

Abstract
We and others have shown that prior exposure to the volatile anesthetic isoflurane induces ischemic tolerance in the brain. Our results also suggest that isoflurane preconditioning reduces cell apoptosis in the penumbral region of rat brain. We designed this study to determine whether isoflurane preconditioning decreased mitochondria-dependent cell apoptosis. Adult male Sprague-Dawley rats were exposed to or not exposed to 2% isoflurane for 30 min at 24 h before the permanent middle cerebral arterial occlusion. Western blotting was used to quantify protein expression in the cytosolic and mitochondrial fractions of non-ischemic brain cortex and brain cortex in the ischemic core and penumbra. Isoflurane preconditioning significantly decreased the infarct volume of cerebral cortex and improved neurological outcome. Isoflurane increased the expression of the antiapoptotic B-cell lymphoma-2 (Bcl-2) proteins in the cerebral cortex of rats without brain ischemia. Rats preconditioned with isoflurane before brain ischemia had increased Bcl-2 expression in the penumbra. Isoflurane preconditioning reduced the release of cytochrome c from the mitochondria and the activation of caspase 3 in the penumbra. However, isoflurane preconditioning did not alter the translocation of Bid and Bax from the cytosol to the mitochondria, identified mechanisms for Bcl-2 to block the release of cytochrome c from the mitochondria. Our results suggest that isoflurane preconditioning increases Bcl-2 expression to block the release of cytochrome c from the mitochondria to decrease the cell apoptosis in the penumbra.
Keywords: isoflurane, preconditioning, neuroprotection, cytochrome c, Bcl-2, mitochondria
1. Introduction
Ischemic brain injury is the underlying pathophysiology contributing to the mortality and morbidity in patients with stroke or head trauma. Although intensive research has been devoted to developing methods to reduce ischemic brain injury, clinically effective methods have not yet been established.
Ischemic preconditioning is a well-known phenomenon in which short episodes of ischemia induce ischemic tolerance (Murry et al., 1986). We and others have shown that clinically commonly used and relatively safe volatile anesthetics, such as isoflurane, can also induce ischemic tolerance (Kapinya et al., 2002; Zhao and Zuo, 2004; Zheng and Zuo, 2003; Zheng and Zuo, 2004). Our study showed that rats pretreated with 2% isoflurane at 24 h before the permanent middle cerebral arterial occlusion (MCAO) reduced terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling positive cell density in the penumbra (Zheng and Zuo, 2004), suggesting that isoflurane preconditioning can reduce cell apoptosis after brain ischemia. However, the mechanisms for this effect are not known.
The release of cytochrome c from the mitochondria to the cytosol is an important mechanism to induce cell apoptosis (Jiang and Wang, 2004). The cytosolic cytochrome c can bind with the protease-activating factor-1 to form the apoptosome that will activate caspase 9 (Zou et al., 1997). The activated caspase 9 will activate caspase 3, which will ultimately cause cell apoptosis (Jiang and Wang, 2004). B-cell lymphoma-2 (Bcl-2) is an anti-apoptotic protein. One of the important mechanisms for Bcl-2 to block apoptosis is to prevent the release of cytochrome c from the mitochondria (Kluck et al., 1997). Although it is not entirely clear how Bcl-2 can block this release, the proposed mechanisms include the inhibition of Bid and Bax (Cahill et al., 2006; Ferrer and Planas, 2003; Gwag et al., 2001) translocation from cytosol to the mitochondria. Both Bid and Bax are predominantly in the cytosol of healthy cells and will translocate to the mitochondria to enhance the cytochrome c release after apoptotic stimulation (Cahill et al., 2006; Ferrer and Planas, 2003; Gwag et al., 2001).
We have shown that isoflurane preconditioning can induce Bcl-2 expression in the human neuroblastoma SH-SY5Y cells (Zuo et al., 2006). Thus, we hypothesize that isoflurane preconditioning can induce the Bcl-2 expression to reduce the release of cytochrome c from the mitochondria in the penumbra to provide neuroprotection against brain ischemia.
2. Materials and methods
The animal protocol was approved by the institutional Animal Care and Use Committee of the University of Virginia (Charlottesville, VA). All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications number 80-23) revised in 1996.
2.1. Materials
A mouse monoclonal anti-Bax antibody (catalog number sc-23959), a rabbit polyclonal anti-Bid antibody (catalog number sc-11423), a mouse monoclonal anti-Bcl-2 antibody (catalog number sc-509), a goat polyclonal anti-caspase-3 p20 antibody (catalog number sc-1225), a horseradish peroxidase (HRP)-conjugated donkey anti-goat IgG secondary antibody (catalog number sc-2020), a HRP-conjugated goat anti-rabbit IgG secondary antibody (catalog number sc-2004) and a HRP-conjugated goat anti-mouse IgG secondary antibody (catalog number sc-2005) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). A mouse monoclonal anti-cytochrome c antibody (catalog number 556433) was purchased from BD Pharmingen™ (San Jose, CA). A rabbit polyclonal anti-actin antibody (Catalog number A2066) and the protease inhibitor cocktail (catalog number P2714) were obtained from Sigma Chemical (Saint Louis, MI). A rabbit polyclonal anti-mitochondrial voltage-dependent anion channel (VDAC) antibody (catalog number 4866) was purchased from Cell Signaling Technology, Inc. (Danvers, MA). Polyvinylidene difluoride membranes for Western blotting and reagents for measuring protein concentrations were purchased from Bio-Rad (Hercules, CA). Enhanced chemiluminescence reagents were obtained from Pharmacia-Amersham Biotech (Piscataway, NJ). Other chemicals were purchased from Sigma.
