

Abstract
Accurate and fast computation of quantitative genetic variance parameters is of great importance in both natural and breeding populations. For experimental designs with complex relationship structures it can be important to include both additive and dominance variance components in the statistical model. In this study, we introduce a Bayesian Gibbs sampling approach for estimation of additive and dominance genetic variances in the traditional infinitesimal model. The method can handle general pedigrees without inbreeding. To optimize between computational time and good mixing of the Markov chain Monte Carlo (MCMC) chains, we used a hybrid Gibbs sampler that combines a single site and a blocked Gibbs sampler. The speed of the hybrid sampler and the mixing of the single-site sampler were further improved by the use of pretransformed variables. Two traits (height and trunk diameter) from a previously published diallel progeny test of Scots pine (Pinus sylvestris L.) and two large simulated data sets with different levels of dominance variance were analyzed. We also performed Bayesian model comparison on the basis of the posterior predictive loss approach. Results showed that models with both additive and dominance components had the best fit for both height and diameter and for the simulated data with high dominance. For the simulated data with low dominance, we needed an informative prior to avoid the dominance variance component becoming overestimated. The narrow-sense heritability estimates in the Scots pine data were lower compared to the earlier results, which is not surprising because the level of dominance variance was rather high, especially for diameter. In general, the hybrid sampler was considerably faster than the blocked sampler and displayed better mixing properties than the single-site sampler.
MOST traits related to adaptation in nature and breeding improvement are influenced by many genes (polygenic traits; Lynch and Walsh 1998). In quantitative genetics, the genetic variation of a polygenic trait is generally estimated as the additive genetic variance (and as the heritability). Estimation of the heritability is of fundamental importance because this ratio affects the response that can be expected to natural and artificial selection in the considered population (Falconer and Mackay 1996). Hence, considerable attention has been devoted to the development of statistical methods for estimation of breeding values and of heritability (Henderson 1984; Searle et al. 1992; Lynch and Walsh 1998).
The genetic variance may be further partitioned into additive genetic and nonadditive genetic components. The dominance variance results from interactions between alleles at the same locus, whereas the epistatic variance is caused by interactions of alleles at different loci. Traditionally, complex crossing designs have been performed to estimate dominance and epistatic variances [e.g., North Carolina (NC)II designs and triple testcrosses; Kearsey and Pooni 1996; Lynch and Walsh 1998]. Henderson (1985a,b) showed how mixed-effects models based on polygenic component and pedigree information (often referred to as animal or individual models) could be used for inference of nonadditive genetic effects and variances in experiments of general design. Given that the additive, dominance, and epistatic genetic effects are uncorrelated in unselected, noninbred populations in linkage equilibrium (Cockerham 1954), it should theoretically be possible to estimate all these genetic effects by multiplication of various combinations of the additive and dominance relationship matrices and using these in the mixed-model equations. For example, the additive-by-additive epistatic relationship matrix would be obtained by elementwise multiplication of two additive genetic relationship matrices. Unfortunately, in practice, the epistatic relationship matrices will be multiples of each other, which can lead to problems of identifiability of the parameters in the statistical model (for a discussion about identifiability see Gelfand and Sahu 1999). Also, one should be careful with models that include a dominance component because the considered pedigree may be too small to lead to identifiable results (Misztal 1997). Estimation of the nonadditive genetic variance components is crucial for several reasons. First of all, it will produce more correct statistical estimates and as a result more accurate selection strategies could be practiced (Du and Hoeschele 2000). This is particularly important in collateral experimental designs (Henderson 1985b). In this case the standard additive model (which assumes that residual errors are uncorrelated with constant variance) can produce biased estimates of the additive genetic values because the simple residual variance structure is erroneous (Lynch and Walsh 1998). It has been found that nonadditive genetic effects can have a considerable effect on the ranking of breeding values (Wall et al. 2005). Second, the dominance itself is of interest because it is coupled to the expected level of inbreeding depression (Cockerham and Weir 1984). Hence, without dominance there would be no inbreeding depression and avoidance of inbreeding would be of less importance in the design of breeding and conservation programs.
Recently, it has also been advocated, from a theoretical perspective, that dominance and epistasis can be converted into additive genetic variance following bottlenecks and inbreeding (e.g., Willis and Orr 1993; Wang et al. 1998; Lopez-Fanjul et al. 2002; Barton and Turelli 2004). These findings have been supported to some extent by empirical studies on model organisms (e.g., García et al. 1994; Fernandez et al. 1995; Whitlock and Fowler 1999). Hallander and Waldmann (2007) investigated the importance of nonadditive genetic interactions when truncation selection was applied to a breeding population. They found that nonadditive variance initially could be converted into additive genetic variance during truncation selection (see also Fuerst et al. 1997). However, these issues need to be further investigated with pedigree-based statistical approaches.
