

Abstract
Dithiocarbamates have a wide spectrum of applications in industry, agriculture and medicine with new applications being actively investigated. One adverse effect of dithiocarbamates is the neurotoxicity observed in humans and experimental animals. Results from previous studies have suggested that dithiocarbamates elevate copper and promote lipid oxidation within myelin membranes. In the current study, copper levels, lipid oxidation, protein oxidative damage and markers of inflammation were monitored as a function of N,N-diethyldithiocarbmate (DEDC) exposure duration in an established model for DEDC-mediated myelinopathy in the rat. Intraabdominal administration of DEDC was performed using osmotic pumps for periods of 2, 4, and 8 weeks. Metals in brain, liver and tibial nerve were measured using ICP-MS and lipid oxidation assessed through HPLC measurement of malondialdehyde in tibial nerve, and GC/MS measurement of F2 isoprostanes in sciatic nerve. Protein oxidative injury of sciatic nerve proteins was evaluated through quantification of 4-hydroxynonenal protein adducts using immunoassay, and inflammation monitored by quantifying levels of IgGs and activated macrophages using immunoassay and immunhistochemistry methods, respectively. Changes in these parameters were then correlated to the onset of structural lesions, determined by light and electron microscopy, to delineate the temporal relationship of copper accumulation and oxidative stress in peripheral nerve to the onset of myelin lesions. The data provide evidence that DEDC mediates lipid oxidation and elevation of total copper in peripheral nerve well before myelin lesions or activated macrophages are evident. This relationship is consistent with copper-mediated oxidative stress contributing to the myelinopathy.
Keywords: Diethyldithiocarbamate, lipid oxidation, myelinopathy, copper
Introduction
Human exposure to dithiocarbamates results from the numerous uses of these compounds. Indirect or unintentional exposures result from residues on food crops, in the occupational setting through their applications as pesticides, and in various industrial processes. Direct or intentional exposures also occur from therapeutic applications of dithiocarbamates. In addition to the use of disulfiram in alcohol aversion therapy (Eneanya et al., 1981), and N,N-diethyldithiocarbamate in the treatment of nickel carbonyl intoxication (Sunderman, 1979), a wide range of new medical applications for dithiocarbamates are currently being explored. Investigations have presented support for potential uses of dithiocarbamates in treating cocaine addiction (Sofuoglu and Kosten, 2005), inflammation (Fang et al., 2005), viral infections (Krenn et al., 2005; Si et al., 2005), and as adjuncts for chemotherapy (Bach et al., 2000). In vitro mechanistic studies have demonstrated the ability of dithiocarbamates to modulate numerous biological processes including apoptosis, oxidative stress, and transcription, providing the molecular basis for these proposed medical applications (Kang et al., 2001; Kimoto-Kinoshita et al., 2004).
One obstacle to the development of new dithiocarbamate-based therapeutic agents is their potential toxicity. Neurotoxicity has been observed in humans and in experimental animals; and at least two independent neuropathies, an axonopathy and a myelinopathy (Johnson et al., 1998; Tonkin et al., 2000) have been reported. Depending upon the chemical structure of the dithiocarbamate and route of exposure, some dithiocarbamates release sufficient CS2 in vivo to produce identical protein cross-linking and morphological changes to those observed in CS2 neurotoxicity following inhalation exposure (Johnson et al., 1998) supporting CS2 as the proximate toxic species responsible for the dithiocarbamate-mediated axonopathy. However, the molecular processes underlying the myelinopathy are not well defined. Previous studies have reported increased levels of copper and lipid oxidation to be associated with dithiocarbamate induced neurotoxicity (Calviello, 2005; Delmaestro, 1995; Tonkin, 2004) and correlative data have shown a relationship between copper levels in peripheral nerve and the severity of myelin injury produced by pyrrolidine dithiocarbamate and N,N-diethyldithiocarbamate (DEDC) in rodent models (Valentine et al., 2006; Valentine et al., 2007). Additionally, proteomic analysis of sciatic nerve proteins using this animal model has demonstrated significant elevations in expression levels of glutathione transferase isozymes within Schwann cells consistent with activation of the antioxidant response element pathways subsequent to DEDC exposure (Viquez et al., 2007). These observations coupled with the affinity of dithiocarbamates for copper have led to the hypothesis that dithiocarbamates bind copper and generate a lipophilic complex that accumulates and promotes lipid oxidation within myelin membranes leading to a subsequent myelinopathy. But because analyses have typically been performed at time points associated with advanced injury, i.e., myelin lesions identifiable by light microscopy, it is not clear whether the observed markers of oxidative stress result from a direct effect of dithiocarbamates and represent processes contributing to the development of myelin injury or alternatively are a result of the injury and associated inflammatory responses that occur with demyelinating injuries in general.
If copper accumulation and increased oxidative stress contribute to the myelin lesions produced by DEDC, then significant changes in these parameters are expected to precede structural changes. In the current study, copper levels, lipid oxidation, protein oxidative damage and markers of inflammation were monitored as a function of DEDC exposure duration in an established model for DEDC-mediated myelinopathy in the rat. Intra-abdominal administration of DEDC was performed using osmotic pumps for periods of 2, 4, and 8 weeks. At the end of each exposure period, tissues including peripheral nerve, liver and brain were collected. Metals in brain, liver and tibial nerve were measured using inductively coupled plasma-mass spectrometry (ICP-MS); and lipid oxidation was assessed through HPLC measurement of malondialdehyde (MDA) in tibial nerve and GC/MS measurement of F2 isoprostanes in sciatic nerve. Protein oxidative injury of sciatic nerve proteins was evaluated through quantification of 4-hydroxynonenal (HNE) protein adducts using immunoassay, and inflammation monitored by quantifying levels of IgGs and activated macrophages using immunoassay and immunohistochemistry methods, respectively. Changes in these parameters were then correlated to the onset of structural lesions, as ascertained by light and electron microscopy, to delineate the temporal relationship of copper accumulation and oxidative stress in peripheral nerve to the onset of myelin lesions.
Materials and methods
Chemicals
2ML4 Alzet® osmotic pumps were obtained from Braintree Scientific (Braintree, MA). Sodium N,N-diethyldithiocarbamate (DEDC) was obtained from Alfa Aesar (Ward Hill, MA). Glutaraldehyde and paraformaldehyde were obtained from Electron Microscopy Sciences (Ft. Washington, PA). Fetal bovine serum was obtained from Mediatech Inc. (Herdon, VA). Proteinase K and bovine serum albumin (BSA) were obtained from Sigma-Aldrich (St. Louis, MO). Dulbecco’s phosphate buffered saline (1X PBS) was purchased from MP Biomedicals (Solon, OH). All solvents used were HPLC grade and purchased from Fisher Scientific (Pittsburgh, PA).