2. 2. Animal groups
Two groups of 2-month-old male Sprague-Dawley rats were studied: rats that were subjected to the permanent right MCAO only and rats that were pretreated with 2% isoflurane for 30 min at 24 h before the MCAO. Two other groups of 2-month-old male Sprague-Dawley rats were exposed or not exposed to 2% isoflurane for 30 min. Their parietal brain tissues were harvested 24 h after the isoflurane exposure for Western analysis of Bcl-2 expression.
2.3. Isoflurane Preconditioning
Anesthesia was induced with isoflurane and then maintained with 2% isoflurane via an endotracheal tube for 30 min. Respiration was controlled by a ventilator to maintain normal end-tidal O2 and CO2 concentrations. The inhaled and exhaled gases were monitored with a Datex infrared analyzer (Capnomac, Helsinki, Finland).
2.4. Permanent Middle Cerebral Artery Occlusion
The rats were anesthetized with isoflurane and then intubated and mechanically ventilated with 60% O2-40% N2 containing 2% isoflurane. The tail artery was cannulated for monitoring arterial blood pressure and blood sampling for measuring blood gases and glucose. Permanent MCAO was accomplished as we described previously (Zheng and Zuo, 2004). The MCAO was achieved by advancing a 3-0 monofilament nylon suture with a rounded tip to the right internal carotid artery via the external carotid artery until slight resistance was felt. Isoflurane anesthesia was stopped once the suture was in place. After recovery from anesthesia, rats were placed back in their cages with ad libitum access to food and water. During anesthesia, temporalis muscle temperature was strictly maintained at 37 ± 0.2°C by warming blanket and lamps. The inhaled and exhaled gases were also monitored to maintain normal end-tidal CO2 concentrations.
2.5. Evaluation of Infarct Volume, Neurological Deficit Scores, and Motor Coordination
The evaluation of infarct volume at 24 h after the permanent MCAO was performed after 2,3,5-triphenyleterazolium chloride staining as described previously (Alessandrini et al., 1999). Briefly, brain hemispheres were cut into 2 mm thick coronal brain slices (usually total 6 slices) in the rostral-caudal direction. The slices were incubated with 2,3,5-triphenyleterazolium chloride for 30 min at 37°C. The infarct areas were quantified using the NIH Image 1.60. To account for the cerebral edema and differential shrinkage resulting from brain ischemia and tissue processing and to correct for the individual difference in brain volume, the percentage of infarct volume in the ipsilateral hemisphere volume was calculated (Swanson et al., 1990).
Neurological deficit scores were evaluated 24 h after the permanent MCAO based on an eight-point scale. Rats were scored as follows: 0, no apparent deficits; 1, failure to extend left forepaw fully; 2, decreased grip of the left forelimb; 3, spontaneous movement in all directions, contralateral circling only if pulled by the tail; 4, circling or walking to the left; 5, walking only if stimulated; 6, unresponsiveness to stimulation and with depressed level of consciousness; 7, dead (Rogers et al., 1997).
Motor coordination was evaluated 24 h before and 24 h after the permanent MCAO. The rats were placed on an accelerating rotarod. The speed of the rotarod was increased from 4 rpm to 40 rpm in 5 min. The latency and the speed of rat’s falling off the rotarod were recorded. Each rat was tested three times. The speed-latency index (latency in seconds × speed in rpm) of each of the three tests was calculated and the mean index of the three trials was used to reflect the motor coordination function of each rat before or after the MCAO. All rats were trained for 3 continuous days before the formal tests.
2.6. Brain tissue sampling and the fractionation of cytosol and mitochondria
The rats were killed by deep isoflurane anesthesia and transcardially perfused by cold phosphate buffered saline (PBS) at 24 h after the permanent MCAO. The frontal cortex area 1 and parietal cortex area 1 from bregma +2 to 0 mm in the right cerebral hemisphere and the corresponding frontal cortex area 1 in the left hemisphere were removed. The frontal cortex area 1 has decreased blood flow and are classified as penumbral areas after MCAO (Memezawa et al., 1992; Nagasawa and Kogure, 1989). The parietal cortex area 1 has a very significant decrease of blood flow, is almost always infarct after MCAO and is considered as ischemic core in this brain ischemic model (Sakai et al., 2007).
The total lysates and the cytosolic and mitochondrial fractions of the harvested brain tissues were prepared as previously described (Hirai et al., 2004). Each brain tissue sample was homogenized in ice-cold lysis buffer [200 mM mannitol, 80 mM HEPES-KOH (pH 7.4), and the protease inhibitor cocktail] by 35 strokes of gentle pounding in a glass tissue grinder. Homogenates were centrifuged at 750 × g for 10 min at 4°C. After removing some of the supernatants that were used as total lysates, the rest of the supernatants were centrifuged at 8,000 × g for 20 min at 4°C. The resulting pellets were washed 3 times and suspended in the lysis buffer as the mitochondrial fraction. Supernatants of 8000 × g centrifugation were centrifuged at 100,000 × g for 1 h at 4°C. The resulting supernatants were cytosolic fraction. The protein concentration of each sample was determined using the Bradford assay.