Bayesian statistical methods have become very popular in genetics (Gianola and Fernando 1986; Shoemaker et al. 1999; Blasco 2001; Walsh 2001; Xu 2003; Beaumont and Rannala 2004) because posterior distributions summarize uncertainty (accuracy) around the point estimate in a probabilistic form. Markov chain Monte Carlo (MCMC) methods, used in Bayesian inference to approximate posterior distributions, were introduced to quantitative genetics in the first half of the 1990s (Wang et al. 1993; Sorensen et al. 1994), facilitated by the development of the Gibbs sampling algorithm (Geman and Geman 1984; Gelfand and Smith 1990). Gibbs sampling has been used for inference of many different quantitative genetic parameters, for example, in estimation of posteriors of additive, permanent environment, and maternal effects, and on multivariate data sets with missing data (Sorensen and Gianola 2002). From a variance component model perspective, Bayesian methods accounting for dominance and epistasis have been developed for finite-locus models (Du and Hoeschele 2000), which can be interpreted as finite-locus approximations to polygenic components. A polygenic component is also present in some Bayesian QTL mapping methods (Yi and Xu 2000; Lee and Van Der Werf 2006).
Whether a model with both additive and dominance components is preferable over a simpler additive model can be evaluated by model selection. Several different methods exist for model selection analysis of alternative statistical models. In frequentist statistics, likelihood-ratio tests (LRTs) and Akaike's information criterion (AIC) are most common, while the Bayes factor, the Bayesian information criterion (BIC), the deviance information criterion (DIC), and the posterior predictive loss statistic are tools proposed in the Bayesian literature. Recent analyses of LRTs and AIC have shown that those methods may yield incorrect results in mixed models (Crainiceanu and Ruppert 2004; Vaida and Blanchard 2005). The Bayesian model comparison approaches are more general, but also need to be further evaluated.
In this article, we formulate a fast Bayesian Gibbs sampling approach for estimation of additive and dominance genetic variances in the infinitesimal animal (or individual) model applied to experiments of general design without inbreeding. We use a hybrid Gibbs sampler, which is a combination of the fast but slow-mixing single-site Gibbs sampling algorithm (e.g., Sorensen and Gianola 2002) and the slow but fast-mixing blocked Gibbs sampling algorithm (Carcía-Cortés and Sorensen 1996). The novelty of our approach is the use of variable transformations, where the covariance structures (i.e., the inverse of the additive and dominance relationship matrices) of the new transformed variables are diagonal, which considerably speeds up the computations of both Gibbs samplers and improves the mixing of the single-site Gibbs sampler. The method is applied to data from two traits (height and trunk diameter) of a previously published quantitative genetic study on Scots pine (Waldmann and Ericsson 2006) and to simulated data from a large NCII design with both high and low dominance variance. Moreover, we use the posterior predictive loss criteria to compare models with different numbers of variance components (Laud and Ibrahim 1995; Gelfand and Gosh 1998).
METHODS
Model:
Henderson (1985a,b) showed how standard linear model methods could be used for combined estimation of additive and dominance genetic variance components. The mixed model
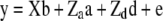 |
(1) |
was used, where y is a vector of phenotypic records on n individuals, b is a vector of fixed effects including environmental covariates, and a (additive) and d (dominance) are normally distributed vectors of random genetic effects with covariances 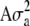 and
and 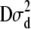 , respectively. Here, e is the vector of error terms, where each component
, respectively. Here, e is the vector of error terms, where each component 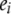 is independently normally distributed with mean zero and variance
is independently normally distributed with mean zero and variance 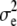 . Further, X,
. Further, X, 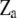 , and
, and 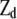 are known incidence matrices associating b, a, and d with y, respectively. A is the additive genetic relationship matrix and it describes additive genetic covariance between relatives (Henderson 1984), and several methods for computation of A and its inverse from general pedigrees exist (e.g., Henderson 1976). It should be emphasized that the actual size of the pedigree q can be larger than the number of individuals with records n (see the discussion for further details). In the absence of inbreeding, the elements of the dominance relationship matrix (D) can be calculated on the basis of the values of A as follows. Let the parents of individual i be indexed with k and l and those of individual j with m and n; then
are known incidence matrices associating b, a, and d with y, respectively. A is the additive genetic relationship matrix and it describes additive genetic covariance between relatives (Henderson 1984), and several methods for computation of A and its inverse from general pedigrees exist (e.g., Henderson 1976). It should be emphasized that the actual size of the pedigree q can be larger than the number of individuals with records n (see the discussion for further details). In the absence of inbreeding, the elements of the dominance relationship matrix (D) can be calculated on the basis of the values of A as follows. Let the parents of individual i be indexed with k and l and those of individual j with m and n; then 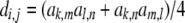 , where
, where 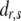 and
and 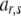 are elements (row r and column s) of matrices D and A, respectively. The elements on the diagonal of D are 1. In the case of inbreeding, the simulation approach of Ovaskainen et al. (2007) can be utilized to infer elements of D. However, the presence of inbreeding induces nonzero covariances that complicate the estimation significantly (see De Boer and Hoeschele 1993). Thus, we consider only the case of no inbreeding here.
