

Abstract
Association mapping (i.e., linkage disequilibrium mapping) is a powerful tool for positional cloning of disease genes. We propose a kernel-based association test (KBAT), which is a composite function of “P-values of single-locus association tests” and “kernel weights related to intermarker distances and/or linkage disequilibria.” The KBAT is a general form of some current test statistics. This method can be applied to the study of candidate genes and can scan each chromosome using a moving average procedure. We evaluated the performance of the KBAT through simulation studies that considered evolutionary parameters, disease models, sample sizes, kernel functions, test statistics, window attributes, empirical P-value estimations, and genetic/physical maps. The results showed that the KBAT had a well-controlled false positive rate and high power compared to existing methods. In addition, the KBAT was also applied to analyze a genomewide data set from the Collaborative Study on the Genetics of Alcoholism. Important genes associated with alcoholism dependence were identified. In summary, the merits of the KBAT are multifold: the KBAT is robust against the inclusion of nuisance markers, is invariant to the map scale, and accommodates different types of genomic data, study designs, and study purposes. The proposed methods are packaged in the user-friendly software, KBAT, available at http://www.stat.sinica.edu.tw/hsinchou/genetics/association/KBAT.htm.
ASSOCIATION study has been broadly implemented to identify disease susceptibility genes related to complex disorders (Cardon and Bell 2001; Hirschhorn and Daly 2005; Wang et al. 2005; Laird and Lange 2006). Several historical milestones of gene mapping that have used association studies include the identifications of the association between late-onset Alzheimer's disease and the APOE-4 allele on 19q13.2 (Corder et al. 1993) and the association between Crohn's disease and NOD2 on 16q21 (Hugot et al. 2001). With the completion of international genetic/genomic projects, such as the Human Genome Project (International Human Genome Mapping Consortium 2001), the HapMap Project (International HapMap Consortium 2003), and the ENCODE Project (Encode Project Consortium 2004), a large number of SNP markers across the human genome have become available for gene association studies. In addition, new SNP array technologies have blossomed (Matsuzaki et al. 2004; Steemers and Gunderson 2007). A great reduction in the cost of genotyping and an increase in the number of genetic markers make genomewide association scans more feasible and efficient. These recent advances have improved the power and resolution of association mapping, thereby providing exquisite genetic dissection of complex disorders and greatly contributing to drug discovery (Wellcome Trust Case Control Consortium 2007).
A key factor in a successful association study is the choice of association tests. Association tests can be divided into single-locus and multilocus tests, according to the number of marker loci involved in a test statistic. Single-locus association tests emphasize marginal effects and are most suitable for studying a locus with a strong main effect on disease manifestation, where the study locus may be causal or highly correlated (indirect association) to genes responsible for disease. Linkage disequilibrium, which reflects allelic association among different loci, plays an important role in indirect mapping. Linkage disequilibrium decays due to chromosomal recombination in meiosis between generations, and hence it exists only within a small chromosome region after many generations in an outbred population. Consequently, association mapping (i.e., linkage disequilibrium mapping) is highly accurate for positional cloning of disease-related genes.
In recent years, multilocus association tests have gained widestream use for association studies identifying disease susceptibility genes related to complex disorders (Hoh and Ott 2003). Multiple genes may be simultaneously involved in a same-disease pathway and act in concert to confer a higher risk of disease. For one thing, multilocus susceptibility models are competent for detection of marginal effects unless a flip-flop phenomenon occurs (Zaykin and Shibata 2008). For another, a multilocus association test provides more information regarding the disease-related gene region and potentially increases the statistical power for gene localization compared with a single-locus inference. The reasons justify the use of multilocus association analyses.
The main multilocus association analyses consist of haplotype inference (Schaid et al. 2002; Zaykin et al. 2002a; Chen and Kao 2006), genotype partition/combination [combinatorial partitioning method (CPM) (Nelson et al. 2001) and multifactor dimensonality reduction (MDR) (Ritchie et al. 2001)], statistic combination tests (Hoh et al. 2001; Hoh and Ott 2003; Wille et al. 2003; Sun et al. 2006), and P-value combinations (Zaykin et al. 2002b; Dudbridge and Koeleman 2003; Yang et al. 2006). Each of these methods has its respective strengths and has proven practical in certain applications. This article focuses on P-value combinations due to two reasons. First, they reflect our research interests. Second, P-value combination has several merits (Zaykin et al. 2002b and discussion and conclusion in this article).
P-value combinations originated with Fisher's product P-value method, equivalent to the sum of log scale of P-values (Fisher 1925). Later, other P-value combination methods were developed (Tippett 1931; Stouffer et al. 1949; Edgington 1972). P-values were assumed to be independent in these methods for the convenience of theoretical development; however, this assumption is too stringent for use in many practical applications, such as candidate gene or genomewide association scans using dense SNP markers. Different computational algorithms have been developed to circumvent the difficulty in deviation of null distributions with dependent P-values, such as permutation, bootstrap, and Monte Carlo (Manly 1998). P-value combination methods have been broadly applied to different fields, such as meta-analysis of linkage mapping (Guerra et al. 1999) and microarray gene expression analysis (Hess and Iyer 2007). Recently, these methods have been extended to association mapping (Zaykin et al. 2002b; Dudbridge and Koeleman 2003; Yang et al. 2006). Some P-value combination methods have been incorporated into popular analysis packages, such as SAS/Genetics version 9.1.3 (Sas Institute 2005).
Although many P-value combination methods have been developed, few incorporate intermarker distances into the algorithm. Two methods were developed to account for intermarker distances, i.e., random intermarker distances and constant intermarker distances. For random distances, mutation processes were formulated by a compound Poisson process, and intermarker distances were assumed to follow an identical exponential distribution independently (Sun et al. 2006). The study found that the Fisher's product P-value method could identify disease gene regions, but the regions were larger than that identified by their scan method. For constant distances, intermarker distances were formulated as fixed constants and served as weights for P-values on different marker loci (Yang et al. 2006). The results showed that the Fisher's product P-value method may lose in power and/or increase in false positive rate when nuisance markers were included in the analysis. This article focuses on the model of constant intermarker distances that do not assume a specific underlying distribution for intermarker distances. The purpose of this article is to propose a new P-value combination method for efficient multilocus association scans and to examine this method using large-scale simulation studies and real data analyses.
