observed for DR3αβ/CLIP (Fig. 2). Thus, stable binding of peptide to class II does not lock the complex in a conformational state that is unable to respond to pH variation. Moreover, the composition of the bound peptide does not affect the binding of ANS to the conformation-sensitive regions of human class II, as observed for murine I-Ek complexes as well (13). This suggests that most ANS-binding sites lie outside the peptide-binding groove and that the pH sensitivity mainly involves regions that do not directly interact with peptide. These pH-induced structural alterations may, however, indirectly affect the conformation of the peptide-binding groove and the hydrogen bonds between bound peptide and class II complex. The overall strength of stabilizing forces between peptide and class II will finally determine if the peptide remains associated. In this respect, Boniface and co-workers (13) suggested for I-Ek molecules a pH-induced destabilization of nonpolar interdomain contacts in the lower (e.g., α2) class II domains or between them and the floor of the peptide-binding groove.
The exposure of nonpolar interior regions of both HLA-DR and HLA-DM at lysosomal pH counteracts the natural tendency of proteins to shield their hydrophobic patches from the aqueous environment. Because this occurs at the pH where the DM–DR complexes are most efficiently isolated (29), we addressed the question whether these regions are involved in DM–DR interaction. Titration studies of increasing amounts of DM in a solution containing either DR3 or the control protein hen egg white lysozyme were performed, using the fluorescence of ANS to monitor if the interaction affected the areas occupied by ANS. Titration of DM at pH 5.0 into a solution containing either 50 nM lysozyme or no protein led, in both cases, to identical steady increases in ANS fluorescence proportional to the increasing amount of DM (Fig. 3A). Thus, the hydrophobic regions available on DM for ANS binding upon titration were undisturbed by the presence of lysozyme, consistent with a lack of interaction involving nonpolar domains. Interestingly, addition of DM to a solution containing 50 nM DR3/CLIP resulted in a steep decrease of the ANS signal at low DM concentrations that levelled off with increasing amounts of DM (Fig. 3A). At 60 nM DM, the signal started to rise with a slope similar to that observed with DM and lysozyme, showing that saturation was achieved. Monitoring of a solution containing DR3/CLIP and ANS alone or titration of hen egg white lysozyme into this solution did not show variation in the emitted signal, demonstrating that the DM-induced decrease in ANS fluorescence was not due to quenching or to changes in DR3/CLIP conformation in time upon addition of an irrelevant protein. Monitoring of the steady-state anisotropy of the solution did not point to aggregation of the proteins in solution, and checking of the pH during and after the experiments showed constant values. From the data it is evident that DM and DR3 interact with each other in a saturable fashion, and that this interaction affects exposed nonpolar regions so that they are no longer available for binding to ANS. This type of association occurred specifically between DM and DR molecules, and a similar DR–DR or DM–DM interaction was not observed. Titration of DM into an ANS solution containing DR3/CLIP at pH 7.3 resulted in a similar saturable interaction between the complexes (Fig. 3B). Interestingly, the total amount of ANS that was displaced as a result of the interaction at lysosomal pH was 6- to 7-fold higher than at pH 7.3 (Fig. 3), indicating that the interaction at neutral pH involves fewer nonpolar regions. Again, using hen egg white lysozyme as a control, titration of DM at neutral pH resulted in a steady increase in fluorescence, although less strong than at pH 5.0 (Fig. 3B).
Since the association between DM and DR was stable over periods of time long enough to generate empty DR complexes by release of CLIP (data not shown), the interaction of DM with these class II molecules was compared with the DM interaction with stable DR/peptide complexes. DM was titrated in a solution containing 50 nM DR3/ApoB(2877–2894) complexes at lysosomal-like pH. This resulted in an interaction between DM and DR3/ApoB(2877–2894) that affected the exposed nonpolar regions as well, as demonstrated by the decrease in fluorescence at low DM concentrations (Fig. 4). The observed decrease in fluorescence was too high to be solely attributed to the estimated residual 12% (6 nM) DR/CLIP in the DR/ApoB(2877–2894) preparation as illustrated by a comparative experiment with DM titration into a 6 nM DR/CLIP solution (data not shown). Thus, DM associates with DR independent of the composition of bound peptide. In spite of the fact that DR3/CLIP and DR3/ApoB(2877–2894) complexes display similar amounts of additional hydrophobic patches upon protonation (Fig. 2), the observed ANS displacement with DM and DR3/ApoB(2877–2894) was considerably lower than observed with DR3/CLIP (Fig. 4). These findings indicate that the DM–DR/CLIP association is more extensive, involving a larger hydrophobic surface area than the interaction between DM and DR stably bound to antigenic peptide. These data may explain recent observations that DM associates less with DR complexed to antigenic peptide than with DR/CLIP (27, 28, 30).
