

Abstract
Background
The development of an effective human immunodeficiency virus (HIV) vaccine is a high global priority. Here, we report the safety, tolerability, and immunogenicity of a replication-defective recombinant adenovirus serotype 5 (rAd5) vector HIV-1 candidate vaccine.
Methods
The vaccine is a mixture of 4 rAd5 vectors that express HIV-1 subtype B Gag-Pol fusion protein and envelope (Env) from subtypes A, B, and C. Healthy, uninfected adults were randomized to receive 1 intramuscular injection of placebo (n = 6) or vaccine at dose levels of 109 (n = 10), 1010 (n = 10), or 1011 (n = 10) particle units and were followed for 24 weeks to assess immunogenicity and safety.
Results
The vaccine was well tolerated but was associated with more reactogenicity at the highest dose. At week 4, vaccine antigen–specific T cell responses were detected in 28 (93.3%) and 18 (60%) of 30 vaccine recipients for CD4+ and CD8+ T cells, respectively, by intracellular cytokine staining assay and in 22 (73%) of 30 vaccine recipients by enzyme-linked immunospot assay. Env-specific antibody responses were detected in 15 (50%) of 30 vaccine recipients by enzyme-linked immunosorbant assay and in 28 (93.3%) of 30 vaccine recipients by immunoprecipitation followed by Western blotting. No neutralizing antibody was detected.
Conclusions
A single injection induced HIV-1 antigen–specific CD4+ T cell, CD8+ T cell, and antibody responses in the majority of vaccine recipients. This multiclade rAd5 HIV-1 vaccine is now being evaluated in combination with a multiclade HIV-1 DNA plasmid vaccine.
More than 40 million people are living with HIV/AIDS. Three million deaths occur annually because of HIV/ AIDS, with ∼5 million new infections occurring in 2005 [1]. Development of an effective vaccine would be an important intervention to help control the expanding global pandemic.
Adenovirus serotype 5 (Ad5) has been developed as a replication-defective recombinant vector (rAd5) to deliver intracellular genes via a number of routes [2]. Vaccination with rAd5 results in transient intracellular gene expression followed by rapid clearance [3]. Cellular and humoral immune responses have been induced in preclinical studies of rAd5 vaccines for HIV-1, simian immunodeficiency virus (SIV), and simian-human immunodeficiency virus (SHIV) [4–7]. This approach builds on previous successes with other infectious disease models, particularly for Ebola virus, against which nonhuman primates have been protected from lethal challenge [8, 9]. Recently, immunization of chimpanzees with rAd expressing hepatitis C virus (HCV) nonstructural genes resulted in T cell–mediated protection against heterologous HCV challenge [10]. These preclinical studies support the concept that gene-based vaccination can induce effective immunity against viral infections in primates.
The development of an HIV vaccine that is effective against multiple circulating viral clades remains a scientific priority and urgent public health need [11]. The rAd5 vaccine evaluated in the present clinical trial was designed to express an HIV-1 clade B Gag-Pol fusion protein and Env glycoproteins from HIV-1 clades A, B, and C. Here, we report the findings from the first phase 1 clinical trial of this multigene, multiclade rAd5 HIV-1 candidate vaccine.
SUBJECTS, MATERIALS, AND METHODS
Study design
Vaccine Research Center (VRC) 006 (National Institutes of Health [NIH] 04-I-0172) was a randomized, double-blinded, placebo-controlled phase 1 trial conducted at the NIH Clinical Center (Bethesda, MD) by the VRC (National Institute of Allergy and Infectious Diseases [NIAID], NIH, Department of Health and Human Services). Enrollment began 19 July 2004, and unblinding occurred on 27 May 2005. Eligibility criteria required that volunteers be HIV uninfected, 18−44 years old, amenable to risk-reduction counseling, and in good general health as determined by medical history, physical examination, and laboratory tests. Thirty-six volunteers were enrolled into 3 dose groups of 12 study subjects and were randomized to receive vaccine or placebo at a 5:1 ratio. Vaccine doses of 109, 1010, and 1011 particle units (PUs; n = 10 subjects/group) and injection of the final formulation buffer as placebo (n = 6 subjects) were evaluated. A 1-mL intramuscular (deltoid) injection was administered on the day of enrollment. The NIAID Data Safety and Monitoring Board completed a safety review before each dose escalation. Safety evaluations included physical examination and monitoring of laboratory parameters. Local (pain, swelling, or redness) and systemic (fever, malaise, myalgia, headache, chills, or nausea) reactogenicity symptoms after vaccination were recorded on a 5-day diary card. Adverse events were graded for severity by use of a preapproved table that incorporated a 5-point scale and were coded by use of Medical Dictionary for Regulatory Activities terminology.
To address a theoretical concern about the interaction of rAd5 with a naturally acquired adenovirus, subjects experiencing upper respiratory tract infection, urinary tract infection, gastroenteritis, or conjunctivitis within 4 weeks of the study injection had a specimen collected for adenoviral cultures. Specimens were cultured for 5 days via shell vial on a human lung epithelial cell line monolayer (Diagnostic Hybrid) and were screened for known adenovirus serotypes by an indirect fluorescent antibody staining method (VRK Bartels Viral Respiratory Screening and Identification Kit, Bartels).
Vaccine
The VRC-HIVADV014−00-VP vaccine is a 3:1:1:1 ratio of recombinant adenovirus vectors that encode for HIV-1 subtype B Gag-Pol fusion protein and Env glycoproteins from clades A, B, and C, respectively. The transgenes were developed at the VRC, and the design is shown in detail in figure 1. Protein expression was optimized by using preferential amino acid sequences found in human cells.
Figure 1.
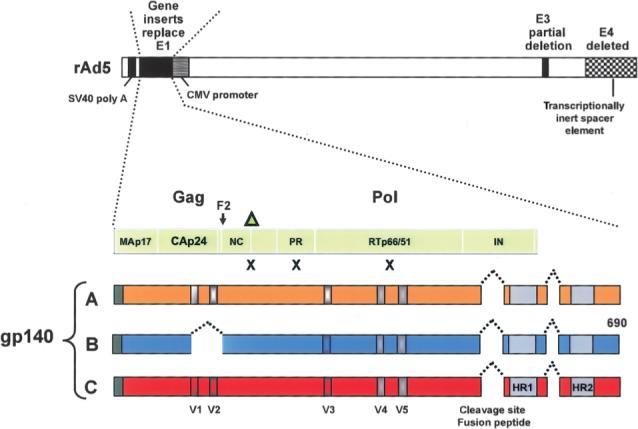
Schematic of the design of the replication-defective recombinant adenovirus serotype 5 (rAd5) vector vaccine. Four separate rAd5 vectors were produced using the same genetic backbone and manufacturing approaches. The HIV-1 vaccine antigen–expression cassettes in the E1 region contained the immediate-early cytomegalovirus (CMV) enhancer/promoter (GenBank accession no. X17403; nucleotide positions 174314−173566), positioned right to left with respect to the viral E1 region. This was followed by an artificial untranslated region of 144 bp and 3′ splice-site sequences, the open reading frame (ORF) of the gene to be expressed, and the simian virus 40 polyadenylation signal (SV40 poly A). The E4 region and a portion of the E3 region were also deleted as shown. A fusion Gag/Pol polyprotein was encoded by a synthetic ORF with nucleotide sequences based on the gag gene from clade B strain HXB2 (GenBank accession no. K03455) and the pol gene (pol/h) from clade B strain NL4−3 (GenBank accession no. M19921). Mutations (indicated by Xs), including the deletion of the carboxy-terminus of Gag (indicated by the triangle), were introduced in the protease and reverse-transcriptase genes to prevent processing of the pol gene products, reducing the potential for functional enzymatic activity [12]. This resulted in a fusion protein that directly reads through the frame shift in Gag (F2). To create synthetic gp140 versions of the Env genes truncated at the transmembrane domain of gp41, sequences from clade A strain 92rw020 (CCR5 tropic; GenBank accession no. U08794), clade B strain HXB2 (X4 tropic; GenBank accession no. K03455) with V1 and V2 deleted and V3 replaced with BaL sequence (GenBank accession no. M68893), and clade C strain 97ZA012 (CCR5 tropic; GenBank accession no. AF286227) were used [13]. In each construct, the cleavage site and fusion peptide at the junction of gp120 and gp41 was deleted, and a portion of the interspace between the 2 heptad-repeat regions in gp41 was deleted. In addition, deletion of the V1/V2 loops from the EnvB construct was required to improve the stability of the vector during manufacturing. HR1–HR2, heptad-repeat regions in gp41; IN, integrase; NC, nucleocapsid; PR, protease; V1–V5, variable regions in envelope.