METHOD
A two-stage procedure:
We introduce a two-stage association mapping strategy to locate genes that influence susceptibility to a complex trait or disorder. Consider a study region [0, T] that contains M SNP markers at positions 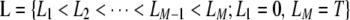 . In the first stage, M single-locus association tests for null hypotheses “H0i: the ith SNP is not associated with the study disease,
. In the first stage, M single-locus association tests for null hypotheses “H0i: the ith SNP is not associated with the study disease, 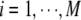 ” are performed, and the observed significance probabilities are summarized in a series of P-values,
” are performed, and the observed significance probabilities are summarized in a series of P-values, 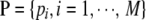 . For instance, allele- and trend-based association tests can be considered for unrelated case–control studies under Hardy–Weinberg equilibrium and disequilibrium, respectively (Sasieni 1997). Family-based association tests (FBATs) (Rabinowitz and Laird 2000; Horvath et al. 2001) can be performed for family-based case–control and quantitative trait mapping studies.
. For instance, allele- and trend-based association tests can be considered for unrelated case–control studies under Hardy–Weinberg equilibrium and disequilibrium, respectively (Sasieni 1997). Family-based association tests (FBATs) (Rabinowitz and Laird 2000; Horvath et al. 2001) can be performed for family-based case–control and quantitative trait mapping studies.
In the second stage, a multilocus association test combining neighboring P-values in sequence P is constructed. The study region [0, T] is scanned using a moving average procedure from the starting SNP at position 0 to the end SNP at position T. The procedure is described as follows. Let an anchor denote a chromosomal position of interest. Given a bandwidth h and an anchor locus t, a window 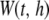 is constructed by choosing all SNPs [i.e.,
is constructed by choosing all SNPs [i.e., 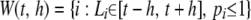 ] or potential SNPs [i.e.,
] or potential SNPs [i.e., 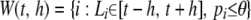 ] within the chromosome region [t − h, t + h] for
] within the chromosome region [t − h, t + h] for 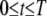 and
and 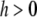 , where constant
, where constant 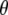 denotes a truncation threshold where P-values greater than the threshold are removed from the window. If all P-values in a window are >
denotes a truncation threshold where P-values greater than the threshold are removed from the window. If all P-values in a window are >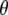 , the window is removed from subsequent analyses. The entire study region is partitioned into contiguous windows by shifting the anchors from the beginning to the end of the study region. The moving windows have a fixed window length (i.e., 2h) except when an anchor is close to the boundary of the study region. Different windows probably contain various numbers of SNPs.
, the window is removed from subsequent analyses. The entire study region is partitioned into contiguous windows by shifting the anchors from the beginning to the end of the study region. The moving windows have a fixed window length (i.e., 2h) except when an anchor is close to the boundary of the study region. Different windows probably contain various numbers of SNPs.
Within each window, we perform a kernel-based association test (KBAT). Given a bandwidth, h, and an anchor at position t, the test statistic for window 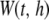 is written as follows:
is written as follows:
 |
(1) |
where
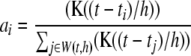 |
(2) |
denotes kernel weight for the ith P-value within window 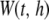 , and function
, and function 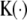 denotes a kernel density satisfying three properties: (1) unimode, (2) symmetry, and (3) integration to one. Three frequently used kernel density functions, Epanechnikov kernel (
denotes a kernel density satisfying three properties: (1) unimode, (2) symmetry, and (3) integration to one. Three frequently used kernel density functions, Epanechnikov kernel (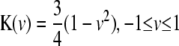 ), triangular kernel (
), triangular kernel (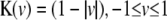 ), and quartic kernel (
), and quartic kernel (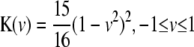 ), were considered in our simulation study. The KBAT elaborated in Equation 1 is constructed under a multiplicative P-value model (Fisher 1925; Zaykin et al. 2002b). Other models, e.g., an additive P-value model, can also be considered. Association between a putative disease locus and an anchor locus is sequentially scanned by shifting the anchor, which is the center of a window, from the start to the end of the study region. The KBAT emphasizes a “local effect,” where higher weights are assigned to single-locus P-values of markers closer to the anchor. Effects of remote marker loci are negligible. This feature pertains to kernel function and kernel weights as mentioned before. Such a local effect is suitable for describing the pattern of linkage disequilibrium, which decays due to historical meiosis recombination.
), were considered in our simulation study. The KBAT elaborated in Equation 1 is constructed under a multiplicative P-value model (Fisher 1925; Zaykin et al. 2002b). Other models, e.g., an additive P-value model, can also be considered. Association between a putative disease locus and an anchor locus is sequentially scanned by shifting the anchor, which is the center of a window, from the start to the end of the study region. The KBAT emphasizes a “local effect,” where higher weights are assigned to single-locus P-values of markers closer to the anchor. Effects of remote marker loci are negligible. This feature pertains to kernel function and kernel weights as mentioned before. Such a local effect is suitable for describing the pattern of linkage disequilibrium, which decays due to historical meiosis recombination.
Sampling distributions of the KBAT statistic and its special cases:
The sampling distribution of the KBAT statistic should be derived for testing disease association. We first discuss the scenario where all P-values within a window are independent. Let 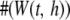 denote the number of P-values within window
denote the number of P-values within window 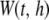 ,
, 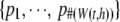 represents the corresponding P-values, and
represents the corresponding P-values, and 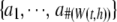 represents the kernel weights. Under the null hypothesis of no association, all P-values follow a uniform distribution.
represents the kernel weights. Under the null hypothesis of no association, all P-values follow a uniform distribution.