are elements (row r and column s) of matrices D and A, respectively. The elements on the diagonal of D are 1. In the case of inbreeding, the simulation approach of Ovaskainen et al. (2007) can be utilized to infer elements of D. However, the presence of inbreeding induces nonzero covariances that complicate the estimation significantly (see De Boer and Hoeschele 1993). Thus, we consider only the case of no inbreeding here.
Bayesian inference on transformation of relationship matrices:
To improve on the speed achieved, we introduce an approach that utilizes transformations of the relationship matrices. By applying such transformations to the original genetic variables the phenotype model (1) can be rewritten as
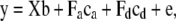 |
(2) |
where 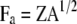 ,
, 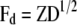 ,
, 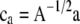 , and
, and 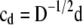 (see the appendix; cf. Mrode and Thompson 1989).
(see the appendix; cf. Mrode and Thompson 1989).
In Bayesian inference the assumptions regarding the statistical phenotype model define a likelihood function and the unknown model parameters are assigned prior distributions. The likelihood function associated with the phenotype model in Equation 2 is
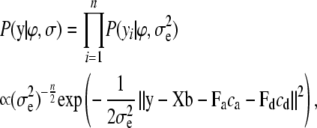 |
(3) |
where 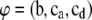 are the unknown location effects,
are the unknown location effects, 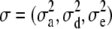 are the variance components, and
are the variance components, and  denotes the Euclidean norm of
denotes the Euclidean norm of  . By Bayes' theorem the joint posterior density of the unknown parameters is proportional to the joint density of the data and parameters that can be further factorized as
. By Bayes' theorem the joint posterior density of the unknown parameters is proportional to the joint density of the data and parameters that can be further factorized as
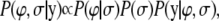 |
(4) |
where 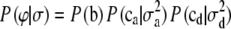 and
and 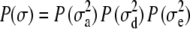 are the prior densities of the location effects and variance components, respectively.
are the prior densities of the location effects and variance components, respectively.
Each term 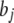 in b is assigned the commonly used improper flat prior distribution
in b is assigned the commonly used improper flat prior distribution
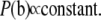 |
(5) |
For the transformed genetic effects we adopt the prior assumption
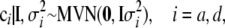 |
(6) |
where 0 is a vector of q zeros. The prior assumption (6) is identical to positing prior 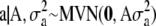 and
and 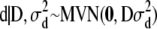 on the original effects, because
on the original effects, because 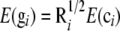 = 0 and
= 0 and 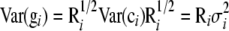 for
for 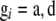 and
and 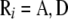 . The scaled inverted chi-square distribution is a practical choice as a prior for the variance components
. The scaled inverted chi-square distribution is a practical choice as a prior for the variance components
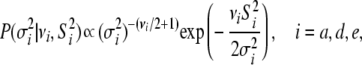 |
(7) |
where 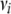 can be interpreted as a degree of belief parameter (larger values indicate higher confidence) and
can be interpreted as a degree of belief parameter (larger values indicate higher confidence) and 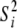 is thought of as a prior value for the appropriate variance (Sorensen and Gianola 2002). We chose to set
is thought of as a prior value for the appropriate variance (Sorensen and Gianola 2002). We chose to set 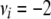 and
and 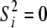 to obtain flat priors (but see below for alternative priors for the simulated data with low dominance variance). Gelman (2006) provides a useful discussion of alternative priors.
to obtain flat priors (but see below for alternative priors for the simulated data with low dominance variance). Gelman (2006) provides a useful discussion of alternative priors.