2.7. Western Analysis
Proteins of 50 μg, 20 μg and 40 μg per lane of the total lysates, mitochondrial and cytosolic fractions, respectively, were subjected to 15% sodium dodecylsulfate-polyacrylamide gel electrophoresis. The proteins were transferred onto a polyvinylidene fluoride membrane. After incubation with PBS containing 5% nonfat milk and 0.1% Tween-20 for 1 h at room temperature, the membranes were probed with various primary antibodies in PBS containing 0.1% Tween-20 (PBST). The primary antibodies used include the anti-Bcl-2 antibody (1:200 dilution), anti-Bax antibody (1:200 dilution), anti-Bid antibody (1:200 dilution), anti-cytochrome c antibody (1:1000 dilution), anti-caspase-3 p20 antibody (1:200 dilution), anti-VDAC antibody (1:1000 dilution) and anti-actin antibody (1:1000 dilution) overnight at 4°C. After being rinsed with PBST, the membranes were incubated with the corresponding HRP-conjugated secondary antibodies for 1 h at room temperature. The protein bands were visualized with the enhanced chemiluminescence methods. Quantitative analysis of the protein bands was performed using an ImageQuant 5.0 GE Healthcare Densitometer (GE Healthcare, Sunnyvale, CA). The densities of Bcl-2, Bax, Bid, cytochrome c and caspase-3 p20 protein bands were normalized to those of actin to control for errors in protein sample loading and transferring during Western analysis. The results from the samples of rats treated with the isoflurane preconditioning and then the MCAO were normalized to the corresponding data of rats with MCAO only on the same film to control for the error caused by different exposure times of films.
2.8. Statistical Analysis
Parametric data are presented as means ± S.D. (n ≥ 5 for each group). Statistical analysis of the results of physiological parameters, speed-latency index ratio, infarct size, and the Western blotting between the MCAO groups and isoflurane preconditioning plus MCAO group was performed by Student’s t test after confirmation of a normal distribution of the data or Mann-Whitney rank sum test when the data are not normally distributed. Neurological deficit scores were analyzed by Mann-Whitney rank sum test. A P ≤ 0.05 was accepted as significant.
3. Results
As we have shown previously (Zheng and Zuo, 2004), rats preconditioned with 2% isoflurane for 30 min had significantly smaller brain infarct volumes than rats without isoflurane pretreatment after the MCAO (Fig 1). This reduction of brain infarct volume was due to the decreased infarct volume of the cerebral cortex. Isoflurane preconditioning did not affect the infarct volumes of the subcortical areas. Consistent with the neuropathological data, isoflurane preconditioning also improved the neurological deficit scores and the motor coordination (Fig 1). However, isoflurane preconditioning did not change the hemodynamics, blood gases and glucose levels during the procedure of MCAO (Table 1).
Fig. 1. Isoflurane preconditioning improves neurological outcome after permanent right middle cerebral arterial occlusion (MCAO).
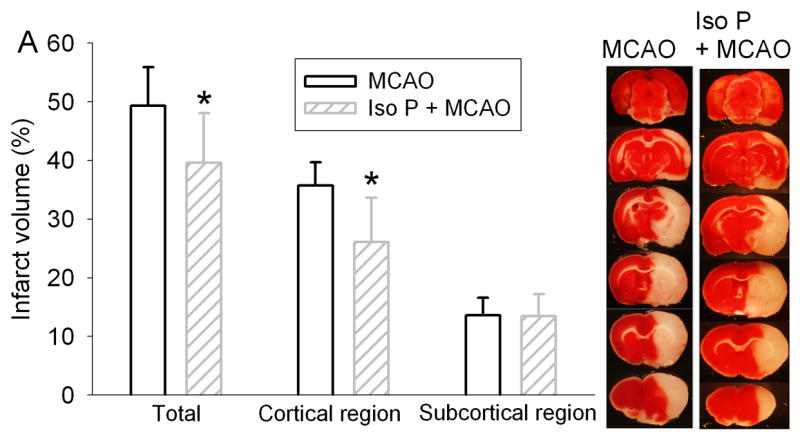
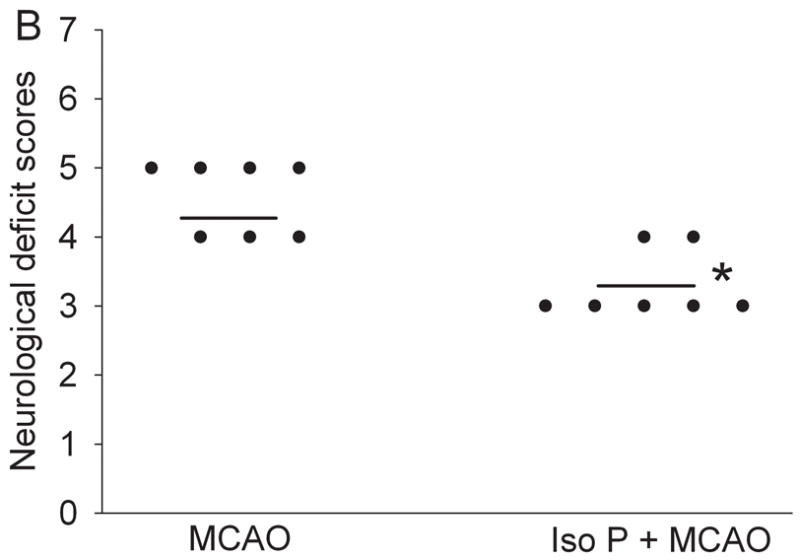
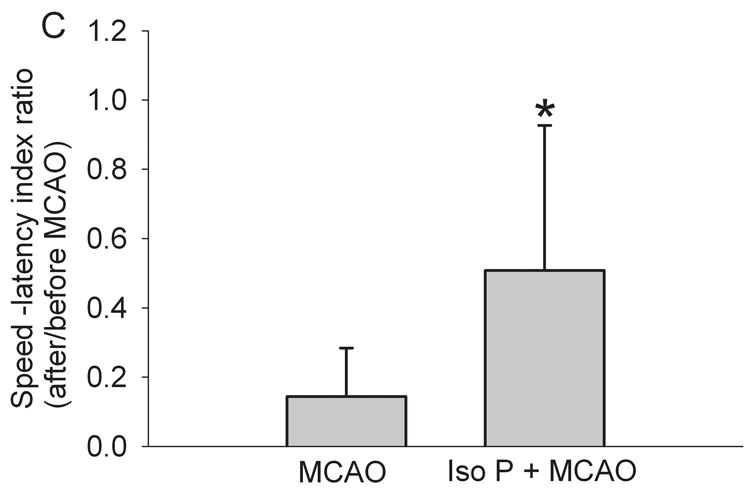
Panel A: Percentage of infarct volume in ipsilateral hemisphere volume is shown in the left panel. Results are the means ± S.D. (n = 7). Brain slices stained with 2,3,5-triphenyleterazolium chloride from representative rats subjected to MCAO only or isoflurane preconditioning plus MCAO are shown in the right panel. Panel B: Neurological deficit scores that were evaluated immediately before the animals were sacrificed for the assessment of infarct sizes (data are presented in Fig. 1A) are shown. Each circle represents the score for a single rat. A horizontal bar indicates the mean value for each group. Panel C: The performance on rotarod. Rats were tested before and 24 h after the MCAO. The speed-latency index ratio of the tests at these two time points is presented. Results are the means ± S.D. (n = 7). * P ≤ 0.05 compared with the corresponding MCAO group. Iso P: isoflurane preconditioning.