The KBAT statistic elaborated in Equation 1 is a general form of the single-locus P-value statistic and the log function of the product P-value statistics. If a kernel function having a single-point mass on an anchor is adopted, then the KBAT statistic reduces to the single-locus P-value statistic. If a rectangle kernel function is adopted and the P-value truncation procedure is not considered (i.e., 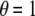 ), then the KBAT statistic reduces to the Fisher's product P-value statistic (Fisher 1925). The commonly used formula, i.e., minus twice the log function of the product P-value statistic, follows a chi-square distribution with a degree of freedom of
), then the KBAT statistic reduces to the Fisher's product P-value statistic (Fisher 1925). The commonly used formula, i.e., minus twice the log function of the product P-value statistic, follows a chi-square distribution with a degree of freedom of 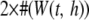 under the null hypothesis. If a rectangle kernel function is adopted and the P-value truncation procedure is considered (i.e.,
under the null hypothesis. If a rectangle kernel function is adopted and the P-value truncation procedure is considered (i.e., 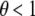 ), then the KBAT statistic reduces to the Zaykin's truncated product P-value statistic whose null distribution has been described previously (Zaykin et al. 2002b). If other kernel functions are used and the P-value truncation procedure is not considered, then the KBAT statistic reduces to the weighted product P-value statistic and its null distribution has been described previously (Good 1955).
), then the KBAT statistic reduces to the Zaykin's truncated product P-value statistic whose null distribution has been described previously (Zaykin et al. 2002b). If other kernel functions are used and the P-value truncation procedure is not considered, then the KBAT statistic reduces to the weighted product P-value statistic and its null distribution has been described previously (Good 1955).
If P-values within a window are statistically dependent, the exact null distribution becomes intractable and relies on the correlation structure of P-values. Monte Carlo (Zaykin et al. 2002b), permutation (Churchill and Doerge 1994; Doerge and Churchill 1996; Dudbridge and Koeleman 2003), and direct simulation methods (Lin 2005; Seaman and Müller-Myhsok 2005) have been proposed to generate the null distribution. All of these algorithms can be applied to yield a null distribution of the KBAT with slight modifications. In this article, we capitalized on the Zaykin's Monte Carlo procedure by applying a five-step algorithm as follows: (1) a correlation matrix was established to define the relationship among P-values; (2) dependent P-values mimicking the original P-values in the real data were generated; (3) the KBAT statistic was recalculated on the basis of each of the Monte Carlo samples; (4) the empirical distribution of the KBAT statistic was constructed; and (5) the empirical P-value was calculated. For the details in each step refer to Yang et al. (2006).
SIMULATION
Simulation conditions:
Using simulations, we evaluated the performance of the KBAT under different conditions. Dichotomous phenotype data (affected case and unaffected control) and SNP genotype data (1/1, 1/0, and 0/0) were generated using HAPSIM software (Curtis et al. 2001). The details of the simulation algorithms are listed in the HAPSIM user manuals. Our simulations considered several parameters/factors including (1) evolutionary parameters (recombination and mutation age); (2) disease models [disease allele frequency (DAF) and penetrance vector (PV)] and sample sizes; (3) kernel functions (Epanechnikov, triangular, and quartic kernels); (4) test statistics [single-locus method, product P-value method (Fisher 1925), truncated product P-value method (Zaykin et al. 2002b), minimum P-value method (Tippett 1931), weighted product P-value method (Yang et al. 2006) and KBATs]; (5) bandwidths and windows (fixed window and moving windows); (6) empirical P-value estimations (Monte Carlo procedure and permutation procedure); and (7) genetic/physical maps (base pairs, centimorgans, morgans, and recombination fraction). In the simulation study, we generated S simulation samples (S = 1000) under each simulation condition. In each simulation sample, a chi-square test statistic was calculated for the single-locus association test in the first stage on the basis of genotype data, and the resulting P-values were used for multilocus association tests in the second stage. Given the P-values from raw data, we calculated correlation matrices of P-values. Then we generated dependent P-values mimicking the original P-values for C times (C = 10,000). In each sequence of dependent P-values, all test statistics were recalculated. Then the empirical distribution of each test statistic was constructed and the empirical P-value was calculated on the basis of the C Monte Carlo samples. The procedure was applied to all S simulation samples. On the basis of the obtained empirical P-values, we calculated the false positive rate for models without disease genes, and we calculated the power in models containing disease genes.
Evolutionary parameters:
We examined the performance of the KBAT using an Epanechnikov kernel under different recombination fractions and mutation ages, which affected the evolutionary process of the disease gene of study. Two recombination fractions were considered: (1) SU-1 and (2) SU-5. Among 31 diallelic markers in the study region, the recombination fraction SU-1 contained 3 middle markers and SU-5 contained 11 middle markers that are highly linked to the true disease locus; the remaining SNPs were not linked to the disease locus (Yang et al. 2006). Two mutation ages were considered: (1) recent polymorphism and (2) historic polymorphism. The first condition considered the number of generations before and after the disease mutation to be 10 and 10, respectively; the second condition considered the number of generations before and after the disease mutation to be 200 and 10, respectively. In the simulation of power study, the (unobservable) disease locus was set close to the center of the study region with 31 markers. For both the affected cases and unaffected controls, the sample size was Na = Nu = 1000. The frequency of disease allele D was DAF = 0.1, and the penetrance vector for genotypes dd, Dd, and DD was PV = (0.1, 0.3, 0.4), respectively. Bandwidths were assigned on the basis of window sizes of 3, 11, 21, and 31.
The results in Figure 1A demonstrate that the recombination fraction had a large effect on the power of the KBAT, but the effect of mutation ages was relatively low. Under the SU-1 recombination fraction containing 3 markers linked to the true disease locus, the power was between 0.42 and 0.45; under the scenario of SU-5 containing 11 markers linked to the true disease locus, the power increased from 0.47 to ∼0.75 when data from all informative markers were capitalized in the KBAT. The effects of window size and bandwidth will be discussed later. The recombination fraction and mutation ages slightly affected the false positive rate of the KBAT, but all false positive rates were controlled between 0.03 and 0.08.
Figure 1.—

Evaluation of effects of evolutionary/genetic/other factors on the KBAT. (A) Effects of recombination fraction and mutation age. (B) Effects of disease model and sample size. (C) Effect of kernel function.
Disease model and sample size:
Two sample sizes were considered: (1) 500 cases and 500 controls and (2) 1000 cases and 1000 controls. Four disease models were considered: (1) high penetrance model with PV = (0, 0.7, 0.7) and DAF = 0.001, (2) modest penetrance model with PV = (0.1, 0.3, 0.4) and DAF = 0.1, (3) modest penetrance model with PV = (0.1, 0.3, 0.4) and DAF = 0.25, and (4) low penetrance model with PV = (0.005, 0.005, 0.03) and DAF = 0.25.