Animals and exposures
All treatments and procedures using animals were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Vanderbilt University. Forty-four adult male Sprague-Dawley rats were obtained from Harlan Bioproducts (Indianapolis, IN) and caged at Vanderbilt University animal facilities in a temperature controlled room (21–22 °C) with a 12 h light–dark cycle, supplied with Purina Lab Diet 5001 and water ad libitum. After a 10-14 day acclimatization period, animals were exposed to DEDC at 0.3 mmol/kg/day for 2, 4 or 8 weeks using intraabdominal 2mL 4 week Alzet osmotic pumps surgically implanted under anesthesia (100 mg/Kg ketamine with 8 mg/Kg xylazine ip). DEDC was delivered as an aqueous solution, the concentration of which was determined from the UV absorbance at 282 nm (ε=13,000 M-1cm-1). For the DEDC 8-week exposure groups, the osmotic pumps were replaced after 4 weeks to extend the exposure period to 8 weeks. Two exposure models were used. The first consisted of one control group and 2, 4, and 8 week exposure groups with n=4 for each group except for the 8-week DEDC exposure group, where n=5. The average starting weight of the 17 animals in the first exposure model was 362.8 ± 9.7 g (SEM). Samples collected from the experimental animals in the first exposure model were used to analyze globin, F2-isoprostanes, metals and peripheral nerve lesions. For the second exposure model 27 rats were assigned to either a control or exposure group of 2, 4, or 8 weeks, n=4 per group, except for the 8-week time point where n=5 (control), and n=6 (DEDC-exposed). The average starting weight of the animals for the second exposure model was 319.4 ± 1.7 g (SEM). Samples collected from the animals in the second exposure model were used to analyze globins, HNE protein adducts, MDA, immunoglobulins (IgG and IgG2a), and macrophages. Sham pump implantation surgey was not performed on control animals.
Collection and analysis of tissue
At the end of each exposure period, control and exposed animals were deeply anesthetized with pentobarbital (100 mg/Kg body weight, ip), and exsanguinated by cardiac puncture. Whole blood (1 mL) was placed into heparinized vials for globin isolation. The animals were then perfused through the left ventricle of the heart with 1X PBS (pH 7.4). For rats in the first exposure model, the left sciatic nerve was removed and immediately frozen in liquid nitrogen and stored at -80 °C for isoprostane analysis. The right sciatic nerve was immersed in 4% glutaraldehyde in 0.1 M PBS and used for histopathology. The liver, brain, and both posterior tibial nerves were immersed in 4% glutaraldehyde in 0.1M PBS (pH 7.4) and stored at 4°C for metal analysis. Metal analysis was performed by ICP–MS at the Diagnostic Center for Population and Animal Health at Michigan State University (East Lansing, MI). Copper, zinc, arsenic, selenium, manganese, molybdenum, iron, cadmium, and mercury were determined. For the rats in the second exposure model, the left sciatic nerve, left half of the brain cut sagitally, the left posterior tibial nerve and one half of the right posterior tibial nerve were removed and immediately frozen in liquid nitrogen and stored at -80 °C until further analysis. A liver section, the right half of the brain and the other half of the right posterior tibial nerve were immersed in 4% glutaraldehyde in 0.1M PBS (pH 7.4) and stored at 4°C. The right sciatic nerve was immersed in 4% paraformaldehyde and stored at 4°C.
Protein Extraction and Quantification
Total proteins from approximately 10 mg of frozen sciatic nerve of control and DEDC-exposed rats were extracted in 0.1 mL chilled MES-SDS-Urea/Thiourea buffer (0.25M 2-morpholino-ethanesulfonic acid (MES), 1% sodium dodecyl sulfate (SDS), 7 M urea, 40 mM dithiothreitol (DTT), 10 mM EDTA and 2 M thiourea, pH 6.0), containing protease inhibitors (P-2714 Sigma-Aldrich, and 80-6501-23 protease inhibitor mix, Amersham Biosciences) and phosphatase inhibitors (P-2850 and P-5726 Sigma-Aldrich), using a sample grinding kit (80-6483-37, Amersham Biosciences, San Francisco, CA). The homogenate was centrifuged at 13,000 rpm for 10 min at 4 °C and the supernatant collected. The extraction step was repeated twice using the remaining pellet and the supernatant collected and combined with the previous extract and stored at -80 °C. Protein concentration in the supernatant of the control and exposed nerve samples was measured by a modified Bradford method (Ramagli, 1999) using BSA as the standard.
RP/HPLC analysis of globin
Globin isolation and analysis to quantify adduct formation was performed as previously described (Erve, 2000; Tonkin, 2004). Blood was separated into plasma and red blood cells, the cells lysed to produce hemolysate, and 100 μL of 1 N ascorbic acid added to each 1 mL hemolysate. The resulting solution was added dropwise to 10 mL of 2.5% oxalic acid in acetone. Globin was allowed to precipitate on ice for 30 min, then centrifuged at 10,000g for 10 min at 4 °C, and washed in 5 mL of ice-cold acetone. The pellet, containing crude globin, was dried under a stream of N2 and stored at -80 °C. Dried globin was solubilized with 0.1% trifluoroacetic acid (TFA) to produce a solution for HPLC analysis. Globin chains were separated by RP-HPLC on a Phenomenex Jupiter 5 m column (150 × 460 mm) using a Waters 2690 liquid chromatograph after adjusting sample concentration to a UV absorption of 1.0 ± 0.2 at 280 nm. Globins were separated using a linear gradient from 56% solvent A and 44% solvent B to 30% solvent A and 70% solvent B over 30 min followed by a linear gradient to 100% solvent B over 10 min. Solvent A was 20:80:0.1 acetonitrile/water/TFA, and solvent B was 60:40:0.08 acetonitrile/water/TFA. The elution of globin peaks was monitored by their UV absorption at 220 nm using a Waters 996 photodiode array detector.