Table 1.
Physiological parameters in rats subjected to permanent middle cerebral arterial occlusion (MCAO).
| MCAO
|
Isoflurane + MCAO
|
|||
|---|---|---|---|---|
| Before MCAO | During MCAO | Before MCAO | During MCAO | |
| MABP (mm Hg) | 97 ± 14 | 98 ± 13 | 99 ± 16 | 101 ± 17 |
| Heart rate (beat/min) | 332 ± 18 | 336 ± 15 | 322 ± 34 | 331 ± 12 |
| Temperature (°C) | 37.1 ± 0.1 | 37.1 ± 0.1 | 37.0 ± 0.2 | 37.0 ± 0.2 |
| pH | 7.29 ± 0.07 | 7.37 ± 0.1 | 7.32 ± 0.1 | 7.27 ± 0.06 |
| PaCO2 (mm Hg) | 50 ± 6 | 45 ± 8 | 46 ± 10 | 53 ± 1 |
| PaO2 (mm Hg) | 216 ± 35 | 196 ± 31 | 223 ± 50 | 341 ± 137 |
| Glucose (mg/dl) | 237 ± 34 | 221 ± 56 | 197 ± 66 | 184 ± 20 |
Arterial samples were taken 10 min before and after the onset of MCAO. Data are mean ± S.D. (n = 3 – 10). There were no statistical differences in these physiological parameters among the rats pretreated with or without 2% isoflurane for 30 min before the MCAO. MABP: mean arterial blood pressure.
As shown in the figure 2, the mitochondrial fractions contained abundant VDAC proteins, a mitochondrial marker. The VDAC proteins were not detected in the cytosolic fractions. These results suggest that we have excellent separation of mitochondrial fractions from cytosolic fractions with our method. Although the amount of actin, the protein used as internal control, in the cytosolic and mitochondrial fractions was not affected by brain ischemia (Fig. 2), cytochrome c in the cytosolic fraction of the ischemic penumbra (right frontal cortex area 1) and core (right parietal cortex area 1) was significantly higher than that in the frontal cortex area 1 contralateral to the ischemic side. Cytochrome c in the mitochondrial fraction of the ischemic penumbra and core was significantly lower than that in the frontal cortex area 1 contralateral to the ischemic side (Fig. 2). These results suggest that ischemia induced cytochrome c release from the mitochondria. This cytochrome c release in the penumbra was significantly reduced by isoflurane preconditioning because more cytochrome c proteins were in the mitochondria and less cytochrome c proteins were in the cytosol in the penumbra of rats preconditioned with isoflurane and then MCAO than rats with MCAO only (Fig 3). Consistent with this result, the active caspase 3 (20 KDa form) in the ischemic penumbra was significantly reduced by isoflurane preconditioning (Fig. 4).
Fig. 2. Brain ischemia induces release of cytochrome c from the mitochondria.

Rats were subjected to the permanent right middle cerebral arterial occlusion (MCAO). The left frontal cortex area 1 (non-ischemia), right frontal cortex area 1 (penumbra) and right parietal cortex area 1 (ischemic core) were harvested. The cytosolic and mitochondrial fractions were prepared for Western blotting. Panel A is a representative Western blot to show the excellent separation of mitochondrial fractions from the cytosolic fractions. Panel B: a representative Western blot is shown on the top panel and the graphic presentation of the cytochrome c and actin protein abundance quantified by integrating the volume of autoradiograms from 5 rats is shown in the bottom panel. Values in graphs are expressed as fold change over non-ischemic brain cortex and presented as the means ± S.D. * P < 0.05 compared with the corresponding value from the non-ischemic brain cortex. VDAC, voltage-dependent anion channel.
Fig. 3. Isoflurane decreases release of cytochrome c from the mitochondria in the ischemic penumbra after the permanent right middle cerebral arterial occlusion (MCAO).


Rats were exposed to or not exposed to 2% isoflurane for 30 min before the MCAO. The left frontal cortex area 1 (non-ischemia), right frontal cortex area 1 (penumbra) and right parietal cortex area 1 (ischemic core) were harvested and the cytosolic and mitochondrial fractions were prepared for Western blotting. Panel A: Cytosolic fraction; Panel B: Mitochondrial fraction. A representative Western blot is shown on the top panel and the graphic presentation of the cytochrome c protein abundance quantified by integrating the volume of autoradiograms from 5 to 7 rats is shown in the bottom panel. Values in graphs are expressed as fold change over the rats subjected to the MCAO only and presented as the means ± S.D. * P < 0.05 compared with the MCAO only. Iso P: isoflurane preconditioning.