The results in Figure 1B demonstrate that the power increased when sample size or penetrance increased. Although the pattern was not surprising, the values of increased power allow for practical study designs. Under the four disease models, the percentage of power gained due to doubling the sample size was 2, 35, 40, and 55%, respectively. Under the same PV (disease models 2 and 3), KBAT had higher power for a disease model with a higher DAF. Under the same DAF (disease models 2 and 3), KBAT had higher power for a disease model with a higher PV.
Without additional descriptions regarding simulation conditions for the subsequent simulation studies, we describe results from the simulation studies, which generated 31 diallelic markers for 1000 cases and 1000 controls under the SU-1 recombination fraction. The number of generations before and after the disease mutation was 10. A disease model of PV = (0.1, 0.3, 0.4) and DAF = 0.1, which had the lowest power among the four previously considered disease models, was discussed. Analyses for other evolutionary/disease models were also performed but results are not shown.
Kernel functions:
We evaluated the impact of kernel functions on the KBAT. Three frequently used kernel density functions were considered: (1) Epanechnikov kernel, (2) triangular kernel, and (3) quartic kernel. All simulation results in Figure 1C show that the power and false positive rates for different kernel functions were consistent. The power for different kernel functions was 0.42, and the false positive rate was 0.03. Therefore, kernel functions have no or only a limited effect on the KBAT. In subsequent simulations, we show only results of the KBAT using an Epanechnikov kernel, which is optimal for density estimation (Epanechnikov 1969).
Test statistics:
We compared the KBAT with existing methods including the single-locus method (SLM), the minimum P-value method (MPM) (Tippett 1931), the product P-value method (PPM) (Fisher 1925), the truncated product P-value method (TPPM) (Zaykin et al. 2002b), and the weighted product P-value method (WPPM) (Yang et al. 2006). Four bandwidths were assigned corresponding to window sizes of 3, 11, 21, and 31. The results in Figure 2, A and B, show simulation results of test statistics with different bandwidths/window sizes. Results of test statistics without P-value truncation are shown in Figure 2A; results of test statistics with P-value truncation are shown in Figure 2B.
Figure 2.—

Comparison of false positive rates and power of different association tests under different bandwidths. (A) Rejection probability of test statistics without P-value truncation. (B) Rejection probability of test statistics with a P-value truncation threshold of 0.05.
In general, test statistics KBAT and WPPM had the highest power, and SLM had the lowest power. Except for PPM, the test statistics controlled false positive rates well. The increase of the false positive rate of PPM should be caused by the interference of nuisance markers. Because inclusion of nuisance markers is sometimes unavoidable in practical gene mapping studies, it is worth discussing the impact of nuisance markers on the test statistics. This simulation study focused on the SU-1 recombination fraction, which contained only the three middle markers linked to the true disease locus. Consequently, the three middle markers were informative, and the others were considered nuisance. Simulation results showed that the power of MPM, PPM, and TPPM was reduced dramatically when nuisance markers were included. The weighted statistics (KBAT and WPPM) properly adjusted the interference of nuisance markers and maintained the power. Inclusion of nuisance markers also resulted in the inflation of false positive rate of PPM. The P-value truncation procedure suggested by Zaykin et al. (2002b) significantly improved inflation of false positive rate, whereas the truncation slightly reduced the power. The weighting procedures recommended in Yang et al. (2006) and in this article also controlled false positive rate well. In summary, KBAT and WPPM had the best performance in both power and false positive rate, i.e., the two test statistics were invariant to the inclusion of nuisance markers.
Bandwidths and windows:
First, we examined the case where the anchor marker was fixed at the center of study markers (scenario of a fixed window). We specified four bandwidths corresponding to window sizes of 3, 11, 21, and 31. Simulation results are shown in Figure 2, A and B. In general, the KBAT and the WPPM performed well with regard to power and false positive rate even although a nonoptimal bandwidth or window size was used (the optimal window size was 3 in this case). MPM, PPM, and TPPM were comparable to KBAT and WPPM when the optimal window size was considered, but the power was dramatically reduced when the incorrect window size was adopted.
We further evaluated the performance of the KBAT using moving anchors (scenario of moving windows). We generated 11 SNP markers in the study region for 1000 cases and 1000 controls under the SU-1 recombination fraction. A true disease locus was located close to one of the following five loci: (1) the starting locus SNP1, (2) the second locus SNP2, (3) the sixth locus SNP6, (4) the tenth locus SNP10, and (5) the last locus SNP11. A “disease-related region” was constructed using markers associated with the true disease locus. Therefore, the disease-related regions under SU-1 were constructed by {SNP1, SNP2}, {SNP1, SNP2, SNP3}, {SNP5, SNP6, SNP7}, {SNP9, SNP10, SNP11}, and {SNP10, SNP11} when the true disease locus was close to the five loci: SNP1, SNP2, SNP6, SNP10, and SNP11, respectively. Two bandwidths (h = 25,000.00001 bp and h = 50,000.000045 bp) were considered. For each true disease location and for each of the two bandwidths, rejection probabilities of three test statistics (SLM, TPPM, and KBAT) with a P-value truncation threshold of 0.05 were calculated sequentially from the first marker locus SNP1 to the last marker locus SNP11. Results are shown in Figure 3, where the vertical axis denotes rejection probability. The location of a true disease locus is signified by a blue inverted triangle. The rejection probability signifies power at a SNP marker if the SNP is located within the disease-related region; the rejection probability signifies a false positive rate at a SNP marker if the SNP is located outside the disease-related region.
Figure 3.—

Comparison of rejection probabilities of the SLM, the TPPM, and the KBAT. In each graph, the horizontal axis denotes SNP locus and the vertical axis denotes rejection probability. Two graphs in each row show results for two different bandwidths (h = 25,000.00001 bp and h = 50,000.000045 bp), given the same location of the true disease gene; 5 graphs in each column show results for five locations of the true disease gene, given a bandwidth. The location of a true disease locus is signified by a blue inverted triangle.