Preparation of tissue for morphology and immunohistochemistry analysis
Dissected sciatic nerves from control and DEDC-exposed animals were immersed in 4% glutaraldehyde in 0.1M PBS buffer or 4% paraformaldehyde in PBS buffer overnight and then transferred to 0.1 M PBS. For morphology, sciatic nerve sections were post-fixed with osmium tetroxide and embedded in Epon. Thick sections (1 μm) were cut and stained with toluidine blue. The thick sections of peripheral nerve were evaluated by light microscopy on an Olympus BX41 microscope equipped with an Optronics Microfire digital camera. One cross section of sciatic nerve was examined per animal and the total number of lesions counted by two observers (WMV and OMV). The lesions quantified were: degenerated axons, axons with thin myelin (g ratio greater than 0.7 (axon/axon with myelin diameter)), intramyelinic edema, and demyelinated axons. Thin (70 nm) sections were prepared from sciatic nerves and evaluated using a Phillips CM-12 electron microscope, 120 keV with a high resolution CCD camera system.
For immunohistochemistry, sciatic nerves fixed with paraformaldehyde from control and DEDC-exposed rats were embedded in paraffin wax; and cross sections of 5 μm were cut and probed for activated macrophages. After deparaffinization in xylene and hydration in graded ethanols from 100% to 70%, antigen retrieval was performed using proteinase K (20 μg/mL) for 10 min at room temperature. Endogenous peroxide was then blocked by incubation with 3% peroxide for 10 min at room temperature. Nonspecific binding sites were quenched by incubation with 10% fetal calf serum (FCS) for 30 min at room temperature. Next, sections were incubated with mouse anti-rat CD68 (MCA-341R, Serotec, Raleigh, NC) at a dilution of 1:200 overnight in a humid chamber at 4 °C. The CD-68 primary antibody is directed to a single chain glycoprotein (110 kDa) that is expressed in macrophages during phagocytic activity. Subsequently, sections were incubated with an HRP goat anti-mouse IgG (A-3682, Sigma, St. Louis, MO) at a dilution of 1:300 for 30 min at room temperature and for 1h at 4 °C. After each step, the sections were washed with 1X PBS (pH 7.4). The peroxidase reaction was developed using 3,3’-diaminobenzidine (DAB) (Dako Corp, Cupertino, CA). Slides were lightly counterstained with 10 % hematoxylin (Sigma-Aldrich), dehydrated in graded ethanols from 70% to 100% and xylene and mounted with Cytoseal ™60 (Richard-Allan Scientific (Kalamazoo, MI). To confirm the specificity of the secondary antibody, two sections per animal were processed identically except the incubation with the primary antibody was omitted. Slides were scored blindly; and four cross-sections of the sciatic nerve were examined by light microscopy for each animal and activated macrophages totaled per nerve (n=4 for each group except for 8-week control (n=5) and DEDC-exposed (n=6)).
Analysis of F2-isoprostanes
Total lipids were extracted from both sciatic nerves from each animal using the modified method of Folch; and isoprostanes were quantified by gas chromatography (GC) followed by detection with negative ion chemical ionization (NICI) mass spectrometry (MS) using selective ion monitoring (SIM) as previously described (Reich et al., 2001; Milatovic et al., 2005). Briefly, frozen nerves were homogenized in CHCl3: MeOH (2:1, v/v) containing 0.05% butylated hydroxytoluene (BHT) as an antioxidant. Then lipid extracts were mixed with 2.0 mL NaCl (0.9%, w/v) and layers separated by centrifugation at 3,000 rpm for 10 minutes at room temperature. Five hundred to one thousand pg of [2 H4-3,3,4,4,]-8-iso PGF2α (iso- PGF2α-III, Cayman Chemicals, Ann Arbor, MI) was added as internal standard. After lipid saponification, isoprostanes were extracted using C-18 and silica Sep-Pak cartridges, and purified by thin layer chromatography. F2-isoprostanes were assayed with GC–NICI-MS and SIM and reported as ng/g tissue (wet weight).
Malondialdehyde (MDA) analysis
Sections of posterior tibial nerves (6-10 mg) from controls and DEDC-exposed rats were weighed in Eppendorf tubes and crushed using a plastic pestle with a tissue grinder in 200 μl of 0.1 M phosphate buffer, pH 7.4. Next, 200 μl of 1,3-diethyl-2-thiobarbituric acid (DETBA), prepared as described (Calviello et al., 2005) was added. The mixture was then heated at 90°C for 30 min, and the MDA derivative extracted with 200 μl of isobutyl alcohol. After centrifugation at 10,000g for 10 min, 60 μl of supernatant was taken for HPLC analysis using a Shimadzu HPLC with a SIL-10AD autosampler with LC10AD pump connected to a SPD10A detector and an SCL-10A System Controller. The LichroCART 4-4 guard column was used before the analytical column (LichroCART 25-4 HPLC Cartridge column filled with Lichrospher® 100 RP-18 (5μm) particles. The MDA derivative was separated using a gradient from 100% solvent A to 90% solvent B with a flow rate of 1 mL/min. Solvent A was 10% acetonitrile in water with 0.1% triethanolamine, and Solvent B was 100% acetonitrile. Under these conditions, the MDA derivative eluted at 8.3 min. A standard curve was obtained using MDA standards prepared by the hydrolysis of 1,1,3,3-tetramethoxy-propane. MDA was expressed as pmol/mg wet weight of tissue. Two separate analyses were performed per nerve(the number of nerves analyzed were n= 26 for controls, n=8 for 2 and 4-week groups and n=12 for the 8-week group), and both were assayed in duplicate by HPLC, giving 4 values per nerve.