Fig. 4. Isoflurane decreases the activation of caspase 3 in the ischemic penumbra after the permanent right middle cerebral arterial occlusion (MCAO).

Rats were exposed to or not exposed to 2% isoflurane for 30 min before the MCAO. The left frontal cortex area 1 (non-ischemia), right frontal cortex area 1 (penumbra) and right parietal cortex area 1 (ischemic core) were harvested for Western blotting of the active caspase 3 (the 20 KDa form). A representative Western blot is shown on the top panel and the graphic presentation of the active caspase 3 protein abundance quantified by integrating the volume of autoradiograms from 5 to 7 rats is shown in the bottom panel. Values in graphs are expressed as fold change over the rats subjected to the MCAO only and presented as the means ± S.D. * P < 0.05 compared with the MCAO only. Iso P: isoflurane preconditioning.
Isoflurane exposure significantly increased the expression of Bcl-2 proteins in the cerebral cortex of rats without brain ischemia (Fig. 5). The expression of Bcl-2 proteins in the penumbral region of rats pretreated with isoflurane was significantly higher than that in rats with MCAO only (Fig. 5). These results suggest that the increased Bcl-2 expression contributes to the reduced cytochrome c release from the mitochondria in the penumbra. We then determined whether this Bcl-2 protective effect was via blocking the translocation of Bid and Bax from the cytosol to the mitochondria. There was no significant difference in the expression of Bid and Bax proteins in the cytosolic and mitochondrial fractions prepared from the penumbra of rats pretreated with or without isoflurane before the MCAO (Figs. 6 and 7), suggesting that Bcl-2 does not affect the translocation of Bid and Bax to block the cytochrome c release from the mitochondria.
Fig. 5. Isoflurane increases the expression of Bcl-2 in the cerebral cortex.
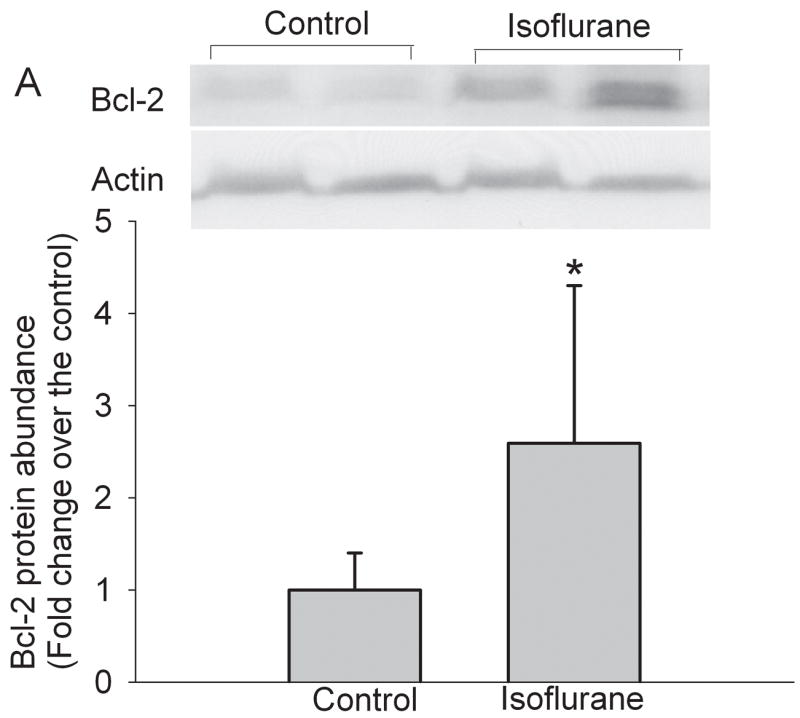
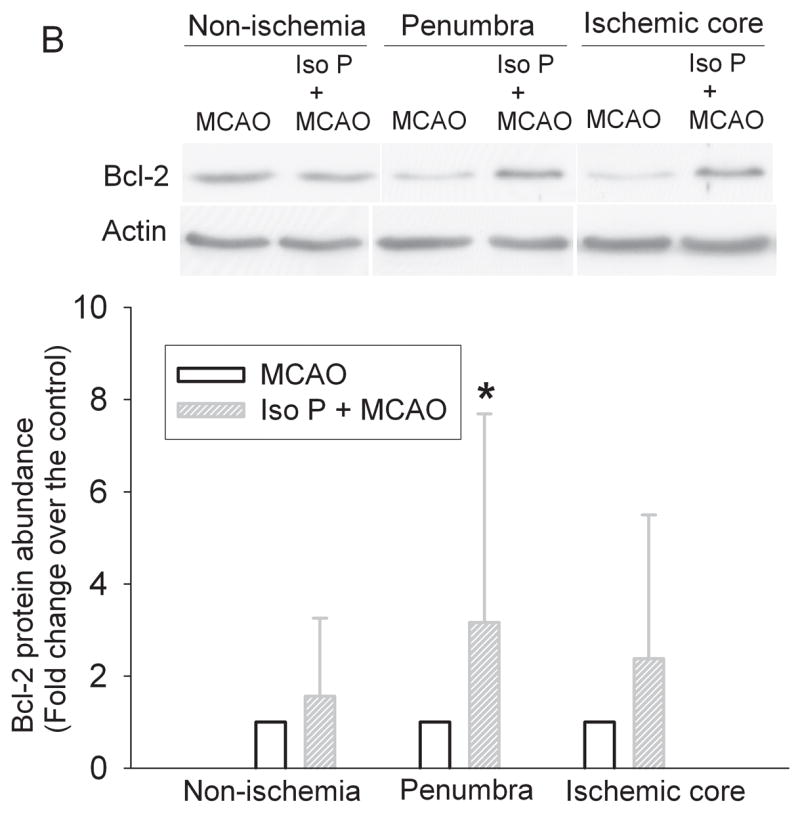
Panel A: Rats were exposed to or not exposed to 2% isoflurane for 30 min and the cerebral cortex was harvested for Western blotting of Bcl-2. A representative Western blot is shown on the top panel and the graphic presentation of the Bcl-2 protein abundance quantified by integrating the volume of autoradiograms from 8 rats is shown at the bottom panel. Values in graphs are expressed as fold change over the control rats and presented as the means ± S.D. * P < 0.05 compared with the control only. Panel B: Rats were exposed to or not exposed to 2% isoflurane for 30 min at 24 h before the MCAO. The left frontal cortex area 1 (non-ischemia), right frontal cortex area 1 (penumbra) and right parietal cortex area 1 (ischemic core) were harvested for Western blotting. A representative Western blot is shown on the top panel and the graphic presentation of the Bcl-2 protein abundance quantified by integrating the volume of autoradiograms from 5 – 7 rats is shown at the bottom panel. Values in graphs are expressed as fold change over the rats subjected to the MCAO only and presented as the means ± S.D. * P < 0.05 compared with the MCAO only. Iso P: isoflurane preconditioning.