Results showed that the KBAT improved power of the SLM due to incorporation of multilocus information and improved false positive rate of the TPPM resulting from integration of proper marker weights. The improvement of false positive rate can also be explained as an improvement in the resolution of association mapping, where the KBAT reliably identified the region linked to the true disease locus as well as accurately determined the width of the disease gene region. In addition, we examined whether the location of a true disease locus affected performance of the proposed methods. When an anchor is close to the boundary of the study region, the corresponding window has an imbalanced number of markers on both sides of the anchor marker. If the power and false positive rate are not influenced by an imbalanced number of markers, we label this property as an “immunity of boundary effect.” Figure 3 shows that the KBAT satisfied this property. These advantages justify the application of the KBAT to association scans.
Empirical P-value estimations:
We compared the two methods of empirical P-value calculation, Monte Carlo procedure (Zaykin et al. 2002b) and permutation procedure (Churchill and Doerge 1994; Doerge and Churchill 1996). A disease model of PV = (0.1, 0.3, 0.4) and DAF = 0.25 was discussed. Four bandwidths were assigned corresponding to window sizes of 3, 11, 21, and 31. KBAT statistics without P-value truncation (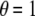 ) and with P-value truncation (
) and with P-value truncation (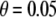 ) were calculated. The total number of simulations was S = 1000. In each simulation, empirical P-values were calculated by using the Monte Carlo procedure and permutation procedure, respectively, where the number of Monte Carlo replications was C = 10,000 and the number of permutation replications was R = 10,000. The power and false positive rate of the KBAT with/without P-value truncation were calculated under different bandwidths. Simulation results of test statistics without P-value truncation are shown in Figure 4A; results of test statistics with P-value truncation are shown in Figure 4B. Results showed that the power and false positive rates from the Monte Carlo procedure and permutation procedure were close. The differences of power of the two procedures across different truncation thresholds and bandwidths were smaller than 0.02; the differences of false positive rates of the two procedures across different truncation thresholds and bandwidths were smaller than 0.03. Two methods of empirical P-value calculation produced the consistent results, suggesting that the calculation of empirical P-value of the KBAT was reliable.
) were calculated. The total number of simulations was S = 1000. In each simulation, empirical P-values were calculated by using the Monte Carlo procedure and permutation procedure, respectively, where the number of Monte Carlo replications was C = 10,000 and the number of permutation replications was R = 10,000. The power and false positive rate of the KBAT with/without P-value truncation were calculated under different bandwidths. Simulation results of test statistics without P-value truncation are shown in Figure 4A; results of test statistics with P-value truncation are shown in Figure 4B. Results showed that the power and false positive rates from the Monte Carlo procedure and permutation procedure were close. The differences of power of the two procedures across different truncation thresholds and bandwidths were smaller than 0.02; the differences of false positive rates of the two procedures across different truncation thresholds and bandwidths were smaller than 0.03. Two methods of empirical P-value calculation produced the consistent results, suggesting that the calculation of empirical P-value of the KBAT was reliable.
Figure 4.—

Comparison of false positive rates and power of KBATs, which used Monte Carlo procedure and permutation procedure for empirical P-value estimation, under different bandwidths. (A) Rejection probability of the KBAT without P-value truncation. (B) Rejection probability of the KBAT with a P-value truncation threshold of 0.05.
Genetic/physical maps:
We examined whether different maps and scales influence the weights used in the KBAT and the WPPM, and therefore, the performance in disease gene mapping. Results showed that weights of the KBAT were not affected by the map scale (Figure 5A), but the weights of the WPPM were affected (Figure 5B). In other words, the KBAT is scale invariant. We also compared the effect of map scale on false positive rates and power of the KBAT and the WPPM. The results are shown in Figure 5C. Because of an invariance to map scale, the false positive rate and power of the KBAT remained constant for different map scales. However, the WPPM was affected greatly. When base pair (bp) was used as the distance scale, the WPPM assigned almost all weight to the anchor marker. Under these circumstances, the WPPM was equivalent to the SLM where the power was reduced while false positive rate was minimized. When recombination fraction (RF) was used, all markers in the window had approximately equal weights. The WPPM, therefore, was approximately equivalent to the PPM. The power decreased and false positive rate increased when nuisance markers were included, i.e., when the number of included markers was >3. When centimorgan (cM) and morgan (M) were used, the KBAT and the WPPM performed similarly. The results showed that the KBAT was invariant to the map scale.
Figure 5.—

Evaluation of the effects of different weighting procedures. (A) Weight values of the proposed kernel-based weighting procedure. (B) Weight values of the distance-based weight procedure (Yang et al. 2006). (C) Rejection probabilities of the KBAT and the WPPM under different distance scales (M, morgan; cM, centimorgan; bp, base pair; and RF, recombination fraction).
REAL DATA ANALYSIS
Alcoholism dependence [Online Mendelian Inheritance in Man (OMIM) no. 103780] is a polygenic and multifactorial disorder characterized by an alcohol craving, alcohol tolerance, and/or aggressive and antisocial behavior. In this study, we analyzed the data from The Collaborative Study on the Genetics of Alcoholism (COGA) provided by Genetic Analysis Workshop 14 (GAW14) (Bailey-Wilson et al. 2005; Edenberg et al. 2005) to illustrate our proposed method. In this study, patients with an alcohol dependency were diagnosed using the DSM-III-R and Feighner criteria. This study collected 143 pedigrees with 1614 samples in total, which corresponded to 643 patients, 285 pure unaffected individuals, and 686 others (“others” contain unknown, never drank, and unaffected with some symptoms). Samples who met the diagnostic criteria of alcohol dependency were treated as affected individuals (cases). The remaining samples were treated as unaffected individuals (controls). Genotyping was performed with the Affymetrix GeneChip Mapping 10K Array (11,560 SNPs) and Illumina Linkage III Panel (4763 SNPs). Our analysis capitalized only on the SNPs in the former platform, having an average intermarker distance of 210 kb, because the former platform provided a larger number of SNP markers. Of 11,120 SNPs on 22 autosomes, 1497 SNPs violating Hardy–Weinberg equilibrium were excluded from our analysis.