The transgenes were inserted into the GV11 (GenVec) adenoviral vector system, which is based on human serotype 5 and contains deletions of the E1 and E4 regions and part of the E3 region, rendering it replication defective. The vectors were as described elsewhere [14], except in the expression cassette contained in the E1 region (figure 1). The vector stocks were serially passaged on complementing mammalian cells (293-ORF6), to generate high-titer stocks of replication-defective adenoviruses [15, 16]. The absence of replication-competent adenovirus was verified by the product-release assays. The adenovirus vectors were purified from the cell substrate by a cesium chloride gradient centrifugation process, dialyzed into final formulation buffer, diluted to the desired concentration, and pooled to form the final vaccine product.
Clinical trial material was manufactured under contract at Molecular Medicine BioServices, under current good manufacturing practice conditions. The Vaccine Clinical Materials Program (operated by Science Applications International Corporation) provided quality-assurance oversight of clinical production and release. In compliance with current US Food and Drug Administration guidance, the vaccine was tested for safety, purity, potency, identity, and quality before release. Placebo consisted of the final formulation buffer, which was custom manufactured by Cambrex.
Flow-cytometric analysis and enzyme-linked immunospot (ELISpot) assays
The frequency of vaccine antigen–specific cells was determined as described elsewhere [17]. Cryopre-served peripheral-blood mononuclear cells (PBMCs) were stimulated by use of peptide pools (15mer overlapping by 11 aa) representing the individual vaccine antigens (6 h with brefeldin A for the intracellular cytokine staining [ICS] assay and overnight for the ELISpot assay). Fixed, permeabilized cells were evaluated by flow cytometry for expression of CD3, CD4, CD8, and interferon (IFN)–γ and/or interleukin (IL)–2 and then analyzed using FlowJo software (Tree Star Software). IFN-γ ELISpot assays were performed using a commercial kit (BD Biosciences); results were read using a CTL ELISpot image analyzer (Cellular Technology) and are expressed as the mean number of spot-forming cells per 106 PBMCs.
Measurement of antibody responses
Standardized research ELISAs were performed to delineate the antibody response to viral antigens encoded in the vaccine. End-point titers of antibodies were determined using 96-well Immulon 2 plates (Dynex Technologies) coated with a preparation of purified recombinant HIV proteins [17]. The end-point titer was calculated as the most dilute serum concentration that gave an optical density reading >0.2 above background. Analysis by immunoprecipitation followed by Western blotting (IP–Western blotting) [13], HIV-1 neutralization [18], and Ad5 neutralization [19] was done. Subjects were screened using a commercial ELISA (Abbott Laboratories HIV-1/HIV-2 rDNA) and Western blotting (Genetic Systems HIV Western blot kit; Bio-Rad Laboratories; performed at the Mayo Laboratory, Rochester, MN).
Statistical methods
T cell data are summarized by positive response rates to individual peptide pools and exact 2-sided 95% confidence intervals (CIs). Positivity criteria for ICS and ELISpot data consisted of a statistical hypothesis test for a difference between stimulated and unstimulated wells and a minimal level of response requirement (i.e., the difference had to be statistically significant and exceed a threshold). For ICS responses, Fisher's exact test (α = .01) was applied to each antigen-specific response versus the negative control response, with a Holm adjustment for multiplicity. The minimum threshold for background-corrected positive-response percentage was 0.0241% for CD4+ T cells and 0.0445% for CD8+ T cells. These thresholds were selected to give a 1% false-positive rate in a VRC ICS validation study that included 34 HIV-1–seronegative individuals and that used 8 HIV peptide pools; only 2 (0.007%) of 272 individuals had responses exceeding the thresholds. For ELISpot responses, a permutation test (α = .05) was applied using the Westfall-Young approach to adjust for multiplicity [20]. The threshold was 50 sfc/106 PBMCs. A variance filter for the antigen-specific responses was also used: samples with a ratio of antigen-well variance to (median + 1) of ≥100 were excluded; no such samples were found. SAS (version 9.1; SAS Institute) and Splus (version 6.0; Insightful) were used for all analyses.
RESULTS
Study population
The mean age of the study participants was 27.4 years (SD, 6.2 years); 56% were men, and 44% were women (table 1). All subjects received the study injection, completed the diary card, and completed 24 weeks of clinical observation. Before vaccination, none of the placebo recipients had a preexisting Ad5 antibody titer ≥1:12, whereas 16 of the 30 vaccine recipients were seropositive for Ad5 antibody: 7 in the 109-PU group, 6 in the 1010-PU group, and 3 in the 1011-PU group.
Table 1.
Subject demographics.
| Category, parameter | Vaccine recipients (n = 30) | Placebo recipients (n = 6) | All subjects (n = 36) |
|---|---|---|---|
| Sex | |||
| Male | 15 (50) | 5 (83.3) | 20 (55.6) |
| Female | 15 (50) | 1 (16.7) | 16 (44.4) |
| Age | |||
| 18−20 years | 0 (0) | 1 (16.7) | 1 (2.8) |
| 21−30 years | 19 (63.3) | 5 (83.3) | 24 (66.7) |
| 31−44 years | 11 (36.7) | 0 (0) | 11 (30.6) |
| Mean ± SD, years | 28.2 ± 6.4 | 23.5 ± 2.4 | 27.4 ± 6.2 |
| Race | |||
| White | 23 (76.7) | 4 (66.7) | 27 (75) |
| Black or African American | 3 (10) | 2 (33.3) | 5 (13.9) |
| Asian | 2 (6.7) | 0 (0) | 2 (5.6) |
| American Indian/Alaskan Native | 0 (0) | 0 (0) | 0 (0) |
| Multiracial | 2 (6.7) | 0 (0) | 2 (5.6) |
| Ethnicity | |||
| Non-Hispanic/Latino | 28 (93.3) | 5 (83.3) | 33 (91.7) |
| Hispanic/Latino | 2 (6.7) | 1 (16.7) | 3 (8.3) |
NOTE. Data are no. (%) of subjects, unless otherwise indicated.