Fig. 6. Isoflurane did not affect the translocation of Bid in the ischemic penumbra after the permanent right middle cerebral arterial occlusion (MCAO).
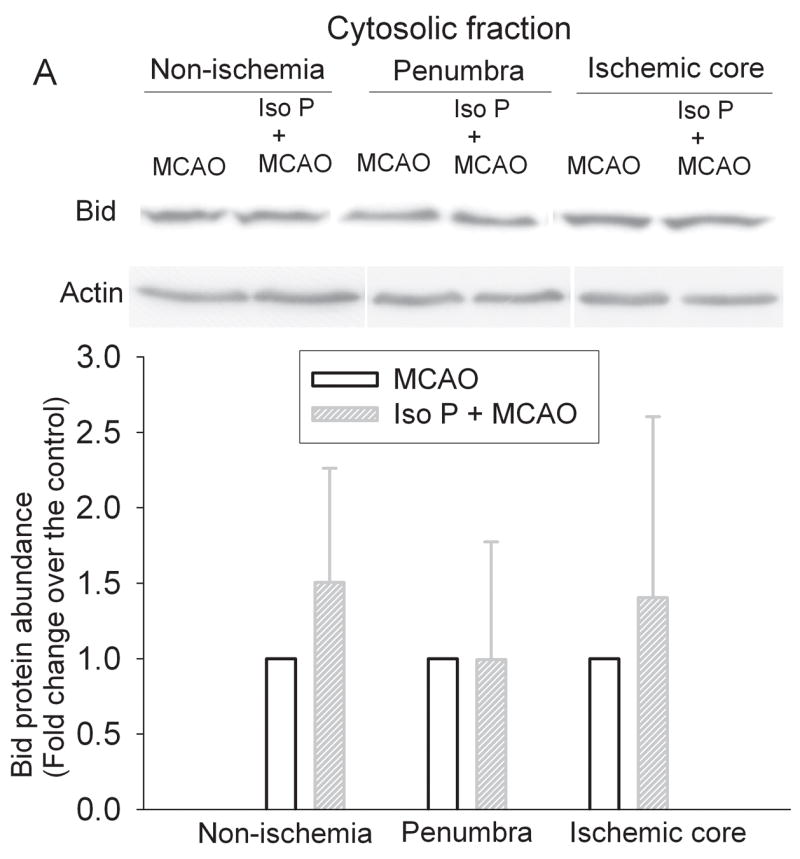

Rats were exposed to or not exposed to 2% isoflurane for 30 min before the MCAO. The left frontal cortex area 1 (non-ischemia), right frontal cortex area 1 (penumbra) and right parietal cortex area 1 (ischemic core) were harvested and the cytosolic and mitochondrial fractions were prepared for Western blotting. Panel A: Cytosolic fraction; Panel B: Mitochondrial fraction. A representative Western blot is shown on the top panel and the graphic presentation of the Bid protein abundance quantified by integrating the volume of autoradiograms from 5 – 7 rats is shown in the bottom panel. Values in graphs are expressed as fold change over the rats subjected to the MCAO only and presented as the means ± S.D. Iso P: isoflurane preconditioning.
Fig. 7. Isoflurane did not affect the translocation of Bax in the ischemic penumbra after the permanent right middle cerebral arterial occlusion (MCAO).