In our analysis, FBATs (Rabinowitz and Laird 2000; Horvath et al. 2001) were conducted to test the null hypothesis of no association in the first stage. False discovery rates (FDR) (Benjamini and Hochberg 1995) of the FBAT are shown in Figure 6 (black dashed line). Results showed that the genomewide single-locus FBAT identified five significant SNPs with FDR < 0.05 (tsc0055322 and tsc0559236 on chromosome 1, tsc0564670 on chromosome 5, tsc0483523 on chromosome 6, and tsc0325449 on chromosome 14). In addition to the single-locus FBAT, we also calculated the multilocus KBAT with bandwidths covering 5, 10, and 20 SNPs on an average on the basis of P-values of the genomewide single-locus FBAT. FDR was applied to the empirical P-values of the KBAT to consider a multiple-test comparison. In this scenario, the regions located by KBATs with the three bandwidths were similar. Therefore, only results based on the bandwidth covering five SNPs on average are shown in Figure 6 (blue solid line). Results showed that all SNPs identified by the genomewide single-locus FBAT were also captured by the KBAT. Nevertheless, the KBAT further identified additional loci over the single-locus FBAT. The genomewide multilocus KBAT identified 24 significant SNPs on chromosomes 1, 2, 3, 5, 6, 7, 14, and 18, where the highest −log10(FDR) on these eight chromosomes were 2.34, 1.62, 1.42, 3.51, 2.90, 1.42, 1.40, and 1.40, respectively. SNPs identified by at least one of three KBATs with different bandwidths are summarized in supplemental Table 1.
Figure 6.—

Genome-wide association scans of alcoholism data. The horizontal axis is the genetic distance in centimorgans. The vertical axis is the false discovery rate (FDR) of the FBAT in −log10. The red reference line denotes the significance threshold of −log10(FDR) = 1.30, i.e., FDR = 0.05. The black dashed line represents −log10 (FDR) of the single-locus FBAT and the blue solid line denotes −log10(FDR) of the multilocus KBAT.
Our analyses confirmed findings in previous genomewide linkage mapping (Hill et al. 2004; Yang et al. 2005a). Our identified strong association signals on chromosomes 1 and 6 were close to the regions mapped by the previous studies (Edenberg et al. 2004; Lappalainen et al. 2004). The highest peak on chromosome 1 was close to the alcoholism gene region between D1S2779 (126.16 cM) and D1S1170 (128.73 cM) identified by Lappalainen et al. (2004). The highest peak on chromosome 6 was close to the human major histocompatibility complex region that covers important alcoholism genes, such as GABRA2 (Edenberg et al. 2004). In addition, KBATs also identified chromosome regions that were neither found by the single-locus FBAT nor reported by other studies. More investigation of biological function and disease etiology of these regions with strong association signals, such as gene regions of FMNL2 (2q23.3), LIMD1 (3p21.3), DDC (7p11), and WDR7 (18q21.1-q22), and regions of 22.0 cM and 49.8–50.0 cM on chromosome 5, is needed.
SOFTWARE
KBAT software was developed on the basis of language R and a user-friendly interface based on R-GUI (See Figure 7). Programs, several illustrated data sets, and a user guide are available at the KBAT web site http://www.stat.sinica.edu.tw/hsinchou/genetics/association/KBAT.htm. Before using KBAT software, it is suggested that users read the user guide for software installation/initialization, function/operation, and format of input/output data.
Figure 7.—

Interface of the KBAT software.
DISCUSSION AND CONCLUSION
This article proposes a nonparametric kernel-based association test. The performance of the proposed methods was examined by simulation studies and a genomewide association study. Simulation results showed that the KBAT performs well with regard to power and false positive rate. In addition, the developed software, KBAT, provides a useful tool for users to analyze their data. In summary, the KBAT is simple in concept and has several main features as follows.
First, the KBAT is scale invariant to marker distances. The KBAT incorporates marker weights to dilute the impact of nuisance markers and to amplify the effect of informative markers. The marker weights in Equation 2 are a ratio of kernel function to marker distances. The basic unit is eliminated. Consequently, kernel weights are invariant to scale change of physical/genetic distances.
Second, the KBAT is able to infer disease association at unassayed loci. Regardless of whether the anchor locus of interest is assayed, a window is constructed by symmetrically extending two regions with a length equal to the bandwidth from the anchor. Continuous kernel function symmetrical to the anchor locus assigns weights to P-values within the window. If the anchor is assayed, the highest weight is assigned to the P-value of the anchor. If the anchor is not assayed, then the continuous kernel function provides an automatic adjustment for the calculation of marker weights. In other words, the weight at the anchor is shared by other nearby assayed markers. Therefore, the KBAT can infer disease association at any specific locus of interest.
Third, the KBAT can incorporate information about the background of linkage disequilibrium. In addition to intermarker distances, information about linkage disequilibrium can also be utilized directly. If genotype data are available, the coefficient of linkage disequilibrium can be calculated (Morton et al. 2001; Shete 2003); otherwise, the information can be gathered from the web site of the International HapMap Project (http://www.hapmap.org/downloads/index.html.en). The information can be used alone or jointly with intermarker distances for weight assignment in the KBAT. This utility was also incorporated into the KBAT software. In our simulation study, the KBAT with a joint weight function of distance and linkage disequilibrium performed similarly to the KBAT and the WPPM with distance-only P-value weights.
The KBAT can also analyze different types of data from different study designs and research purposes:
The KBAT can be applied to genetic association studies without genotype data, such as pooled DNA multilocus association tests (Sham et al. 2002; Yang and Fann 2007). In such a study, the loss of individual genotype information limits the capitalization of haplotype-based or genotyped-based multilocus association tests in such studies. The KBAT can easily apply to perform pooled DNA multilocus association tests by incorporating pooled DNA single-locus association tests (Visscher and Le Hellard 2003; Yang et al. 2005b). In addition, the KBAT can be used for meta-analysis (Glass 1976) where only P-value data are collected. P-value sequence data of disease association from different sources, e.g., several studies or related publications, are merged and analyzed by the KBAT to provide an integrated conclusion.
The KBAT can be applied to genetic studies with different study designs. For example, the KBAT can easily adapt to unrelated case–control studies, family-based case–control studies, and quantitative trait studies once P-values from proper single-locus association tests are collected.