Vaccine safety
Local and systemic signs and symptoms (i.e., reactogenicity) increased in frequency and severity with vaccine dose but were never of more than moderate (grade 2) severity. Local reactogenicity was reported by 2 (20%) of the 10 vaccine recipients in the 109-PU group, 8 (80%) of the 10 vaccine recipients in the 1010-PU group, and 10 (100%) of the 10 vaccine recipients in the 1011-PU group; by comparison, 2 (33%) of the 6 placebo recipients reported having at least 1 local symptom. Pain was the most frequently reported symptom; all local reactogenicity was recorded as being of mild (grade 1) severity, except for 1 report of moderate local pain by a vaccine recipient who received the 1011-PU dose (table 2). Systemic reactogenicity was reported by 2 (20%) of the 10 vaccine recipients in the 109-PU group, 6 (60%) of the 10 vaccine recipients in the 1010-PU group, and 9 (90%) of the 10 vaccine recipients in the 1011-PU group; by comparison, 2 (33%) of the 6 placebo recipients reported having at least 1 systemic symptom. Systemic symptoms did not exceed mild severity at the 109- and 1010-PU doses, and all vaccine recipients remained afebrile. At the 1011-PU dose, 6 (60%) of the 10 vaccine recipients reported having at least 1 systemic symptom of moderate severity (table 3). Of these 6, 4 reported having fever (3 mild and 1 moderate in severity) beginning 18−24 h after injection. These 4 vaccine recipients reported having moderate headache 1 day after vaccination, and 3 of these vaccine recipients reported having at least 1 other moderate systemic symptom (malaise, myalgia, or chills). The fevers resolved within 24 h, and other symptoms decreased in severity within a day, although mild symptoms persisted for up to 4 days in some subjects.
Table 2.
Local reactogenicity.
| Vaccine recipients |
||||
|---|---|---|---|---|
| Placebo recipients | 109 PUs | 1010 PUs | 1011 PUs | |
| Local symptoms, intensity | (n = 6) | (n = 10) | (n = 10) | (n = 10) |
| Pain/tenderness | ||||
| None | 4 (66.7) | 8 (80) | 3 (30) | 0 (0) |
| Mild | 2 (33.3) | 2 (20) | 7 (70) | 9 (90) |
| Moderate | 0 (0) | 0 (0) | 0 (0) | 1 (10) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Swelling | ||||
| None | 6 (100) | 10 (100) | 9 (90) | 10 (100) |
| Mild | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Moderate | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Erythema | ||||
| None | 6 (100) | 10 (100) | 7 (70) | 9 (90) |
| Mild | 0 (0) | 0 (0) | 3 (30) | 1 (10) |
| Moderate | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Any local symptom | ||||
| None | 4 (66.7) | 8 (80) | 2 (20) | 0 (0) |
| Mild | 2 (33.3) | 2 (20) | 8 (80) | 9 (90) |
| Moderate | 0 (0) | 0 (0) | 0 (0) | 1 (10) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
NOTE. Data are no. (%) of subjects. Each subject was counted once for the worst severity recorded during the 5 days after vaccination. PUs, particle units.
Table 3.
Self-assessed maximum systemic reactogenicity summary.
| Vaccine recipients |
Placebo recipients (n = 6) | ||||
|---|---|---|---|---|---|
| Systemic symptoms, intensity | 109 PUs (n = 10) | 1010 PUs (n = 10) | 1011 PUs (n = 10) | All (n = 30) | |
| Malaise | |||||
| None | 10 (100) | 6 (60) | 2 (20) | 16 (53.3) | 6 (100) |
| Mild | 0 (0) | 4 (40) | 4 (40) | 8 (26.7) | 0 (0) |
| Moderate | 0 (0) | 0 (0) | 4 (40) | 4 (13.3) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Myalgia | |||||
| None | 10 (100) | 7 (70) | 4 (40) | 21 (70) | 5 (83.3) |
| Mild | 0 (0) | 3 (30) | 3 (30) | 6 (20) | 1 (16.7) |
| Moderate | 0 (0) | 0 (0) | 3 (30) | 3 (10) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Headache | |||||
| None | 8 (80) | 7 (70) | 4 (40) | 19 (63.3) | 4 (66.7) |
| Moderate | 0 (0) | 0 (0) | 4 (40) | 4 (13.3) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Chills | |||||
| None | 10 (100) | 10 (100) | 6 (60) | 26 (86.7) | 6 (100) |
| Mild | 0 (0) | 0 (0) | 3 (30) | 3 (10) | 0 (0) |
| Moderate | 0 (0) | 0 (0) | 1 (10) | 1 (3.3) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Nausea | |||||
| None | 9 (90) | 8 (80) | 6 (60) | 23 (76.7) | 5 (83.3) |
| Mild | 1 (10) | 2 (20) | 3 (30) | 6 (20) | 1 (16.7) |
| Moderate | 0 (0) | 0 (0) | 1 (10) | 1 (3.3) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Fever | |||||
| None | 10 (100) | 10 (100) | 6 (60) | 26 (86.7) | 6 (100) |
| Mild | 0 (0) | 0 (0) | 3 (30) | 3 (10) | 0 (0) |
| Moderate | 0 (0) | 0 (0) | 1 (10) | 1 (3.30) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Any systemic symptom | |||||
| None | 8 (80) | 4 (40) | 1 (10) | 13 (43.3) | 4 (66.7) |
| Mild | 2 (20) | 6 (60) | 3 (30) | 11 (36.7) | 2 (33.3) |
| Moderate | 0 (0) | 0 (0) | 6 (60) | 6 (20) | 0 (0) |
| Severe | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
NOTE. Data are no. (%) of subjects. Systemic reactogenicity, reported by subjects on diary cards, is listed by dose group. PUs, particle units.
Other adverse events that occurred after vaccination were either mild or moderate in severity except for a seizure that occurred 64 days after vaccination. This event was assessed as being unrelated to vaccination, on the basis of the timing and of a medical history of a seizure (3 years prior). There were 3 grade 2 adverse events assessed as being possibly related to vaccination, as follows: (1) asymptomatic neutropenia occurring 21 days after vaccination in a subject in the 109-PU group who had had documented transient mild neutropenia before vaccination; (2) an episode of diarrhea (1-day duration) occurring 3 days after vaccination in a subject in the 1011-PU group; and (3) steatohepatitis (fatty liver) in a subject in the 1011-PU group that was diagnosed by ultrasound after observation of a persistent grade 1 elevated alanine aminotransferase level noted 25 days after vaccination and lasting for ∼5 months. This last event was assessed by a hepatologist as most likely being related to recent rapid weight gain and alcohol consumption.
Eight specimens were obtained from 6 vaccine recipients after the onset of upper respiratory tract infections (pharynx; n = 7) or a urinary tract infection (urine; n = 1) for adenovirus culture. All cultures were negative for adenovirus. A stool sample could not be obtained from 1 subject with transient diarrhea.