Slot blot immunoassay
The content of HNE protein adducts, IgG and IgG2a in rat sciatic nerve were determined using an immunoassay method. Total sciatic nerve protein, a BSA standard curve and HNE-BSA proteins were applied to each polyvinylidene difluoride (PVDF) membrane analyzed using a slot blot apparatus (Bio-Dot SF, BioRad, Hercules, CA). To calculate the amount of protein present in each spot, densitometric analysis was performed on stained membranes using the MemCode Reversible Protein Stain Kit for PVDF (Pierce, Rockford, IL) and compared to the BSA standard curve on the same PVDF membrane. Nonspecific binding sites were blocked in blocking buffer (5% nonfat powdered skim milk in Tris buffered saline (TBS), pH 7.4) at room temperature for 1 h. Membranes were then incubated with primary antibodies against IgG (A-5795, anti-rat IgG peroxidase conjugate, Sigma, St. Louis, MO, dilution 1:5,000), IgG-2a (R-0761, monoclonal anti-rat IgG2a clone R2A-2, Sigma, St. Louis, MO, dilution 1:1,000) or HNE adducts (rabbit anti-HNE, HNE1-S, Alpha Diagnostic, San Antonio, TX, dilution 1:2,500) overnight at 4°C. After washing with TBS/T (1X TBS containing 0.05% tween-20), the membranes were incubated at room temperature for 1 h with an anti-rabbit peroxidase conjugated secondary antibody (A-8275, Sigma, St. Louis, MO, dilution 1:10,000) for 1 h at room temperature. Following further washes in TBS/T, the membranes were incubated with Western Lightning Chemiluminescence Reagent Plus substrate (Perkin-Elmer LAS, Inc., Boston, MA) according to the manufacturer’s directions, and exposed to Kodak X-Omat Blue XB-1 film. For quantification, films were scanned with a GS-700 densitometer (Bio-Rad, CA) and analyzed using Quantity One 1-D Analysis Program version 4.1 (Bio-Rad, CA). Total IgG, IgG2a and HNE adduct content in rat sciatic nerve proteins were determined by densitometry and expressed as trace optical density (OD) × mm2 per microgram of total sciatic nerve protein. Two membranes per analysis were evaluated containing triplicate slots for each control and DEDC-exposed rat on the same membrane.
Statistical Analysis
One-way analysis of variance (ANOVA), Tukey–Kramer’s Multiple Comparisons Test, Dunnett Multiple Comparisons Test, Pearson Correlation Test, the unpaired one-tailed t-test, and One-sided Fisher’s Exact Test were performed using Prism 4 (Graphpad Software, Inc.). Lesion counts were square root transformed to normalize their distribution and equalize variances prior to statistical comparisons by ANOVA and Tukey-Kramer’s Multiple Comparisons Test. Statistical significance was taken to be p< 0.05 unless otherwise noted.
Results
Globin Analysis and Osmotic Pump Delivery
The level of DEDC exposure was monitored by analysis of globin preparations using RP–HPLC. Globin preparations from all animals exposed to DEDC contained an additional HPLC peak that was not observed in globin preparations from control animals. Earlier work demonstrated that this additional peak is produced by carbamylation of Cys-125 of a ß-globin chain by a metabolite of DEDC to produce S-(diethylaminocarbonyl)-cysteine adducts on Cys-125 and is correlated to dose level (Erve et al., 2000; Tokin et al., 2000). At the end of 2, 4 and 8 weeks, the mean levels ± SEM of modified ß-globin expressed as a percent of total peak area for the DEDC-exposed groups for the first exposure model was 0% (control), 12.6 ± 1.0% (2-week), 19.2 ± 0.8% (4-week), and 16.3 ± 0.6% (8-week), and for the second exposure model was 0% (all controls), 11.8 ± 0.6% (2-week), 16.9 ± 1.4% (4-week), and 16.6 ± 0.3% (8-week), respectively.
Visual inspection of the osmotic pumps for blockage and measurement of the remaining volume in the pumps after their removal at the end of the exposure periods to calculate the volume delivered was performed. The coefficient of variation for the volumes delivered by the 2-week pumps (n=8) and all the 4-week pumps used for the 4-week and 8-week exposure groups (n=30) were 5% and 4%, respectively.
Analysis of Tissue Metal Levels
Tissue copper levels are shown in Fig.1. Copper levels in peripheral nerve and brain in all DEDC-exposed groups were significantly elevated over the controls and increased as a function of exposure time. The liver copper levels were significantly elevated at the 2, 4 and 8-week exposure times relative to the controls; however, no time-dependent increases in liver copper levels were seen, with values remaining relatively constant subsequent to the 2-week exposure. No significant differences were seen in levels of zinc, iron, cadmium, manganese, selenium, arsenic, molybdenum or mercury (data not shown) in the brain, liver and peripheral nerve as compared to the controls.
Figure 1.

Copper levels in posterior tibial nerve, liver and brain for controls (0-week) and each DEDC exposure duration (n=4, except for DEDC 8-week n=5) were determined by ICP-MS. Values are means ± SEM., and reported as ppm dry weight of nerve. * p < 0.05, **p < 0.01 and ***p < 0.001, One-way ANOVA and Tukey-Kramer Multiple Comparisons Test as compared to zero time point.
Lipid oxidation assays
MDA levels in the posterior tibial nerves are shown in Fig. 2A. MDA levels were significantly increased in the 2-week DEDC exposed group as compared to the controls. No significant differences in MDA were seen for the 4-week or 8-week tibial nerve samples.
Figure 2.

Lipid oxidation products measured in control and DEDC-exposed rat nerves. (A) MDA levels were determined in posterior tibial nerves by HPLC MDA values are means ± SEM for each time point (the number of nerves analyzed were n=26 for the control zero time point, n=8 for the 2 and 4-week time points and n= 12 for the 8-week time point) two separate analyses were performed per nerve and both were assayed in duplicate by HPLC, giving 4 values per nerve.. (B) F2-Isoprostane levels in sciatic nerve were determined by the modified method of Folch and quantified by GC/MS. Isoprostane values are means ± SEM for each time point (n=4, except for DEDC 8-week n=5). *p < 0.05 and+p < 0.01, One-way ANOVA and Dunnett Multiple Comparisons Test as compared to zero time point.
F2-isoprostane levels in sciatic nerve were significantly elevated in the 2-week DEDC-exposed group over controls (Fig. 2B). Although there were no significant changes at the 4- and 8-week time points, F2-isoprostane levels in all these DEDC-exposure groups showed greater mean values over controls.
HNE Protein Adducts
HNE protein adducts measured in sciatic nerve proteins extracted from control and DEDC-exposed rats are shown in Fig. 3. The data indicated a significant increase of HNE adducts in the 8-week DEDC sciatic nerves relative to 8-week controls. The HNE adducts were positively correlated to the duration of exposure (p < 0.05 Pearson correlation test r2 = 0.88) and continued to increase over the entire 8-week period examined.
Figure 3.

Relative amounts of HNE protein adducts in sciatic nerve proteins from DEDC-exposed groups and their corresponding controls. Total sciatic nerve proteins (0.2 μg) from each control and DEDC treatment group (n=4, except for 8-week time point, control (n=5) and DEDC-exposed (n=6)) were applied in triplicate to a PVDF membrane using a slot blot apparatus and evaluated for HNE adducts by immunoassay (see methods section) using a rabbit anti-HNE antibody. Data are expressed as means ± SEM of four membranes for each animal. Values were normalized so that optical density of controls was 1.0. * p < 0.05 by unpaired one-tailed t test as compared to the corresponding control group.