Taken together, the conformations of both DM and DR are sensitive to pH variation in that the dimers expose a higher degree of nonpolarity upon protonation. Interaction between DM and DR affects the pH-sensitive nonpolar areas at both lysosomal and neutral pH. These regions may become buried indirectly through interaction-induced conformational changes in both complexes. More likely, however, a direct association of these areas takes place, stabilizing the conformations of both DM and DR by shielding their exposed nonpolar regions from the aqueous environment. At lysosomal pH, the uncovering of additional interior nonpolar patches drives both complexes to associate over a larger hydrophobic surface area, as reflected in the stronger displacement of ANS from the complexes. This effect thus explains the enhanced DM–DR association observed at low pH in vivo (29) and may account for the physical mechanism behind both the catalytic action of DM on class II peptide loading and the chaperone function of DM. We envisage that the more extensive DM–DR association at low pH stabilizes the pH-induced conformational changes of the class II complex, thus catalyzing peptide loading and simultaneously preventing self-aggregation of the DR molecules by occupying (part of) the otherwise exposed hydrophobic areas (27, 28). Naturally, the behavior of DM–class II complexes may differ, depending on the products of particular class II alleles (28). The recent finding of few free energy differences between empty and peptide-loaded murine I-Ek molecules may indicate that the observed interaction of DM with class II molecules causes catalysis of peptide loading by affecting a kinetic (transition state) energy barrier rather than equilibrium thermodynamics (42). It remains to be determined why the DM–DR/CLIP (or DM-empty DR) association involves a larger hydrophobic surface area than the DM–DR/ApoB(2877–2894) interaction, with both DR complexes responding to protonation with a similar degree of exposure of nonpolar regions. It suggests, however, that the exposed hydrophobic regions, which for DR most likely lie outside of the peptide-binding cleft, have a reduced accessibility for the bulky DM complex, but not for the small ANS molecules, after binding of an antigenic peptide to the class II-binding groove. This reduced accessibility may explain why empty DR complexes have a higher tendency to self-aggregate than DR molecules complexed to antigenic peptides (43–45).
We have not yet addressed how release of DM from the DM–DR complex is achieved, resulting in the cell surface expression of peptide-loaded class II molecules and the intracellular retention of DM. On the basis of our data, we propose that release of DM from the DM–DR complex occurs through reduction of the available interactive surface. This may be achieved in two ways. First, complexing of class II molecules with a stably binding peptide in MIICs will result in less extensive association of DM and its subsequent release, allowing DR to make its way to the cell surface. Alternatively, DM may stay associated with the DR complex and the two molecules comigrate to the cell surface in low-pH compartments. Upon fusion of the acidic vesicles with the cell membrane (as recently observed; ref. 46), the DM–DR complexes meet with a neutral-pH environment. This would result in burying of the majority of exposed hydrophobic regions of both DM and DR. Consequently, the interactive surface area between DM and DR is minimized, resulting in release of DM from DR and subsequent, immediate reinternalization of DM by means of a targeting signal in the DMβ chain (47, 48). The dynamic processing of peptide exchange of class II molecules would thus be continued until cell surface exposure of DR is achieved and DM is released and recycled to continue its catalytic role. These two models need not be mutually exclusive and, though consistent with the data, have to be consolidated by future experiments.
Acknowledgments
We gratefully acknowledge P. Marrack for providing the vector pBacp10pol, P. Cresswell for the T2.DR3 cells, and G. Malcherek for the ApoB(2877–2894) peptide. We thank our colleagues from our laboratories for helpful suggestions, discussions, and reagents. This work was supported by an institutional fellowship from the European Community (ERB-CHBG-CT93-0463) and a Pioneer Grant for S.M.v.H. and by the Boehringer Ingelheim Funds for U.G.