Overall, 18 (60%) of the 30 vaccine recipients had vaccine-induced HIV-1 antibody detected by commercial ELISA at the first postvaccination testing time point (week 12), and all antibody responses persisted through week 24. The frequency of diagnostic ELISA positivity increased with vaccine dose: 3 (30%) of 10 in the 109-PU group, 6 (60%) of 10 in the 1010-PU group, and 9 (90%) of 10 in the 1011-PU group (table 4). Western blots were indeterminate or positive at week 24 in 0 (0%) of the 10 vaccine recipients in the 109-PU group, 4 (40%) of the 10 vaccine recipients in the 1010-PU group, and 8 (80%) of the 10 vaccine recipients in the 1011-PU group.
Table 4.
Commercial assay results for HIV-1 antibody at study week 24.
| ELISA |
Western blot |
|||||
|---|---|---|---|---|---|---|
| Group | Negative | Positive | Negative | Indeterminate | Positive | Uninterpretable |
| Placebo (n = 6) | 6 | 0 | ... | ... | ... | ... |
| 109 PUs (n = 10) | 7 | 3 | 2 | 0 | 0 | 1 |
| 1010 PUs (n = 10) | 4 | 6 | 2 | 4 | 0 | 0 |
| 1011 PUs (n = 10) | 1 | 9 | 1 | 4 | 4 | 0 |
NOTE. The Abbott HIVAB HIV-1/HIV-2 rDNA kit was used for routine commercial HIV ELISA testing. Western blot analysis was performed only if the ELISA result was positive. The Western blot analyses were done at the Mayo Laboratory by use of the Genetic Systems HIV Western Blot kit (BioRad Laboratories). A positive Western blot required a band at p24 in addition to a band for at least 1 of the Env glycoproteins (gp41, gp120, or gp160). If there were bands present that did not meet the positivity criteria, the result was reported as indeterminate. If a blot had a high level of nonspecific background staining, it was reported as uninterpretable. HIV-1 infection did not occur in any study subjects, as confirmed by polymerase chain reaction. PUs, particle units.
Vaccine-specific antibody responses
At week 4, IP-Western blotting (figure 2A and 2B) indicated that 28 (93.3%) of the 30 vaccine recipients had developed antibody to EnvB (figure 2C). By research ELISAs, 15 (50%) of the 30 vaccine recipients had developed an antibody response to 1 or more vaccine antigen by week 4, with the greatest frequency of response being to EnvC. The geometric mean reciprocal titer of antibody to Env in the 1011-PU group at week 4 among responders was 200, 310, and 1010, for EnvA, EnvB, and EnvC, respectively (figure 2D). The ELISA titer for each Env antigen was significantly higher at the 1011-PU dose than at either the 109- or 1010-PU dose (P<.001, Wilcoxon rank sum test). Weak antibody responses to Gag were noted in 1 (10%) of the 10 vaccine recipients in the 1010-PU group and 5 (50%) of the 10 vaccine recipients in the 1011-PU group. There was a greater magnitude of antibody response to EnvC than to EnvA (P = .022, Wilcoxon rank sum test) and a trend toward greater responses to both of these antigens compared with responses to EnvB (P = .051, Wilcoxon rank sum test).
Figure 2.
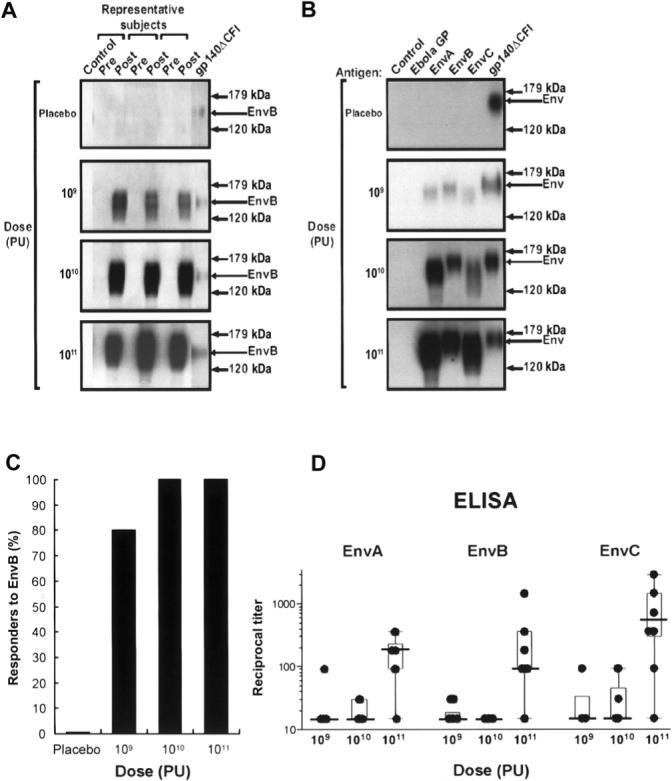
Induction, specificity, and dose response of vaccine-induced antibody to clades A, B, and C Env proteins. A, Western blot analysis of the antigen captured by immunoprecipitation using prevaccination (pre) and week 4 (post) serum samples from 3 representative subjects in the placebo group and in the 3 dose groups receiving 109, 1010, or 1011 particle units (PUs). The analysis shows that Env-specific antibody is induced by the vaccine; arrows indicate the EnvB-specific band. B, Recognition of vaccine-induced antibody. Serum from 1 subject representing each of the dose groups shows that vaccine-induced antibody recognized all 3 Env subtypes but not the Ebola virus glycoprotein–negative control (Ebola GP). Arrows indicate the Envspecific band. No positive bands were detected in any of the placebo recipients at any time point in the study. C, Frequency of positive antibody responders to EnvB at week 4 as measured by immunoprecipitation followed by Western blotting, for each dose group. D, Geometric means of the reciprocal dilution of antibody to purified gp140 for EnvA, EnvB, and EnvC for each subject at week 4, by dose group. Titers were determined by endpoint titration ELISA. Error bars represent SDs. The dilution series began at 1:30, and negative samples were assigned a value of 1:15. The proteins used for ELISA were between 85% and 90% pure as determined by Western blotting and polyacrylamide gel electrophoresis.
No HIV-1–specific neutralizing antibody was detected. Serum samples obtained from vaccine recipients at week 4 and week 24 after vaccination and diluted 1:10 did not neutralize viruses HXB2 or SF162.
Vaccine-induced T cell responses
Antigen-specific CD8+ T cell responses were detected 2 weeks after vaccination in most vaccine recipients and peaked at week 4 (figure 3). T cell responses were observed by both ICS assay (IFN-γ and/or IL-2) and ELISpot assay (IFN-γ). The Env antigens elicited the most frequent response (figure 4A and 4B).
Figure 3.