Analysis of IgG and IgG2a
Total tissue IgG and IgG2a levels for each time point are presented in Fig. 4. IgG2a levels in peripheral nerve for the 4 and 8-week DEDC exposed groups were significantly elevated over their controls; and total IgG was significantly elevated at the 8-week time point.
Figure 4.

IgG and IgG2a levels in sciatic nerve proteins of control and DEDC-exposed groups. Total sciatic nerve protein (0.2 μg) from each control and DEDC-exposed rat were evaluated for IgG and IgG2a levels using a slot blot apparatus and immunoassay (see methods for details). (n=4, except for 8-week time point, control (n=5) and DEDC-exposed (n=6)) Values are means ± SEM. of four membranes with individual animals run in duplicate on each membrane. Values were normalized so that optical density of controls was 1.0. *p < 0.05, unpaired two-tailed t test as compared to the corresponding exposure duration controls.
Analysis of Macrophages
Four sciatic nerve cross-sections from each exposed and control animal were analyzed to quantify CD68-positive macrophages, and representative cross-sections are shown in Fig. 5. No activated macrophages were observed in the controls or the 2 and 4-week DEDC exposure groups. The incidence of nerves with activated macrophages (4 out of 6 animals) was significantly increased in the 8-week DEDC-exposed group relative to controls (p < 0.05 by One-sided Fischer’s Exact Test). The number of activated macrophages observed per nerve ranged from 0 to 210 with a mean square root (SEM) of 4.3 (2.3) for the 8-week DEDC exposed group.
Figure 5.
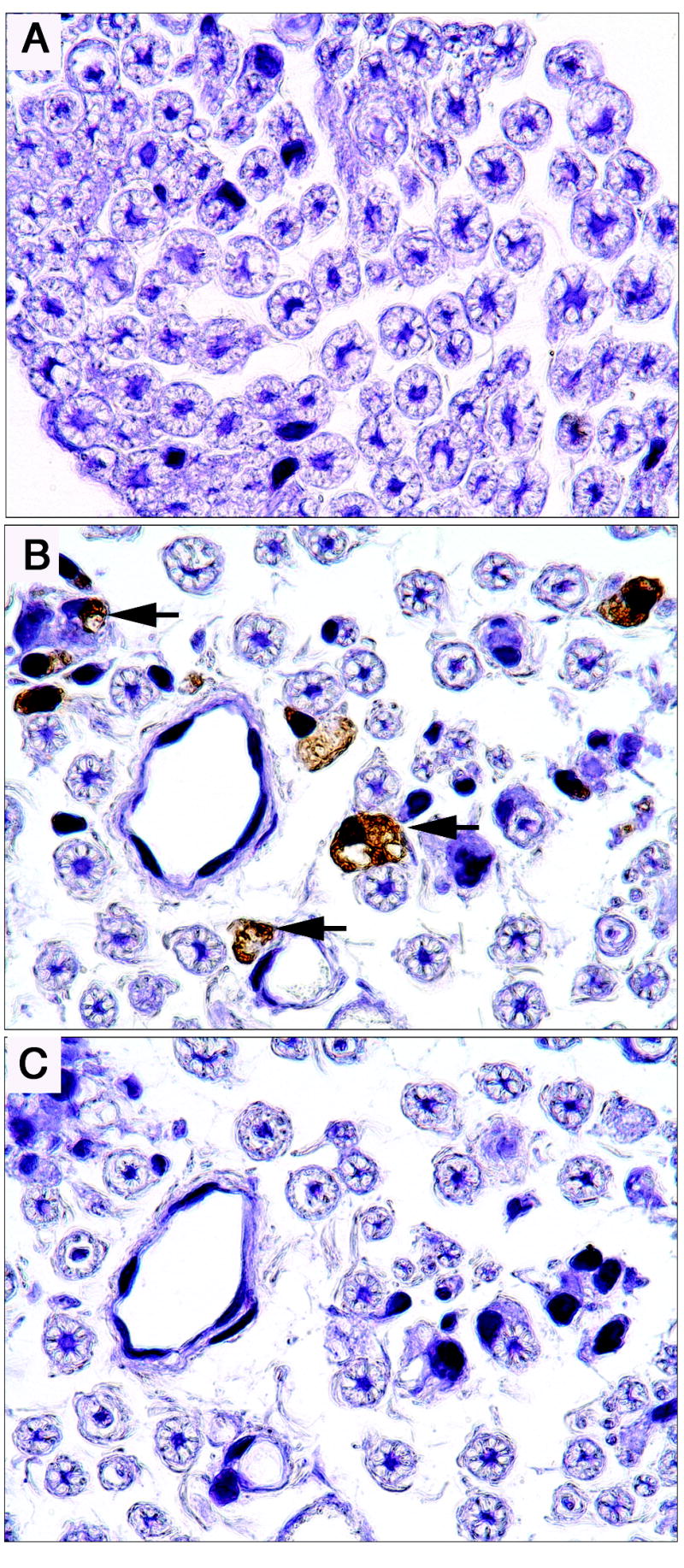
Immunolocalization of activated macrophages in representative cross-sections of sciatic nerves from an 8-week control rat (A) and an 8-week DEDC exposed rat (B) probed with mouse-anti-rat CD68 and counterstained with hematoxylin. Some activated macrophages are identified with arrows. (C) A serial cross-section to that shown in B from the 8-week DEDC exposed rat that was processed identically to the sections in A and B, but without mouse-anti-rat CD68 primary antibody.
Peripheral Nerve Morphology
Representative sections of sciatic nerve obtained from controls and animals exposed to DEDC are shown at the light microscope and electron microscope level in Fig. 6 and Fig. 7 respectively. No significant difference in the occurrence or severity of axonal degeneration was seen between the controls and DEDC-exposed groups. However, axons exhibiting thin myelin, demyelination and intramyelinic edema were only observed in the 8-week DEDC-exposed group with the incidence of thin myelin and intramyelinic edema significantly elevated at this time point (Table 1).
Figure 6.

Morphology of sciatic nerve cross-sections stained with toluidine blue. (A) Sciatic nerve from a control rat showing axons surrounded by normal compact myelin. (B) Cross section of a nerve obtained from an 8-week DEDC-exposed rat showing examples of thinly myelinated (arrows) and demyelinated axons (concave arrow head) and intramyelinic edema (arrow heads).
Figure 7.