The KBAT has potential for different study purposes. In addition to disease gene association mapping, the KBAT also has potential to identify genetic linkage and detection of chromosomal aberrations, e.g., copy number change and allelic imbalance.
The KBAT can be applied to study marker loci violating Hardy–Weinberg equilibrium, which is the fundamental assumption of many multilocus association tests. The KBAT can circumvent this restriction by choosing a proper single-locus association test that is valid under Hardy–Weinberg disequilibrium. For example, a trend test (Armitage 1955) for a case control association study can be applied.
The KBAT has several other interesting qualities. The KBAT is a Nadaraya–Watson-type statistic (Nadaraya 1964; Watson 1964) with an underlying model—the local constant regression model. The model is a special case of local polynomials having advantages of minimax efficiency, absence of boundary effect, and flexible fluctuation data fitting (Fan and Gijbels 1996). The determination of the degree of polynomials is a trade-off. A large degree of polynomials improves the accuracy of curve fitting but also increases its variability and computational time. Further investigation of the KBAT under this extended model is worth pursuing. Additionally, the KBAT is nonparametric. This strength is flexible in data analysis because this approach is not restricted by specific parametric assumptions. However, the calculation of empirical P-values may take longer relative to parametric approaches. The computational time required by the KBAT is reasonable, but intensive time is demanded for a large-scale, whole genome scan. Further refinement of an efficient computational algorithm to derive null distribution is underway.
Acknowledgments
Data of alcoholism dependence analysis were provided by the Collaborative Study on the Genetics of Alcoholism (U10AA008401). This work was partially supported by a National Science Council grant of Taiwan (NSC 96-2314-B-001-005) and a National Research Program for Genomic Medicine grant of Taiwan (NSC 97-3112-B-001-027).
References
- Armitage, P., 1955. Tests for linear trends in proportion and frequencies. Biometrics 11 375–386. [Google Scholar]
- Bailey-Wilson, J. E., L. Almasy, M. De Andrade, J. Bailey, H. Bickeboller et al., 2005. Genetic Analysis Workshop 14: microsatellite and single-nucleotide polymorphism marker loci for genome-wide scans. BMC Genet. 6 S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57 289–300. [Google Scholar]
- Cardon, L. R., and J. I. Bell, 2001. Association study designs for complex diseases. Nat. Rev. Genet. 2 91–99. [DOI] [PubMed] [Google Scholar]
- Chen, Y. H., and J. T. Kao, 2006. Multinomial logistic regression approach to haplotype association analysis in population-based case-control studies. BMC Genet. 7 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder, E. H., A. M. Saunders, W. J. Strittmatter, D. E. Schmechel, P. C. Gaskell et al., 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261 921–923. [DOI] [PubMed] [Google Scholar]
- Curtis, D., B. V. North and P. C. Sham, 2001. Use of an artificial neural network to detect association between a disease and multiple marker genotypes. Ann. Hum. Genet. 65 95–107. [DOI] [PubMed] [Google Scholar]
- Doerge, R. W., and G. A. Churchill, 1996. Permutation tests for multiple loci affecting a quantitative character. Genetics 142 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge, F., and B. P. Koeleman, 2003. Rank truncated product of p values, with application to genomewide association scans. Genet. Epidemiol. 25 360–366. [DOI] [PubMed] [Google Scholar]
- Edenberg, H. J., D. M. Dick, X. Xuei, H. Tian, L. Almasy et al., 2004. Variations in GABRA2, encoding the alpha 2 submit of the GABAA receptor, are associated with alcohol dependence and with brain oscillations. Am. J. Hum. Genet. 74 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg, H. J., L. J. Bierut, P. Boyce, M. Cao, S. Cawley et al., 2005. Description of the data from the Collaborative Study on the Genetics of Alcoholism (COGA) and single-nucleotide polymorphism genotyping for Genetic Analysis Workshop 14. BMC Genet. 6 S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington, E. S., 1972. An additive model for combining probability values from independent experiments. J. Psychol. 80 351–363. [Google Scholar]
- Encode Project Consortium, 2004. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 22 636–640. [DOI] [PubMed] [Google Scholar]
- Epanechnikov, V. A., 1969. Non-parametric estimation of a multivariate probability density. Theory Probab. Appl. 14 153–158. [Google Scholar]
- Fan, J., and I. Gijbels, 1996. Local Polynomial Modeling and Its Applications. Chapman & Hall, London.
- Fisher, R. A., 1925. Statistical Methods for Research Workers. Oliver & Boyd, London.
- Glass, G. V., 1976. Primary, secondary, and meta-analysis of research. Educ. Res. 5 3–8. [Google Scholar]
- Good, I. J., 1955. On the weighted combination of significance tests. J. R. Stat. Soc. B 17 264–265. [Google Scholar]
- Guerra, R., C. J. Etzel, D. R. Goldstein and S. R. Sain, 1999. Meta-analysis by combining p-values: simulated linkage studies. Genet. Epidemiol. 17 S605–S609. [DOI] [PubMed] [Google Scholar]
- Hess, A., and H. Iyer, 2007. Fisher's combined p-value for detecting differentially expressed genes using Affymetrix expression arrays. BMC Genomics 8 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, S. Y., S. Shen, N. Zezza, E. K. Hoffman, M. Perlin et al., 2004. A genome wide search for alcoholism susceptibility genes. Am. J. Med. Genet. (Neuropsychiat. Genet.) B 128 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn, J. N., and M. J. Daly, 2005. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6 95–108. [DOI] [PubMed] [Google Scholar]
- Hoh, J., and J. Ott, 2003. Mathematical multi-locus approaches to localizing complex human trait genes. Nat. Rev. Genet. 4 701–709. [DOI] [PubMed] [Google Scholar]
- Hoh, J., A. Wille and J. Ott, 2001. Trimming, weighting, and grouping SNPs in human case-control association studies. Genome Res. 11 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, S. X. Xu, and N. Laird, 2001. The family based association test method: strategies for studying general genotype-phenotype associations. Euro. J. Hum. Genet. 9 301–306. [DOI] [PubMed] [Google Scholar]
- Hugot, J. P., M. Chamaillard, H. Zouali, S. Lesage, J. P. Cezard et al., 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411 599–603. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium, 2003. The International HapMap Project. Nature 426 789–796. [DOI] [PubMed] [Google Scholar]
- International Human Genome Mapping Consortium, 2001. A physical map of human genome. Nature 409 934–941. [DOI] [PubMed] [Google Scholar]
- Laird, M. N., and C. Lange, 2006. Family-based designs in the age of large-scale gene-association studies. Nat. Rev. Genet. 7 385–394. [DOI] [PubMed] [Google Scholar]
- Lappalainen, J., H. R. Kranzler, I. Petrakis, L. K. Somberg, G. Page et al., 2004. Confirmation and fine mapping of the chromosome 1 alcohol dependence risk locus. Mol. Psychiat. 9 312–319. [DOI] [PubMed] [Google Scholar]
- Lin, D., 2005. An efficient Monte Carlo approach to assessing statistical significance in genomic studies. Bioinformatics 21 781–787. [DOI] [PubMed] [Google Scholar]
- Manly, B. J. F., 1998. Randomization, Bootstrap and Monte Carlo Methods in Biology, Ed. 2. Chapman & Hall, New York.