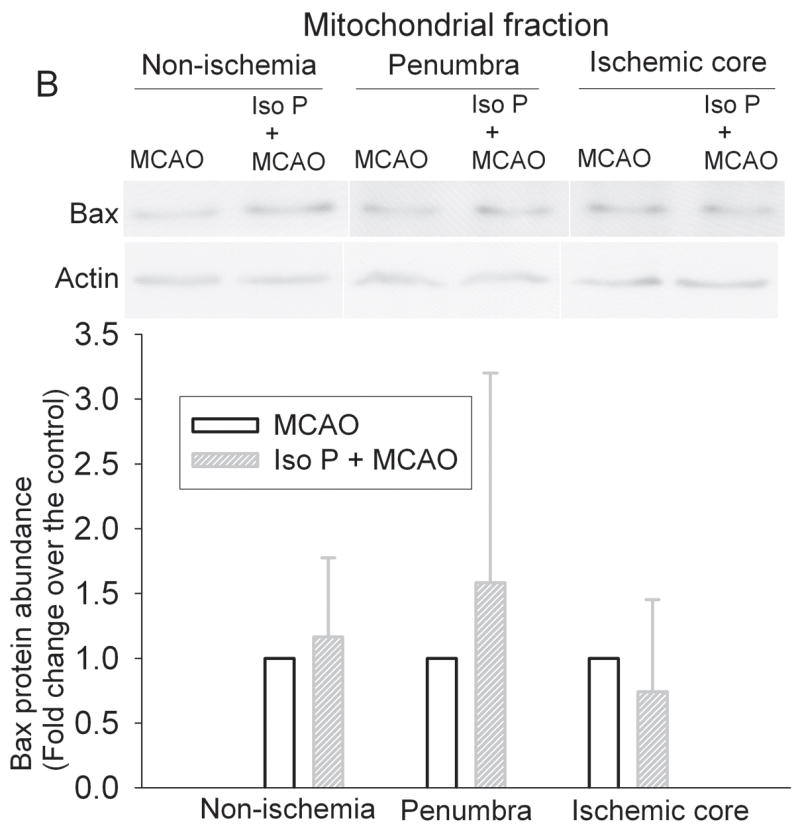
Rats were exposed to or not exposed to 2% isoflurane for 30 min before the MCAO. The left frontal cortex area 1 (non-ischemia), right frontal cortex area 1 (penumbra) and right parietal cortex area 1 (ischemic core) were harvested and the cytosolic and mitochondrial fractions were prepared for Western blotting. Panel A: Cytosolic fraction; Panel B: Mitochondrial fraction. A representative Western blot is shown on the top panel and the graphic presentation of the Bax protein abundance quantified by integrating the volume of autoradiograms from 5 – 7 rats is shown in the bottom panel. Values in graphs are expressed as fold change over the rats subjected to the MCAO only and presented as the means ± S.D. Iso P: isoflurane preconditioning.
4. Discussion
Our study showed that isoflurane preconditioning reduced brain infarct volumes and improved neurological functions after the MCAO. This protective effect is mainly from the reduction of the infarct volume in the cerebral cortex. Since we used permanent MCAO and the collateral circulation to the striatum is very poor in rats after MCAO, it is not surprising to observe that isoflurane preconditioning did not reduce the subcortical infarct volume. Similarly, the parietal cortex area 1 has a significant decrease of blood flow and is considered as ischemic core after MCAO (Sakai et al., 2007). However, frontal cortex area 1 has decreased blood flow and are classified as penumbral areas after MCAO (Memezawa et al., 1992; Nagasawa and Kogure, 1989). Thus, our findings are consistent with the idea that isoflurane preconditioning reduces the infarction/cell death in the penumbral regions. We observed about 20 – 25% reduction in infarct brain volumes by isoflurane preconditioning. This relatively small reduction may be related to the use of a severe brain ischemia model, permanent MCAO, in this study. Nevertheless, we still observed improvement of neurological functions by isoflurane preconditioning in this model.
Our previously published data suggest that isoflurane preconditioning reduced cell apoptosis in the penumbral region (Zheng and Zuo, 2004). We have also shown that isoflurane induced Bcl-2 expression in the human neuroblastoma SH-SY5Y cells (Zuo et al., 2006). Thus, it is possible that isoflurane preconditioning increases Bcl-2 in the penumbral region to decrease cell apoptosis. Our results showed that a 30-min isoflurane exposure increased Bcl-2 expression in the rat cerebral cortex at 24 h after the isoflurane exposure. Isoflurane preconditioning also increased the Bcl-2 protein expression in the penumbral region at 24 h after the MCAO. Since Bcl-2 is a well-known antiapoptotic molecule (Kluck et al., 1997; Yang et al., 1997), our results suggest a role of Bcl-2 in the isoflurane preconditioning-induced decrease of cell apoptosis.
One of the important mechanisms for Bcl-2 to block cell apoptosis is to decrease cytochrome c release from the mitochondria (Kluck et al., 1997; Yang et al., 1997). Consistent with this idea, our results showed that the release of cytochrome c from the mitochondria in the penumbra was inhibited by isoflurane preconditioning. Once cytochrome c is released into cytosol, it will form the apoptosome to activate caspase 9 and then caspase 3 to induce cell apoptosis (Jiang and Wang, 2004). Our results also showed that the active caspase 3 in the penumbra was decreased by isoflurane preconditioning. These results suggest that isoflurane preconditioning provides protection in the penumbra by increasing Bcl-2 expression to block the activation of caspase 3. Consistent with our findings, a recent study has shown that ischemic preconditioning reduced the number of apoptotic neurons, cytochrome c release from the mitochondria and the activation of caspase 9 in the hippocampal CA1 region of rats after global brain ischemia (Liu et al., 2007).
It has been suggested that Bcl-2 can block the translocation of Bid and Bax from cytosol to the mitochondria to decrease the release of cytochrome c from the mitochondria (Cahill et al., 2006; Ferrer and Planas, 2003; Gwag et al., 2001). However, our results suggest that isoflurane preconditioning does not change the translocation of Bid or Bax in the penumbra. In addition to affecting the translocation of Bid and Bax to block the release of cytochrome c, it has been proposed that Bcl-2, by binding to the mitochondrial membrane, can directly block the release of cytochrome c from the mitochondria (Kluck et al., 1997).