Electron micrographs of sciatic nerve cross-sections from control and DEDC-treated rats (black bars = 2μm). (A) Myelinated axon (a) in a control animal surrounded by normal compact myelin. (B-D) Sections from a rat exposed to DEDC for 8-weeks showing lesions that were quantified in Table 1. (B) A myelinated axon (a) surrounded by abnormally thin compact myelin. (C) Myelinated axon (a) with fluid separating the inner lamellae of myelin (*) (intramyelinic edema). (D) A demyelinated axon (a) from an 8-week DEDC exposed rat is surrounded by a Schwann cell containing a non-membrane vacuole. Two nucleated cells possessing basal lamina consistent with Schwann cells are also present (s).
TABLE 1.
Incidence and Severity of Sciatic Nerve Lesionsa
| Degenerated axons | Thin myelinb | Demyelination | Intramyelinic edema | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment group | Incidencec | Severityd | Incidencec | Severityd | Incidencec | Severityd | Incidencec | Severityd | |
| Controls | 4/4 | 2.5 (0.3) | 0/4 | 0 (0) | 0/4 | 0 (0) | 0/4 | 0 (0) | |
| Rangee | (2 - 3) | ||||||||
| DEDC- 2wks- Exposed | 4/4 | 3.0 (0.7) | 0/4 | 0 (0) | 0/4 | 0 (0) | 0/4 | 0 (0) | |
| Rangee | (1 – 4) | ||||||||
| DEDC- 4wks- Exposed | 4/4 | 3.5 (0.9) | 0/4 | 0 (0) | 0/4 | 0 (0) | 0/4 | 0 (0) | |
| Rangee | (1 – 5) | ||||||||
| DEDC- 8wks- Exposed | 5/5 | 15 (6) | 4/5f | 25.4 (24.6) | 2/5 | 7.6 (7.3) | 4/5f | 28 (22.5) | |
| Rangee | (2 – 35) | (0 - 124) | (0 – 37) | (0 – 115) | |||||
Slides were scored blindly by two observers (WMV and OMV). One cross-section of the sciatic nerve was examined by light microscopy for each animal and the lesions totaled per nerve. (n=4 for each group except for DEDC 8-week n=5)
Defined as having a g ratio (axon diameter/axon with myelin diameter) greater than 0.7.
Number of animals with positive observations/number of animals in treatment group.
Mean square root value of axons with lesion (SEM).
The least to greatest number of lesions observed for an individual nerve within a treatment group.
p < 0.05 by one-sided Fisher’s exact test relative to Controls.
Discussion
Previous investigations have demonstrated that DEDC produces significant elevations of copper in the central (CNS) and peripheral (PNS) nervous systems (Delmaestro and Trombetta, 1995; Tonkin et al., 2004), and that the ability of DEDC to disrupt metal homeostasis appears specific for copper and predominantly affects the nervous system (Tonkin et al., 2004; Valentine et al., 2006). The current study supplements those previous studies through demonstrating that significant elevations in copper substantially precede the onset of myelin structural changes produced by DEDC in peripheral nerve. The myelin lesions including intramyelinic edema, thin myelin and demyelination were not observed until 8 weeks of DEDC exposure; whereas significant elevations in total copper where detected in both the CNS and PNS at the earliest time point examined, i.e., two weeks exposure. The copper levels within the nervous system were positively correlated to the duration of exposure (p < 0.05 by the Pearson correlation test, r2 = 0.99 brain and r2 = 0.85 nerve). As observed in previous studies, the magnitude of the increase in total copper was substantially greater for the CNS relative to the PNS (Tonkin et al., 2004). In contrast, although liver copper levels were significantly elevated at the two-week exposure period, there appeared to be no further increase with longer exposures to DEDC. Furthermore, the magnitude of the increase in liver was similar to that observed previously (Tonkin et al., 2004) and was substantially below what would be expected to effect hepatotoxicity (Cisternas et al., 2005). Thus the cumulative increases in copper levels in the nervous system appear to be a direct and specific effect of DEDC.
Significant elevations in lipid oxidation were also observed prior to the onset of myelin lesions. The levels of F2-isoprostanes and MDA in tibial nerve were significantly increased at the 2-week exposure. MDA and F2-isoprostanes showed a similar relationship to exposure duration with the greatest levels observed at two weeks. Previous measurements of MDA in tibial nerve (Caviello et al., 2005) in rats treated for two weeks with pyrrolidine dithiocarbamate showed a significant increase over the control group, and values obtained were consistent with the measurements made in this paper. In addition, the 8-week F2-isoprostane levels measured in tibial nerve are consistent with the levels obtained for the same exposure and duration reported previously for sciatic nerve (Tonkin et al., 2004), supporting the reproducibility of this measurement. Since malondialdehyde and F2-isoprostanes are lipid oxidation products of polyunsaturated fatty acids and arachidonic acid (Montuschi et al., 2004; Uchida, 2006), respectively, these data suggest that the greatest level of lipid oxidation occurred at the 2-week exposure time point. Up regulation of the antioxidant response element pathway may have contributed to the subsequent decrease in these parameters. Evidence for activation of the antioxidant response element by DEDC has been provided by the observed elevations in expression levels of several isoforms of glutathione transferase subsequent to DEDC exposure (Viquez et al., 2007). Additionally, if the observed lipid oxidation is dependent upon redox active copper species, increased expression of copper chaperones or metallothioneins may have also diminished the level of redox active copper within nerve subsequent to the 2-week time point. With exposures longer than four weeks it is possible that copper may accumulate to levels that exceed the cellular antioxidant defense mechanisms and the development of myelin lesions..
In contrast to the response observed for lipid oxidation, protein oxidative injury monitored by HNE adducts was positively correlated to the duration of exposure and continued to increase over the entire 8-week period examined. HNE being an α,β-unsaturated aldehyde can react with protein nucleophiles to generate a variety of covalent protein modifications including Schiff bases and Michael adducts on cysteine, histidine, arginine or lysine that vary considerably in their chemical stabilities (Sayre et al., 2006). The antibody used in this study was a polyclonal antibody raised against protein reacted with HNE, and thus was expected to recognize a variety of adducts including the most stable species. Whereas the relatively small molecular weight MDA and isoprostane markers of lipid oxidation are subject to metabolism and excretion, at least some of the HNE adducts would be expected to exhibit considerable biological persistence and be better able to integrate the level of oxidative injury occurring over the entire exposure period resulting in the observed cumulative response.