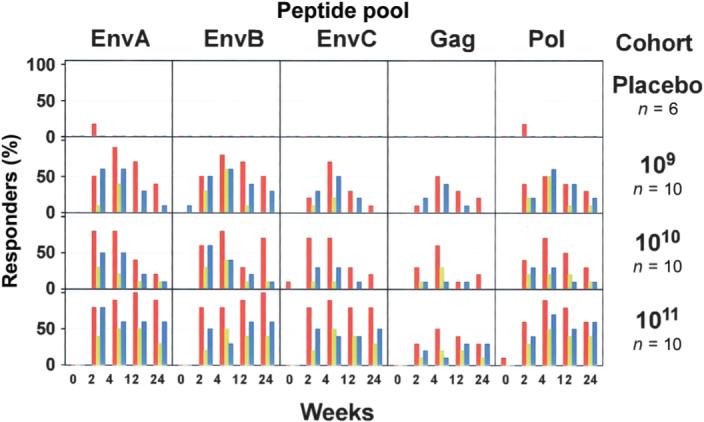
Frequency of subjects with detectable T cell responses. T cell responses to each peptide pool in all dose groups are shown. Each box shows the entire time course for each T cell assay. The Y-axis of each box shows the frequency of positive responders to the respective peptide pool for each assay as percentage of subjects in a dose group (0%−100%). Red bars indicate CD4+ T cell responses as measured by intracellular cytokine staining (ICS) assay, green bars indicate CD8+ T cell responses as measured by ICS assay, and blue bars indicate CD4+ or CD8+ T cell responses as measured by enzyme-linked immunospot assay (ELISpot).
Figure 4.
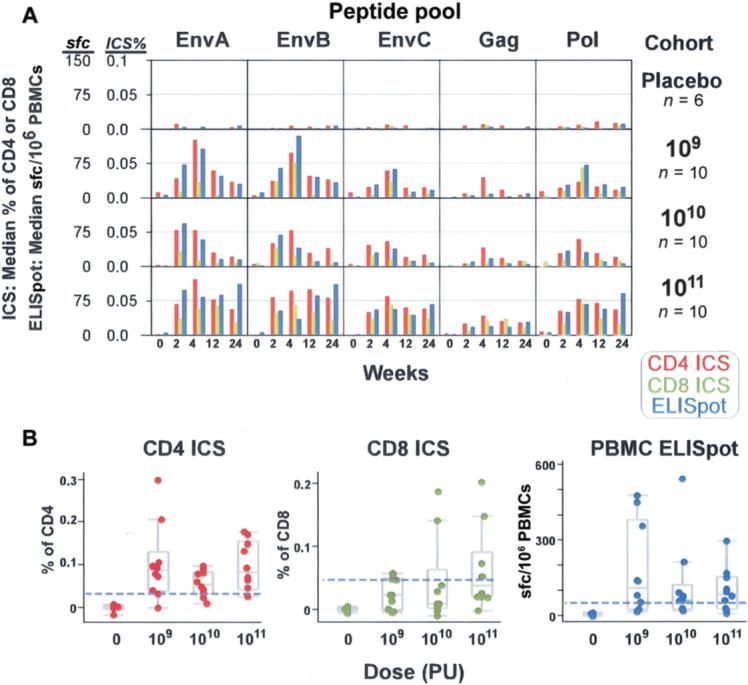
Magnitude of T cell responses to specific vaccine components. T cell responses to each peptide pool for each dose group were measured by intracellular cytokine staining (ICS) assay to detect interferon (IFN)–γ and/or interleukin-2 and by IFN-γ enzyme-linked immunospot (ELISpot) assay for all placebo and vaccine recipients. A, Median magnitudes of peptide pool–specific responses, shown as a percentage of total CD4+ or CD8+ T cells for the ICS assay (scale 0−0.1) and as the no. of spot-forming cells per 106 peripheral-blood mononuclear cells (PBMCs) for the ELISpot assay (scale 0−150), by dose group. Both values are plotted on a linear scale. Each box shows the 24-week time course of the study for each T cell assay. Red bars indicate CD4+ T cell responses as measured by ICS assay, green bars indicate CD8+ T cell responses as measured by ICS assay, and blue bars indicate CD4+ or CD8+ T cell responses as measured by ELISpot assay. B, Magnitudes of EnvA-specific T cell responses at study week 4 for each subject as measured by 3 assays. Shown are the percentages of CD4+ or CD8+ T cells producing cytokine as measured by ICS assay and the no. of spot-forming cells per 106 PBMCs as measured by ELISpot assay, by dose group. The box plots indicate the median, 25th, and 75th percentiles for each dose level, and the error bars show the 5th and 95th percentile. The horizontal dashed line on each plot indicates the threshold of positivity.
Twenty-eight (93.3% [95% CI, 77.9%−99.2%]) of the 30 vaccine recipients had a positive CD4+ T cell response to at least 1 Env peptide pool by week 4. Twenty-seven (90% [95% CI, 73.5%−97.9%]) of the 30 had a CD4+ T cell response to at least 1 Env peptide pool, 15 (50% [95% CI, 31.3%−68.7%]) of the 30 had responses to Gag, and 16 (53.3% [95% CI, 34.3%− 71.7%]) of the 30 had responses to the Pol peptide pools by week 4 (figure 3). The magnitude of CD4+ T cell responses ranged from 0.0013% to 0.15%, and the median response peaked at week 4 (figure 4A). The magnitude of the peak response did not appear to be affected by vaccine dose (figure 4B). However, at week 24, there was a trend toward a dose effect with respect to the persistence of CD4+ T cells responses (table 5).
Table 5.
Frequency of subjects with positive T cell responses to the dose levels of 109 particle units (PUs), 1010 PUs, and 1011 PUs at 4 and 24 weeks after vaccination, as measured by intracellular cytokine staining (ICS) assay in CD4+ and CD8+ T cells and by enzyme-linked immunospot (ELISpot) assay in CD4+ or CD8+ T cells.
| Week 4 |
Week 24 |
|||||
|---|---|---|---|---|---|---|
| Peptide pool | 109 PUs | 1010 PUs | 1011 PUs | 109 PUs | 1010 PUs | 1011 PUs |
| EnvA | 90, 40, 70 | 80, 20, 60 | 80, 50, 70 | 40, 0, 20 | 20, 10, 10 | 80, 30, 60 |
| EnvB | 80, 60, 60 | 70, 40, 40 | 80, 50, 40 | 40, 0, 40 | 70, 10, 10 | 80, 40, 70 |
| EnvC | 60, 20, 60 | 60, 10, 30 | 90, 50, 60 | 10, 0, 10 | 20, 0, 10 | 80, 30, 60 |
| Gag | 50, 0, 40 | 40, 30, 20 | 60, 20, 10 | 20, 0, 0 | 20, 0, 0 | 30, 10, 30 |
| Pol-1a | 30, 40, 50 | 20, 10, 20 | 40, 20, 30 | 0, 0, 20 | 0, 0, 10 | 10, 30, 40 |
| Pol-2a | 30, 10, 40 | 40, 10, 20 | 40, 20, 30 | 10, 10, 10 | 10, 0, 0 | 30, 10, 30 |
| Any | 90, 60, 80 | 90, 50, 70 | 100, 70, 70 | 40, 10, 60 | 80, 10, 10 | 100, 60, 90 |
NOTE. Data are percentages for CD4+ T cells (ICS assay), CD8+ T cells (ICS assay), and CD4+ or CD8+ T cells (ELISpot assay) (n = 10 subjects/group).
Pol protein is too large to include all peptides in 1 pool; therefore, the Pol peptides were divided into 2 pools.