ABBREVIATIONS
- ANS
8-anilino-1-naphthalenesulfonic acid
- ApoB(2877–2894) apolipoprotein B-(2877–2894) peptide
CLIP, class II-associated invariant chain peptides
- TRFA
time-resolved fluorescence anisotropy
- Ii
invariant chain
- MIIC
major histocompatibility complex class II compartment
- OG
n-octyl β-d-glucopyranoside
References
- 1.Neefjes J J, Stollorz V, Peters P J, Geuze H J, Ploegh H L. Cell. 1990;61:171–183. doi: 10.1016/0092-8674(90)90224-3. [DOI] [PubMed] [Google Scholar]
- 2.Peters P J, Neefjes J J, Oorschot V, Ploegh H L, Geuze H J. Nature (London) 1991;349:669–676. doi: 10.1038/349669a0. [DOI] [PubMed] [Google Scholar]
- 3.Bakke O, Dobberstein B. Cell. 1990;63:707–716. doi: 10.1016/0092-8674(90)90137-4. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh P, Amaya M, Mellins E, Wiley D C. Nature (London) 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 5.Roche P A, Cresswell P. Proc Natl Acad Sci USA. 1991;88:3150–3154. [PubMed] [Google Scholar]
- 6.Riberdy J M, Newcomb J R, Surnam M J, Barbosa J A, Cresswell P. Nature (London) 1992;360:474–477. doi: 10.1038/360474a0. [DOI] [PubMed] [Google Scholar]
- 7.Jensen P E. J Exp Med. 1991;174:1111–1120. doi: 10.1084/jem.174.5.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriquez G M, Diment S. J Immunol. 1992;149:2894–2898. [PubMed] [Google Scholar]
- 9.Riese R J, Wolf P R, Brömme D, Natkin L R, Villadangos J A, Ploegh H L, Chapman H A. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 10.Avva R R, Cresswell P. Immunity. 1994;1:763–774. doi: 10.1016/s1074-7613(94)80018-9. [DOI] [PubMed] [Google Scholar]
- 11.Boniface J J, Allbritton N L, Reay P A, Kantor R M, Stryer L, Davis M M. Biochemistry. 1993;32:11761–11768. doi: 10.1021/bi00095a003. [DOI] [PubMed] [Google Scholar]
- 12.Fremont D H, Hendrickson W A, Marrack P, Kappler J. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
- 13.Boniface J J, Lyons D S, Wettstein D A, Allbritton N L, Davis M M. J Exp Med. 1996;183:119–126. doi: 10.1084/jem.183.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Runnels H A, Moore J C, Jensen P E. J Exp Med. 1996;183:127–136. doi: 10.1084/jem.183.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fling S P, Arp B, Pious D. Nature (London) 1994;368:554–558. doi: 10.1038/368554a0. [DOI] [PubMed] [Google Scholar]
- 16.Morris P, Shaman J, Attaya M, Amaya M, Goodman S, Bergman C, Monaco J J, Mellins E. Nature (London) 1994;368:551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 17.Miyazaki T, Wolf P, Tourne S, Waltzinger C, Dierich A, Barois N, Ploegh H, Benoist C, Mathis D. Cell. 1996;84:531–541. doi: 10.1016/s0092-8674(00)81029-6. [DOI] [PubMed] [Google Scholar]
- 18.Martin W D, Hicks G G, Mendiratta S K, Leva H I, Ruley H E, Van Kaer L. Cell. 1996;84:543–550. doi: 10.1016/s0092-8674(00)81030-2. [DOI] [PubMed] [Google Scholar]
- 19.Kelly A P, Monaco J J, Cho S, Trowsdale J. Nature (London) 1991;353:571–573. doi: 10.1038/353571a0. [DOI] [PubMed] [Google Scholar]
- 20.Sanderson F, Kleijmeer M J, Kelly A P, Verwoerd D, Tulp A, Neefjes J J, Geuze H J, Trowsdale J. Science. 1994;266:1566–1569. doi: 10.1126/science.7985027. [DOI] [PubMed] [Google Scholar]
- 21.Sloan V S, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller D M. Nature (London) 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 22.Sherman M A, Weber D A, Jensen P E. Immunity. 1995;3:197–205. doi: 10.1016/1074-7613(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 23.Denzin L K, Cresswell P. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 24.Kropshofer H, Vogt A B, Moldenhauer G, Hammer J, Blum J S, Hämmerling G J. EMBO J. 1996;15:6144–6154. [PMC free article] [PubMed] [Google Scholar]