It is not known yet how isoflurane can induce the expression of Bcl-2. We have shown that isoflurane preconditioning-induced neuroprotection may be p38 activation-dependent (Zheng and Zuo, 2004). Signal transducer and activator of transcription 3 (STAT3) increases Bcl-2 expression (Alas et al., 2001). Inhibition of p38 has been shown to inhibit STAT3-increased Bcl-2 expression (Vega et al., 2004). We and others have shown that isoflurane can induce the expression of inducible nitric oxide synthase (Kapinya et al., 2002; Zhao and Zuo, 2004). Nitric oxide produced by inducible nitric oxide synthase can activate STAT3 (Hierholzer et al., 2004). Future studies will be designed to determine whether p38, STAT3 and inducible nitric oxide synthase are involved in the isoflurane-induced Bcl-2 expression.
In summary, our results showed that isoflurane preconditioning reduced cell death in the penumbral region. This protection may involve Bcl-2 to block the cytochrome c release from the mitochondria and, therefore, to decrease cell apoptosis in the penumbra.
Acknowledgments
This study was supported by grants (R01 GM065211 and R01 NS045983 to Z Zuo) from the National Institute of Health, Bethesda, Maryland.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alas S, Emmanouilides C, Bonavida B. Inhibition of interleukin 10 by rituximab results in down-regulation of bcl-2 and sensitization of B-cell non-Hodgkin’s lymphoma to apoptosis. Clin Cancer Res. 2001;7:709–723. [PubMed] [Google Scholar]
- Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. PNAS. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26:1341–1353. doi: 10.1038/sj.jcbfm.9600283. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol. 2003;62:329–339. doi: 10.1093/jnen/62.4.329. [DOI] [PubMed] [Google Scholar]
- Gwag BJ, Won SJ, Kim DY. Excitotoxicity, oxidative stress, and apoptosis in ischemic neuronal death. In: Lin CS, editor. New Concepts in Cerebral Ischemia. CRC Press; New York: 2001. pp. 79–112. [Google Scholar]
- Hierholzer C, Kalff JC, Billiar TR, Bauer AJ, Tweardy DJ, Harbrecht BG. Induced nitric oxide promotes intestinal inflammation following hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2004;286:G225–233. doi: 10.1152/ajpgi.00447.2002. [DOI] [PubMed] [Google Scholar]
- Hirai K, Hayashi T, Chan PH, Zeng J, Yang GY, Basus VJ, James TL, Litt L. PI3K inhibition in neonatal rat brain slices during and after hypoxia reduces phospho-Akt and increases cytosolic cytochrome c and apoptosis. Brain Res Mol Brain Res. 2004;124:51–61. doi: 10.1016/j.molbrainres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Kapinya KJ, Lowl D, Futterer C, Maurer M, Waschke K, Isaev NK, Dirnagl U. Tolerance Against Ischemic Neuronal Injury Can Be Induced by Volatile Anesthetics and Is Inducible NO Synthase Dependent. Stroke. 2002;33:1889–1898. doi: 10.1161/01.str.0000020092.41820.58. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sercombe R, Xie D, Liu K, Chen L. Inhibition of caspase-9 activation and apoptosis is involved in ischemic preconditioning-induced neuroprotection in rat brain. Neurol Res. 2007;29:855–861. doi: 10.1179/016164107X223575. [DOI] [PubMed] [Google Scholar]
- Memezawa H, Minamisawa H, Smith M-L, Siesjo BK. Ischemic penumbra in a model of reversible middle cerebral artery occlusion in the rat. Exp Brain Res. 1992;89:67–78. doi: 10.1007/BF00229002. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nagasawa H, Kogure K. Correlation between cerebral blood flow and histologic changes in a new rat model of middle cerebral artery occlusion. Stroke. 1989;20:1037–1043. doi: 10.1161/01.str.20.8.1037. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Campbell CA, Stretton JL, Mackay KB. Correlation between motor impairment and infarct volume after permanent and transient middle cerebral artery occlusion in the rat. Stroke. 1997;28:2060–2065. doi: 10.1161/01.str.28.10.2060. [DOI] [PubMed] [Google Scholar]
- Sakai H, Sheng H, Yates RB, Ishida K, Pearlstein RD, Warner DS. Isoflurane Provides Long-term Protection against Focal Cerebral Ischemia in the Rat. Anesthesiology. 2007;106:92–99. doi: 10.1097/00000542-200701000-00017. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume [see comments] J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Vega MI, Huerta-Yepaz S, Garban H, Jazirehi A, Emmanouilides C, Bonavida B. Rituximab inhibits p38 MAPK activity in 2F7 B NHL and decreases IL-10 transcription: pivotal role of p38 MAPK in drug resistance. Oncogene. 2004;23:3530–3540. doi: 10.1038/sj.onc.1207336. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zhao P, Zuo Z. Isoflurane preconditioning induces neuroprotection that is inducible nitric oxide synthase-dependent in the neonatal rats. Anesthesiology. 2004;101:695–702. doi: 10.1097/00000542-200409000-00018. [DOI] [PubMed] [Google Scholar]
- Zheng S, Zuo Z. Isoflurane preconditioning reduces Purkinje cell death in an in vitro model of rat cerebellar ischemia. Neuroscience. 2003;118:99–106. doi: 10.1016/s0306-4522(02)00767-4. [DOI] [PubMed] [Google Scholar]
- Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of p38 mitogen-activated protein kinase. Mol Pharmacol. 2004;65:1172–1180. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Wang Y, Huang Y. Isoflurane preconditioning protects human neuroblastoma SH-SY5Y cells against in vitro simulated ischemia-reperfusion through the activation of extracellular signal-regulated kinases pathway. Eur J Pharmacol. 2006;542:84–91. doi: 10.1016/j.ejphar.2006.05.027. [DOI] [PubMed] [Google Scholar]