Previous proteomic investigations of peripheral nerve have shown the greatest increases in protein expression to be associated with members of the IgG class (Viquez et al., 2007). In particular IgG2a demonstrated increases greater than six fold. The observation that the IgG fraction of serum can produce complement-dependent demyelination in experimental models (Appel and Bornstein, 1961; Halstead et al., 2005; Saida et al., 1979) raises the question as to whether the elevation in IgGs could represent an autoimmune component in DEDC myelinopathy. Autoantibodies could possibly be produced from the generation of non-self oxidatively damaged proteins or from release of normally sequestered nerve proteins into the general circulation. Alternatively, the increase in IgG proteins may just result from passive influx of plasma proteins due to a compromise in the blood-nerve barrier. Unfortunately, this question cannot be resolved by the present study. The greatest levels of IgG were measured coincident with the lesions at 8 weeks suggesting that passive influx could account for the elevated levels of IgG. However a significant, albeit lesser, increase in IgG2a was also detected at four weeks prior to the detection of lesions by light microscopy allowing for the potential role of autoimmune processes in the development of the myelin lesions.
Resident endoneurial macrophages are present in peripheral nerve and these resident macrophages together with infiltrating hematogenous macrophages recruited during injury help to phagocytize and remove degenerating myelin (Mueller et al., 2001). The ED1 monoclonal primary antibody used in this study recognizes the rat homologue of human CD68, a glycoprotein expressed on lysosomal membranes of myeloid cells (Damoiseaux et al., 1994). Although ED1 cannot distinguish between resident and hematogenous macrophages to help ascertain the integrity of the blood-nerve barrier, it is selective for activated macrophages and provides a marker to help identify the onset of inflammatory changes. The absence of activated macrophages and myelin lesions prior to the 8-week exposure period suggests that the increased level of lipid oxidation observed at earlier time points was not mediated through inflammation. Additionally, the appearance of activated macrophages coincident with myelin injury is not consistent with demyelination as occurs in some autoimmune diseases, e.g., Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy (Dyck et al., 2002; Sheikh et al., 2004), although examination of exposure durations closer to the onset of lesions, i.e., 5 through 7 weeks, would be required to conclusively rule out this possibility.
The goal of the current study was to delineate the temporal relationship of copper accumulation, oxidative stress and inflammatory changes to the development of myelin lesions in DEDC peripheral neuropathy. The data provide evidence that DEDC mediates lipid oxidation and elevation of total copper before myelin lesions or activated macrophages are evident. This relationship is consistent with copper-mediated oxidative stress contributing to the myelinopathy. Further investigation is needed to determine whether copper is required for the observed lipid oxidation in nerve and if so, through what mechanism it promotes oxidative injury. In addition, important questions remain regarding the subcellular location, oxidation state, and redox activity of the excess copper, and details about its molecular structure, i.e. whether excess copper is free, associated with protein or dithiocarbamates or in a free ionic state. Defining the mechanism of DEDC-induced myelinopathy and the essential structural properties of dithiocarbamates required for neurotoxicity will facilitate the formulation of accurate risk assessments as well as the design of safer and more effective dithiocarbamates for the many current and proposed applications of this class of compounds in agriculture, industry and medicine.
Acknowledgments
This work was supported by NIEHS Grant ES06387 and by the Center in Molecular Toxicology Grant P30 ES00267. Experiments were performed in part through the use of the VUMC Research EM Resource (sponsored by NIH Grants DK20539 and DK58404). We would like to thank Sandra Olson and Tracie Moss for advice and assistance in preparing the tissues for immunohistochemical analyses of macrophages, and Dr. Justin Zyskowski at the MSU DCPAH for his expertise in ICP-MS tissue metal analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appel SH, Bornstein MB. The application of tissue culture to the study of experimental allergic encephalomyelitis. II. Serum factors responsible for demyelination. J Exp Med. 1961;119:303–312. doi: 10.1084/jem.119.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach SP, Chinery R, O’Dwyer ST, Potten CS, Coffey RJ, Watson AJ. Pyrrolidinedithiocarbamate increases the therapeutic index of 5-fluorouracil in a mouse model. Gastroenterology. 2000;118:81–89. doi: 10.1016/s0016-5085(00)70416-1. [DOI] [PubMed] [Google Scholar]
- Calviello G, Filippi GM, Toesca A, Palozza P, Maggiano N, Nicuolo FD, Serini S, Azzena GB, Galeotti T. Repeated exposure to pyrrolidine-dithiocarbamate induces peripheral nerve alterations in rats. Toxicol Lett. 2005;158:61–71. doi: 10.1016/j.toxlet.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Cisternas FA, Tapia G, Arredondo M, Cartier-Ugarte D, Romanque P, Sierralta WD, Vial MT, Videla LA, Araya M. Early histological and functional effects of chronic copper exposure in rat liver. Biometals. 2005;18:541–551. doi: 10.1007/s10534-005-1244-1. [DOI] [PubMed] [Google Scholar]
- Damoiseaux JG, Dopp EA, Calame W, Chao D, MacPherson GG, Dijkstra CD. Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology. 1994;83:140–147. [PMC free article] [PubMed] [Google Scholar]
- Delmaestro E, Trombetta LD. The effects of disulfiram on the hippocampus and cerebellum of the rat brain; A study of oxidative stress. Toxicol Lett. 1995;75:235–243. doi: 10.1016/0378-4274(94)03187-c. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Dyck PJB, Giannini C, Sahenk Z, Windebank AJ, Engelstad J. Peripheral nerves. In: Graham DI, Lantos PL, editors. Greenfield’s Neuropathology. Seventh. Vol. 2. Arnold Publishers; London: 2002. pp. 615–619. [Google Scholar]