At week 4, 20 (66.7% [95% CI, 47.2%−82.7%]) of the 30 vaccine recipients had a CD8+ T cell response stimulated by at least 1 Env peptide pool, 5 (16.7% [95% CI, 5.6%−34.7%]) of the 30 had responses to Gag, and 12 (40% [95% CI, 22.7%−59.4%]) of the 30 had responses to the Pol peptide pools (figure 3). The magnitude of the CD8+ T cell responses ranged from 0.0019% to 0.69%, and the median response peaked at week 4 (figure 4A). By week 24, there was a trend toward better persistence of CD8+ T cell responses in the highest dose group (table 5).
ELISpot responses (the IFN-γ end point in unfractionated PBMCs) peaked at week 4, with 22 (73.3%) of the 30 vaccine recipients showing a response to 1 or more of the vaccinespecific peptide pools. The frequency of ELISpot responses to specific peptide pools at week 4 were 20 (66.7%) of 30 for EnvA, 14 (46.7%) of 30 for EnvB, 5 (50%) of 30 for EnvC, 10 (33%) of 30 for Pol-1, 9 (30%) of 30 for Pol-2, and 7 (23.3%) of 30 for Gag (figure 3). The geometric (background-corrected) means at week 4 for all dose groups combined, in order of magnitude, were 68 sfc/106 PBMCs for EnvA, 60 sfc/106 PBMCs for EnvB, 49 sfc/106 PBMCs for EnvC, 23 sfc/106 PBMCs for Pol-1, 23 sfc/106 PBMCs for Pol-2, and 13 sfc/106 PBMCs for Gag. The magnitude (figure 4A) and frequency (table 5) of antigen-specific ELISpot responses were consistent with the pattern observed by ICS assay.
The overall magnitude of the T cell responses and the kinetics of the responses for CD4+ and CD8+ T cells show that the peak responses occurred during week 4 after vaccination (figure 5A). The magnitude of the ELISpot response in the 1011-PU group was maintained through the 24 weeks of the study.
Figure 5.
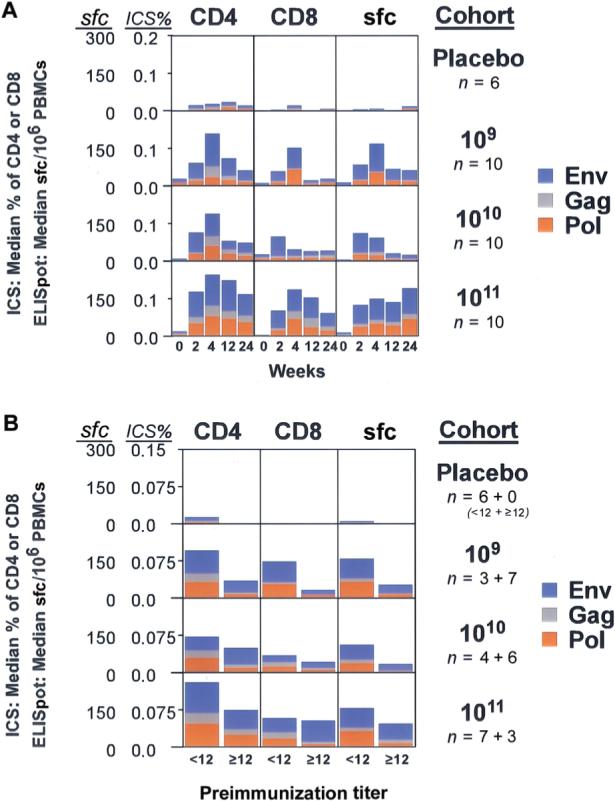
Combined cellular responses to vaccine antigens. The highest Env response to a single subtype was added to the Gag and Pol responses for each subject as a measure of the total vaccine-induced T cell response, and the median for each dose group was plotted on a linear scale for the intracellular cytokine staining (ICS) and the enzyme-linked immunospot (ELISpot) assays. A, Median magnitudes for maximum Env (blue), Gag (grey), and Pol (brown) are shown as a percentage of total CD4+ or CD8+ T cells producing cytokine for the ICS assay and as the no. of spot-forming cells per 106 peripheral-blood mononuclear cells (PBMCs) for the ELISpot assay. Each box represents the 24-week time course; the scale is 0%−0.2% of the total T cell subset for the ICS data and 0−300 sfc/106 PBMCs for the ELISpot data. B, Median magnitudes of total T cell response at week 4 as measured by the ICS assay (percentage of total CD4+ or CD8+ T cells producing cytokine) and the ELISpot assay (no. of spot-forming cells per 106 PBMCs). Subjects are grouped by preimmunization 90% adenovirus serotype 5 (Ad5) neutralization titers (<1:12 or ≥1:12); at screening, the proportion of subjects with an Ad5 antibody titer <1:12 was 6 (100%) of 6 in the placebo group, 3 (30%) of 10 in the group receiving 109 particle units (PUs), 4 (40%) of 10 in the 1010-PU group, and 7 (70%) of 10 in the 1011-PU group. The scale is 0%−0.15% of the total T cell subset for the ICS data and 0−300 sfc/106 PBMCs for the ELISpot data.
Vaccine recipients who were seropositive for Ad5 antibody before vaccination frequently mounted cellular responses to the vaccine antigens (figure 5). However, evaluation of the number of total spot-forming cells (sum of highest Env response plus the Gag and the Pol responses) revealed a significant effect of preexisting Ad5 neutralizing antibody for all rAd5 doses at weeks 4, 12, and 24 (P<.002 for all, linear mixed effects model). Overall, for subjects with a preexisting reciprocal 90% Ad5 neutralization titer<1:12, the total ELISpot response was 3.29 times higher than that in subjects who were seropositive for Ad5 antibody (figure 5B).
DISCUSSION
The induction of significant T cell and antibody responses in nonhuman primates by use of a multiclade rAd5 prototype and the conferment of protection from SHIV and SIV challenge provided the rationale for the present clinical trial. rAd5 vectors have several attractive features, including (1) scalable manufacturing in stable cell lines approved for other human biologics; (2) efficient gene delivery to antigen-presenting cells [21]; (3) potent transient protein expression; (4) induction of innate immune responses; (5) induction of antibody responses; (6) intracellular production of antigen, allowing induction of both CD8+ and CD4+ T cell responses [22]; and (7) induction of immunity after a single injection.
The present study identified a delivery approach and dose of a candidate multiclade HIV-1 rAd5 vaccine that was assessed as safe and immunogenic. Vaccination did not cause severe adverse reactions. The highest dose of rAd5 caused a moderate, short-lived syndrome of headache, myalgia, malaise, and fever that began within 24 h after vaccination. Therefore, it is expected that dose-dependent systemic reactogenicity may follow rAd5 vaccination.
The induction of HIV-specific cellular immunity is a major goal for HIV vaccine development. HIV-specific CD8+ T cell responses clear virus-infected cells and appear during the declining viremia that follows acute infection [23, 24]. High levels of CD8+ T cells present during chronic HIV-1 infection suggest their importance in the control of viremia [25–34]. HIV-specific CD4+ T cell responses also peak early during infection; however, this response diminishes soon after seroconversion [35, 36]. Maintenance of a functional HIV-specific CD4+ T cell response correlates with long-term nonprogression of HIV disease [37]. In addition, nonhuman primate models of lentivirus infection have shown that either CD8+ T cell depletion or mutation of key CD8+ T cell epitopes within the circulating virus results in increased levels of viremia and disease progression [32, 38]. These observations suggest that HIV-1–specific CD4+ and CD8+ T cells are important for controlling HIV replication and preventing disease progression.