- Matsuzaki, H., S. Dong, H. Loi, X. Di, G. Liu et al., 2004. Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat. Methods 1 109–111. [DOI] [PubMed] [Google Scholar]
- Morton, N. E., W. Zhang, P. Taillon-Miller, P. Y. Kwok and A. Collins, 2001. The optimal measure of allelic association. Proc. Natl. Acad. Sci. USA 98 5217–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadaraya, E. R., 1964. On estimating regression. Theory Prob. Appl. 9 141–142. [Google Scholar]
- Nelson, M. R., S. L. R. Kardia, R. E. Ferrell and C. F. Sing, 2001. A combinatorial partitioning method to identify multilocus genotypic partitions that predict quantitative trait variation. Genome Res. 11 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz, D., and N. M. Laird, 2000. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum. Hered. 50 211–223. [DOI] [PubMed] [Google Scholar]
- Ritchie, M. D., L. W. Hahn, N. Roodi, L. R. Bailey, W. D. Plummer et al., 2001. Multifactor dimensionality reduction reveals high-order interactions among estrogen metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 69 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sas Institute, 2005. SAS/Genetics User's Guide. Cary, NC.
- Sasieni, P. D., 1997. From genotypes to genes: doubling the sample size. Biometrics 53 1253–1261. [PubMed] [Google Scholar]
- Schaid, D. J., C. M. Rowland, D. E. Tines, R. M. Jacobson and G. A. Poland, 2002. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 70 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman, S. R., and B. Müller-Myhsok, 2005. Rapid Simulation of P value for product methods and multiple testing adjustment in association studies. Am. J. Hum. Genet. 76 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham, P., J. S. Bader, I. Craig, M. O'Donovan and M. Owen, 2002. DNA pooling: a tool for large-scale association studies. Nat. Rev. Genet. 3 862–871. [DOI] [PubMed] [Google Scholar]
- Shete, S., 2003. A note on the optimal measure of allelic association. Ann. Hum. Genet. 67 189–191. [DOI] [PubMed] [Google Scholar]
- Steemers, F. J., and K. L. Gunderson, 2007. Whole genome genotyping technologies on the BeadArray platform. Biotechnol. J. 2 41–49. [DOI] [PubMed] [Google Scholar]
- Stouffer, S. A., E. A. Suchman, L. C. Devinney, S. A. Star and R. M. Williams, Jr., 1949. The American Soldier, Vol. 1: Adjustment During Army Life. Princeton University Press, Princeton, NJ.
- Sun, Y. V., A. M. Levin, E. Boerwinkel, H. Robertson and S. L. R. Kardia, 2006. A scan statistic for identifying chromosomal patterns of SNP association. Genet. Epidemiol. 30 627–635. [DOI] [PubMed] [Google Scholar]
- Tippett, L. H., 1931. The Methods of Statistics. Williams and Norgate, London.
- Visscher, P. M., and S. Le Hellard, 2003. Simple method to analyze SNP-based association studies using DNA pools. Genet. Epidemiol. 24 291–296. [DOI] [PubMed] [Google Scholar]
- Wang, W. Y. S., B. J. Barratt, D. G. Clayton and J. A. Todd, 2005. Genome-wide association studies: theoretical and practical concerns. Nat. Rev. Genet. 6 109–118. [DOI] [PubMed] [Google Scholar]
- Watson, G. S., 1964. Smooth regression analysis. Sankhya A 26 359–372. [Google Scholar]
- Wellcome Trust Case Control Consortium, 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille, A., J. Hoh and J. Ott, 2003. Sum statistics for the joint detection of multiple disease loci in case-control association studies with SNP markers. Genet. Epidemiol. 25 350–359. [DOI] [PubMed] [Google Scholar]
- Yang, H. C., and C. S. J. Fann, 2007. Association mapping using pooled DNA, pp. 161–176 in Linkage Disequilibrium and Association Mapping, edited by A. Collins. Humana Press, Clifton, NJ. [DOI] [PubMed]
- Yang, H. C., C. C. Chang, C. Y. Lin, C. L. Chen, C. Y. Lin et al., 2005. a A genome-wide scanning and fine mapping study of COGA data. BMC Genet. 6 S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. C., C. C. Pan, R. C. Y. Lu and C. S. J. Fann, 2005. b New adjustment factors and sample size calculation in a DNA-pooling experiment with preferential amplification. Genetics 169 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. C., C. Y. Lin and C. S. J. Fann, 2006. A sliding-window weighted linkage disequilibrium test. Genet. Epidemiol. 30 531–545. [DOI] [PubMed] [Google Scholar]
- Zaykin, D. V., and K. Shibata, 2008. Genetic flip-flop without an accompanying change in linkage disequilibrium. Am. J. Hum. Genet. 82 794–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin, D. V., P. H. Westfall, S. S. Young, M. A. Karnoub, M. J. Wagner et al., 2002. a Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum. Hered. 53 79–91. [DOI] [PubMed] [Google Scholar]
- Zaykin, D. V., L. A. Zhivotovsky, P. H. Westfall and B. S. Weir, 2002. b Truncated product method for combining P values. Genet. Epidemiol. 22 170–185. [DOI] [PubMed] [Google Scholar]