- Eneanya DI, Bianchine JR, Duran DO, Andresen BD. The actions and metabolic fate of disulfiram. Ann Rev Pharmacol Toxicol. 1981;21:575–596. doi: 10.1146/annurev.pa.21.040181.003043. [DOI] [PubMed] [Google Scholar]
- Erve JC, Jensen ON, Valentine HS, Amarnath V, Valentine WM. Disulfiram generates a stable N,N-diethylcarbamoyl adduct on Cys-125 of rat hemoglobin beta-chains in vivo. Chem Res Toxicol. 2000;13:237–244. doi: 10.1021/tx990191n. [DOI] [PubMed] [Google Scholar]
- Fang IM, Yang CH, Lin CP, Yang CM, Chen MS. Effects of pyrrolidine dithiocarbamate, an NF-kappaB inhibitor, on cytokine expression and ocular inflammation in experimental autoimmune anterior uveitis. J Ocul Pharmacol Ther. 2005;21:95–106. doi: 10.1089/jop.2005.21.95. [DOI] [PubMed] [Google Scholar]
- Halstead SK, Humphreys PD, Goodfellow JA, Wagner ER, Smith RA, Willison HJ. Complement inhibition abrogates nerve terminal injury in Miller Fisher syndrome. Ann Neurol. 2005;58:203–210. doi: 10.1002/ana.20546. [DOI] [PubMed] [Google Scholar]
- Johnson DJ, Graham DG, Amarnath V, Amarnath K, Valentine WM. Release of carbon disulfide is a contributing mechanism in the axonopathy produced by N,N-diethyldithiocarbamate. Toxicol Appl Pharmacol. 1998;148:288–296. doi: 10.1006/taap.1997.8344. [DOI] [PubMed] [Google Scholar]
- Kang JH, Wei YM, Zheng RL. Effects of diethyldithiocarbamate on proliferation, redifferentiation, and apoptosis in human hepatoma cells. Acta Pharmacol Sin. 2001;22:785–792. [PubMed] [Google Scholar]
- Kimoto-Kinoshita S, Nishida S, Tomura TT. Diethyldithiocarbamate can induce two different types of death: apoptosis and necrosis mediating the differential MAP kinase activation and redox regulation in HL60 cells. Mol Cell Biochem. 2004;265:123–132. doi: 10.1023/b:mcbi.0000044366.32073.2d. [DOI] [PubMed] [Google Scholar]
- Krenn BM, Holzer B, Gaudernak E, Triendl A, van Kuppeveld FJ, Seipelt J. Inhibition of polyprotein processing and RNA replication of human rhinovirus by pyrrolidine dithiocarbamate involves metal ions. J Virol. 2005;79:13892–13899. doi: 10.1128/JVI.79.22.13892-13899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatovic D, VanRollins M, Li K, Montine KS, Montine TJ. Suppression of murine cerebral F2-isoprostanes and F4-neuroprostanes from excitotoxicity and innate immune response in vivo by alpha- or gamma-tocopherol. J Chromatogr B Anal Technol Biomed Life Sci. 2005;827:88–93. doi: 10.1016/j.jchromb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Montuschi P, Barnes PJ, Roberts LJ. Isoprostanes: markers and mediators of oxidative stress. Faseb J. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- Mueller M, Wacker K, Ringelstein EB, Hickey WF, Imai Y, Kiefer R. Rapid response of identified resident endoneurial macrophages to nerve injury. Am J Pathol. 2001;159:2187–2197. doi: 10.1016/S0002-9440(10)63070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramagli LS. Quantifying protein in 2-D PAGE solubilization buffers. Methods Mol Biol. 1999;112:99–103. doi: 10.1385/1-59259-584-7:99. [DOI] [PubMed] [Google Scholar]
- Reich EE, Markesbery WR, Roberts LJ, 2nd, Swift LL, Morrow JD, Montine TJ. Brain regional quantification of F-ring and D-/E-ring isoprostanes and neuroprostanes in Alzheimer’s disease. Am J Pathol. 2001;158:293–297. doi: 10.1016/S0002-9440(10)63968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saida T, Saida K, Brown MJ, Silberberg DH. Peripheral nerve demyelination induced by intraneural injection of experimental allergic encephalomyelitis serum. J Neuropath Exper Neurol. 1979;38(5):498–518. doi: 10.1097/00005072-197909000-00005. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and ONE. Drug Metab Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Zhang G, Gong Y, Schnaar RL, Griffin JW. An anti-ganglioside antibody-secreting hybridoma induces neuropathy in mice. Ann Neurol. 2004;56:228–239. doi: 10.1002/ana.20173. [DOI] [PubMed] [Google Scholar]
- Si X, McManus BM, Zhang J, Yuan J, Cheung C, Esfandiarei M, Suarez A, Morgan A, Luo H. Pyrrolidine dithiocarbamate reduces coxsackievirus B3 replication through inhibition of the ubiquitin-proteasome pathway. J Virol. 2005;79:8014–8023. doi: 10.1128/JVI.79.13.8014-8023.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofuoglu M, Kosten TR. Novel approaches to the treatment of cocaine addiction. CNS Drugs. 2005;19:13–25. doi: 10.2165/00023210-200519010-00002. [DOI] [PubMed] [Google Scholar]
- Sunderman FW. Efficacy of sodium diethyldithiocarbamate (dithiocarb) in acute nickel carbonyl poisoning. Ann Clin Lab Sci. 1979;9:1–10. [PubMed] [Google Scholar]
- Tonkin EG, Erve JC, Valentine WM. Disulfiram produces a non-carbon disulfide-dependent schwannopathy in the rat. J Neuropathol Exp Neurol. 2000;59:786–797. doi: 10.1093/jnen/59.9.786. [DOI] [PubMed] [Google Scholar]
- Tonkin EG, Valentine HL, Milatovic DM, Valentine WM. N,N-Diethyldithiocarbamate produces copper accumulation, lipid peroxidation, and myelin injury in rat peripheral nerve. Toxicol Sci. 2004;81:160–171. doi: 10.1093/toxsci/kfh190. [DOI] [PubMed] [Google Scholar]
- Uchida K. Lipofuscin-like fluorophores originated from malondialdehyde. Free Radical Res. 2006;40:1335–1338. doi: 10.1080/10715760600902302. [DOI] [PubMed] [Google Scholar]
- Valentine HL, Amarnath K, Amarnath V, Valentine WM. Dietary copper enhances the peripheral myelinopathy produced by oral pyrrolidine dithiocarbamate. Toxicol Sci. 2006;89:485–494. doi: 10.1093/toxsci/kfj047. [DOI] [PubMed] [Google Scholar]
- Valentine HL, Does MD, Marshall V, Tonkin EG, Valentine WM. Multicomponent T(2) analysis of dithiocarbamate-mediated peripheral nerve demyelination. Neurotoxicology. 2007;28:645–654. doi: 10.1016/j.neuro.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez OM, Valentine HL, Friedman DB, Olson SJ, Valentine WM. Peripheral nerve protein expression and carbonyl content in N,N-diethlydithiocarbamate myelinopathy. Chem Res Toxicol. 2007;20:370–379. doi: 10.1021/tx6003453. [DOI] [PMC free article] [PubMed] [Google Scholar]