The present study demonstrates that a single rAd5 immunization can induce HIV-specific CD4+ and CD8+ T cell responses in the majority of vaccinated subjects (response rates by week 24 were 28/30 [93.3%] and 20/30 [60%], respectively). The peak cellular response occurred 4 weeks after vaccination, and the frequency of detectable responses diminished during the 24-week follow-up period. The peak cellular immune response was not dose related, although there was a greater frequency of responses in the higher dose groups and greater frequency of detectable cellular responses for 24 weeks in the 1011-PU group. The frequency and magnitude of both CD4+ and CD8+ T cell responses to rAd5 immunization as measured by the ICS and ELISpot assays were similar to those induced by 3 doses of plasmid DNA vaccine expressing matching vaccine antigens [17]. Defining the duration of response more precisely will require larger studies, which are currently under way.
Generating a neutralizing antibody response to HIV-1 may be the greatest challenge to the development of a protective HIV vaccine [39]. Although rAd5 vaccination induced modest levels of HIV-specific antibodies as measured by ELISA (∼10-fold higher magnitude than in DNA-primed subjects [17]), it did not induce significant HIV-1 neutralizing antibodies to HXB2 or SF162. Typically, neutralizing activity is not detected in the serum of HIV-1–infected humans or SIV-infected macaques until the ELISA titer is >1:100,000. By ELISA, there was a greater frequency and titer of HIV-specific antibodies in the 1011-PU group (figure 2D) than in the 109- or 1010-PU group, suggesting a threshold effect for antibody induction. The magnitude of antibody responses to EnvC was greater than that to EnvA (P = .022). However, it is not known whether this was related to the immunogenicity of EnvC in the vaccine or to enhanced antigenicity of the EnvC protein used in the ELISA. The rAd5 vaccine induced HIV-1 Env–specific antibodies to multiple clades, but the induction of neutralizing antibodies remains an elusive goal.
The existence of neutralizing antibodies to Ad5 in adults has led to questions regarding the utility of rAd5-based vaccines in humans. Nonhuman primate studies have suggested that preexisting Ad5 immunity diminishes the cellular response to vaccine antigens [40] and that higher doses of rAd5 are required to overcome the immune inhibiting effects of preexisting Ad5 immunity. Selected vaccine recipients in the present study with high levels of Ad5 neutralizing antibody mounted significant HIV-specific humoral and cellular immune responses, indicating that preexisting neutralizing antibodies do not preclude the induction of an immune response to the recombinant antigens expressed by rAd5. However, overall the subjects with preexisting Ad5 immunity had T cell responses that were ∼3-fold lower in magnitude than those of the Ad5-seronegative subjects.
The present clinical study identified 1010 PUs as a well-tolerated rAd5 dose that can stimulate a multiclade HIV-1 immune response in subjects with a range of preexisting Ad5 antibody titers for further evaluation as a booster vaccine to be used in combination with a multiclade HIV-1 DNA vaccine candidate [17]. Although some vaccine recipients at the 1011-PU dose experienced a short-lived and self-limited syndrome of headache, myalgia, malaise, and fever, they also had better induction of HIV-1–specific antibody and a trend toward better maintenance of T cell responses. Therefore, additional studies evaluating dose are in progress concomitantly with the expanded studies evaluating the 1010-PU dose as a booster vaccine. The present clinical trial demonstrates the safety and immunogenicity of a rAd-based multigene, multi-clade HIV-1 vaccine in humans and may represent a step toward the development of a globally relevant vaccine regimen.
VACCINE RESEARCH CENTER (VRC) 006 STUDY TEAM
Brenda Larkin, Margaret McCluskey, Sarah Hubka, LaSonji Holman, Ingelise Gordon, Pamela Edmonds, Steve Rucker, Janie Parrino, and Joseph Casazza of the VRC, National Institute of Allergy and Infectious Diseases (NIAID), and Alan Fix of the Division of AIDS, NIAID.
Acknowledgments
We thank the study volunteers and the many community organizations that support the importance of being involved in the clinical evaluation of HIV candidate vaccines. We also thank the National Institutes of Health Clinical Center staff, the Clinical Center Pharmacy staff (Judith Starling, Hope Decederfelt, and others), the National Institute of Allergy and Infectious Diseases (NIAID) staff, the Patient Recruitment and Public Liaison Office and Office of Communication and Public Liaison staff, the members of the NIAID Data Safety and Monitoring Board, the members of the Capital Area Vaccine Effort, the EMMES Corporation (Phyllis Zaia, Lihan Yan, and others), GenVec (Douglas Brough, Victoria Haque, Alena Lizonova, Perry Newton, and Mike Sowers), and other supporting staff (Tiffany Alley, Erica Eaton, Richard Jones, Monique Young, and Ariella Blejer) who made this work possible. We are grateful as well for the advice and important preclinical contributions of Vaccine Research Center investigators and key staff, including Daniel Douek, Doria (Woody) Dubois, Jennifer Fischer, Yue Huang, Wing-Pui Kong, Peter Kwong, Laurence Lemiale, Norman Letvin, Abraham Mittelman, Laurie Lamoreaux, Adrienne McNeil, Mara Abashian, Steve Perfetto, Srini Rao, John Rathmann, Robert Seder, Judith Stein, Ellen Turk, Jessica Wegman, Richard Wyatt, Ling Xu, and Zhi-yong Yang.
Financial support: National Institute of Allergy and Infectious Diseases intramural research program.
Footnotes
Potential conflicts of interest: G.J.N. and B.K.C. are named on patent applications for this vaccine concept. B.T.B., J.G.D.G., and C.R.K. are employees of GenVec, Inc. All other authors report no conflicts of interest.