- 25.van Ham S M, Grüneberg U, Malcherek G, Bröker I, Melms A, Trowsdale J. J Exp Med. 1996;184:2019–2024. doi: 10.1084/jem.184.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber D A, Evavold B D, Jensen P E. Science. 1996;274:618–620. doi: 10.1126/science.274.5287.618. [DOI] [PubMed] [Google Scholar]
- 27.Denzin L K, Hammond C, Cresswell P. J Exp Med. 1996;184:2153–2165. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kropshofer H, Arndt S O, Moldenhauer G, Hämmerling G J, Vogt A B. Immunity. 1997;6:293–302. doi: 10.1016/s1074-7613(00)80332-5. [DOI] [PubMed] [Google Scholar]
- 29.Sanderson F, Thomas C, Neefjes J, Trowsdale J. Immunity. 1996;4:1–20. doi: 10.1016/s1074-7613(00)80301-5. [DOI] [PubMed] [Google Scholar]
- 30.Vogt A B, Kropshofer H, Moldenhauer G, Hämmerling G J. Proc Natl Acad Sci USA. 1996;93:9724–9729. doi: 10.1073/pnas.93.18.9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozono H, White J, Clements J, Marrack P, Kappler J. Nature (London) 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 32.Riberdy J M, Cresswell P. J Immunol. 1992;148:2586–2590. [PubMed] [Google Scholar]
- 33.Malcherek G, Gnau V, Jung G, Rammensee H-G, Melms A. J Exp Med. 1995;181:527–536. doi: 10.1084/jem.181.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blum H, Beier H, Gross H J. Electrophoresis. 1987;8:93–99. [Google Scholar]
- 35.Doering K, Konermann L, Surrey T, Jähnig F. Eur Biophys. 1996;23:423–432. doi: 10.1007/BF00196829. [DOI] [PubMed] [Google Scholar]
- 36.Doering K, Beck W, Konermann L, Jähnig F. Biophys J. 1997;72:326–334. doi: 10.1016/S0006-3495(97)78671-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Libertini L J, Small E W. Anal Biochem. 1984;138:314–318. doi: 10.1016/0003-2697(84)90814-5. [DOI] [PubMed] [Google Scholar]
- 38.Memming R. Z Phys Chem. 1961;28:168–189. [Google Scholar]
- 39.van der Meer W, Pottel H, Herreman W, Ameloot M, Hendricks H, Schröder H. Biophys J. 1984;46:515–523. doi: 10.1016/S0006-3495(84)84049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beechem J M, Brand L. Annu Rev Biochem. 1985;54:43–71. doi: 10.1146/annurev.bi.54.070185.000355. [DOI] [PubMed] [Google Scholar]
- 41.Stryer L. J Mol Biol. 1965;13:482–495. doi: 10.1016/s0022-2836(65)80111-5. [DOI] [PubMed] [Google Scholar]
- 42.Reich Z, Altman J D, Boniface J J, Lyons D S, Kozono H, Ogg G, Morgan C, Davis M M. Proc Natl Acad Sci USA. 1997;94:2495–2500. doi: 10.1073/pnas.94.6.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stern L J, Wiley D C. Cell. 1992;68:465–477. doi: 10.1016/0092-8674(92)90184-e. [DOI] [PubMed] [Google Scholar]
- 44.Germain R, Rinker A G., Jr Nature (London) 1993;363:725–728. doi: 10.1038/363725a0. [DOI] [PubMed] [Google Scholar]
- 45.Sadegh-Nasseri S, Stern L J, Wiley D C, Germain R N. Nature (London) 1994;370:647–650. doi: 10.1038/370647a0. [DOI] [PubMed] [Google Scholar]
- 46.Wubbolts R, Fernandez-Borja M, Oomen L, Verwoerd D, Janssen H, Calafat J, Tulp A, Dusseljee S, Neefjes J. J Cell Biol. 1996;135:611–622. doi: 10.1083/jcb.135.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindstedt R, Liljedahl M, Peleraux A, Peterson P A, Karlsson L. Immunity. 1995;3:561–572. doi: 10.1016/1074-7613(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 48.Marks M S, Roche P A, van Donselaar E, Woodruff L, Peters P J, Bonifacino J S. J Cell Biol. 1995;131:351–369. doi: 10.1083/jcb.131.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]