References
- 1.UNAIDS/WHO . AIDS epidemic update: December 2005. UNAIDS; Geneva: 2005. [Google Scholar]
- 2.Barouch DH, Nabel GJ. Adenovirus vector-based vaccines for human immunodeficiency virus type 1. Hum Gene Ther. 2005;16:149–56. doi: 10.1089/hum.2005.16.149. [DOI] [PubMed] [Google Scholar]
- 3.Clark KR, Johnson PR. Gene delivery of vaccines for infectious disease. Curr Opin Mol Ther. 2001;3:375–84. [PubMed] [Google Scholar]
- 4.Shiver JW, Emini EA. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Ann Rev Med. 2004;55:355–72. doi: 10.1146/annurev.med.55.091902.104344. [DOI] [PubMed] [Google Scholar]
- 5.Shiver JW, Fu TM, Chen L, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415:331–5. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 6.Casimiro DR, Tang A, Chen L, et al. Vaccine-induced immunity in baboons by using DNA and replication-incompetent adenovirus type 5 vectors expressing a human immunodeficiency virus type 1 gag gene. J Virol. 2003;77:7663–8. doi: 10.1128/JVI.77.13.7663-7668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mascola JR, Sambor A, Beaudry K, et al. Neutralizing antibodies elicited by immunization of monkeys with DNA plasmids and recombinant adenoviral vectors expressing human immunodeficiency virus type 1 proteins. J Virol. 2005;79:771–9. doi: 10.1128/JVI.79.2.771-779.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan NJ, Sanchez A, Rollin PE, Yang ZY, Nabel GJ. Development of a preventive vaccine for Ebola virus infection in primates. Nature. 2000;408:605–9. doi: 10.1038/35046108. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan NJ, Geisbert TW, Geisbert JB, et al. Accelerated vaccination for Ebola virus haemorrhagic fever in nonhuman primates. Nature. 2003;424:681–4. doi: 10.1038/nature01876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Folgori A, Capone S, Ruggeri L, et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat Med. 2006;12:190–7. doi: 10.1038/nm1353. [DOI] [PubMed] [Google Scholar]
- 11.Nabel G, Makgoba W, Esparza J. HIV-1 diversity and vaccine development. Science. 2002;296:2335. doi: 10.1126/science.296.5577.2335. [DOI] [PubMed] [Google Scholar]
- 12.Huang Y, Kong WP, Nabel GJ. Human immunodeficiency virus type 1-specific immunity after genetic immunization is enhanced by modification of Gag and Pol expression. J Virol. 2001;75:4947–51. doi: 10.1128/JVI.75.10.4947-4951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakrabarti BK, Kong WP, Wu BY, et al. Modifications of human immunodeficiency virus envelope glycoprotein enhance immunogenicity for genetic immunization. J Virol. 2002;76:5357–68. doi: 10.1128/JVI.76.11.5357-5368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasmussen H, Rasmussen C, Lempicki M, et al. TNFerade biologic: preclinical toxicology of a novel adenovector with a radiation-inducible promoter, carrying the human tumor necrosis factor alpha gene. Cancer Gene Ther. 2002;9:951–7. doi: 10.1038/sj.cgt.7700518. [DOI] [PubMed] [Google Scholar]
- 15.Brough DE, Lizonova A, Hsu C, Kulesa VA, Kovesdi I. A gene transfer vector-cell line system for complete functional complementation of adenovirus early regions E1 and E4. J Virol. 1996;70:6497–501. doi: 10.1128/jvi.70.9.6497-6501.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butman BT, Lizonova A, Brough DE, et al. Comprehensive characterization of the 293-ORF6 cell line. In: Petricciani J, Sheets R, editors. Vaccine cell substrates 2004: developments in biologicals. Vol. 123. Karger; Rockville, MD: 2006. pp. 225–33. [PubMed] [Google Scholar]
- 17.Graham BS, Koup RA, Roederer M, et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 DNA candidate vaccine. J Infect Dis. 2006;194:1650–60. doi: 10.1086/509259. in this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Gao F, Mascola JR, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79:10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sprangers MC, Lakhai W, Koudstaal W, et al. Quantifying adenovirus-neutralizing antibodies by luciferase transgene detection: addressing preexisting immunity to vaccine and gene therapy vectors. J Clin Microbiol. 2003;41:5046–52. doi: 10.1128/JCM.41.11.5046-5052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moodie Z, Huang Y, Gu L, Hural J, Self SG. Statistical positivity criteria for the analysis of ELISpot assay data in HIV-1 vaccine trials. J Immunol Methods. 2006;315:121–32. doi: 10.1016/j.jim.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 21.Bruna-Romero O, Schmieg J, Del Val M, Buschle M, Tsuji M. The dendritic cell-specific chemokine, dendritic cell-derived CC chemokine 1, enhances protective cell-mediated immunity to murine malaria. J Immunol. 2003;170:3195–203. doi: 10.4049/jimmunol.170.6.3195. [DOI] [PubMed] [Google Scholar]
- 22.Harvey BG, Maroni J, O'Donoghue KA, et al. Safety of local delivery of low- and intermediate-dose adenovirus gene transfer vectors to individuals with a spectrum of morbid conditions. Hum Gene Ther. 2002;13:15–63. doi: 10.1089/10430340152712638. [DOI] [PubMed] [Google Scholar]
- 23.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–10. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koup RA, Safrit JT, Cao Y, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–5. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pantaleo G, Demarest JF, Soudeyns H, et al. Major expansion of CD8+ T cells with a predominant V beta usage during the primary immune response to HIV. Nature. 1994;370:463–7. doi: 10.1038/370463a0. [DOI] [PubMed] [Google Scholar]
- 26.Musey L, Hu Y, Eckert L, Christensen M, Karchmer T, McElrath MJ. HIV-1 induces cytotoxic T lymphocytes in the cervix of infected women. J Exp Med. 1997;185:293–303. doi: 10.1084/jem.185.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musey L, Hughes J, Schacker T, Shea T, Corey L, McElrath MJ. Cytotoxic-T-cell responses, viral load, and disease progression in early human immunodeficiency virus type 1 infection. N Engl J Med. 1997;337:1267–74. doi: 10.1056/NEJM199710303371803. [DOI] [PubMed] [Google Scholar]
- 28.Altman JD, Moss PA, Goulder PJ, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–6. [PubMed] [Google Scholar]
- 29.Yang O, Kalams SA, Rosenzweig M, et al. Efficient lysis of human immunodeficiency virus type 1-infected cells by cytotoxic T lymphocytes. J Virol. 1996;70:5799–806. doi: 10.1128/jvi.70.9.5799-5806.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein MR, van Baalen CA, Holwerda AM, et al. Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infection: a longitudinal analysis of rapid progressors and long-term asymptomatics. J Exp Med. 1995;181:1365–72. doi: 10.1084/jem.181.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moss PA, Rowland-Jones SL, Frodsham PM, et al. Persistent high frequency of human immunodeficiency virus-specific cytotoxic T cells in peripheral blood of infected donors. Proc Natl Acad Sci USA. 1995;92:5773–7. doi: 10.1073/pnas.92.13.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitz JE, Kuroda MJ, Santra S, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–60. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 33.Jin X, Bauer DE, Tuttleton SE, et al. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. 1999;189:991–8. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nixon DF, Townsend AR, Elvin JG, Rizza CR, Gallwey J, McMichael AJ. HIV-1 gag-specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature. 1988;336:484–7. doi: 10.1038/336484a0. [DOI] [PubMed] [Google Scholar]
- 35.Rosenberg ES, Billingsley JM, Caliendo AM, et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia [see comments]. Science. 1997;278:1447–50. doi: 10.1126/science.278.5342.1447. [DOI] [PubMed] [Google Scholar]
- 36.McNeil AC, Shupert WL, Iyasere CA, et al. High-level HIV-1 viremia suppresses viral antigen-specific CD4+ T cell proliferation. Proc Natl Acad Sci USA. 2001;98:13878–83. doi: 10.1073/pnas.251539598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pontesilli O, Carotenuto P, Kerkhof-Garde SR, et al. Lymphoproliferative response to HIV type 1 p24 in long-term survivors of HIV type 1 infection is predictive of persistent AIDS-free infection. AIDS Res Hum Retroviruses. 1999;15:973–81. doi: 10.1089/088922299310485. [DOI] [PubMed] [Google Scholar]
- 38.Barouch DH, Kunstman J, Glowczwskie J, et al. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J Virol. 2003;77:7367–75. doi: 10.1128/JVI.77.13.7367-7375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burton DR, Desrosiers RC, Doms RW, et al. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5:233–6. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- 40.Casimiro DR, Chen L, Fu TM, et al. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J Virol. 2003;77:6305–13. doi: 10.1128/JVI.77.11.6305-6313.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]