For the fixed and transformed genetic effects 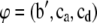 the fully conditional posterior distribution is
the fully conditional posterior distribution is
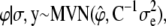 |
(8) |
where
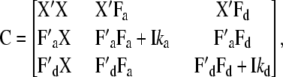 |
(9) |
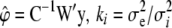 , and
, and 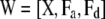 . The fully conditional posterior distribution of
. The fully conditional posterior distribution of 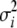 is
is
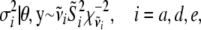 |
(10) |
where 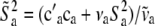 ,
, 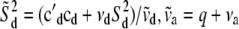 , and
, and 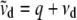 . For the residual variance,
. For the residual variance, 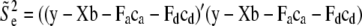 +
+ 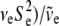 , where
, where 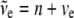 .
.
García-Cortés and Sorensen (1996) described a blocked Gibbs sampling approach for Gaussian linear models. In general, blocked Gibbs sampling has a faster convergence rate and better mixing when correlated parameters (e.g., because of family relations) are present in the data. The García-Cortés and Sorensen (1996) approach avoids calculation of the matrix inverse of C in each iteration, but the requirement for solutions to large equation systems still makes this algorithm slow. Instead, we combine the faster single-site Gibbs sampler (Sorensen and Gianola 2002) and the blocked Gibbs sampler (García-Cortés and Sorensen 1996) into a hybrid Gibbs sampler that is described in the appendix. The benefit of presenting the model in the transformed form is that the new variables are a priori statistically independent, which enables huge savings (at least fivefold) in the implementation of the Gibbs sampler of García-Cortés and Sorensen (1996). For example, sampling from a multivariate normal distribution reduces to sampling independent one-dimensional normal components (see the appendix). An important property of the approach is that the variance components 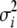 are the same in both approaches and therefore the estimates obtained using the transformed variables can be directly interpreted as those of the original variables. Thus the same prior distributions are assigned to the variances and also the likelihood function stays the same. Further, if needed, estimates of the original breeding values (effect sizes) can be attained from the transformed counterparts using the back-transformations as
are the same in both approaches and therefore the estimates obtained using the transformed variables can be directly interpreted as those of the original variables. Thus the same prior distributions are assigned to the variances and also the likelihood function stays the same. Further, if needed, estimates of the original breeding values (effect sizes) can be attained from the transformed counterparts using the back-transformations as 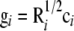 . This allows complete MCMC paths of the original effect sizes to be derived ex post facto, with only one set of matrix multiplications.
. This allows complete MCMC paths of the original effect sizes to be derived ex post facto, with only one set of matrix multiplications.
Posterior predictive loss criteria:
Many different methods exist to evaluate and perform a selection among competing statistical models. See the discussion for more information about different model comparison approaches. We decided to use the posterior predictive loss approach that has been found to work well for most exponential family models (Laud and Ibrahim 1995; Gelfand and Gosh 1998). Prediction is here based on generating new replicates 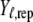 of the observed data,
of the observed data, 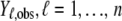 . The criterion is calculated as
. The criterion is calculated as
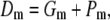 |
(11) |
where
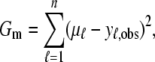 |
(12) |
and
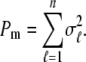 |
(13) |
Here 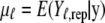 is the posterior predictive mean, and
is the posterior predictive mean, and 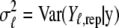 is the corresponding variance.
is the corresponding variance. 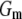 is a goodness-of-fit term, whereas
is a goodness-of-fit term, whereas 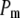 is a penalty term. For poor models one would expect large predictive variance and poor fit. When the model improves, both terms should get better (i.e., smaller). Eventually, once the model becomes overfitted,
is a penalty term. For poor models one would expect large predictive variance and poor fit. When the model improves, both terms should get better (i.e., smaller). Eventually, once the model becomes overfitted, 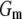 will still decrease but the variance will become inflated (i.e.,
will still decrease but the variance will become inflated (i.e., 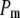 increases). Hence, the model with smallest
increases). Hence, the model with smallest 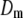 is preferred. Computation of
is preferred. Computation of 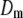 is described in the appendix. For comparative purposes, we derived restricted maximum-likelihood (REML) estimates of the variance components and calculated AIC (on the basis of the rank of the incidence matrix of the fixed effects plus the number of variance components) statistics using ASReml software (Gilmour et al. 2006).
is described in the appendix. For comparative purposes, we derived restricted maximum-likelihood (REML) estimates of the variance components and calculated AIC (on the basis of the rank of the incidence matrix of the fixed effects plus the number of variance components) statistics using ASReml software (Gilmour et al. 2006).
We also compared the breeding values of the additive and additive plus dominance models by ranking the mode of the posterior distribution of the additive effects for both traits of the Scots pine data. We used the mode of the posterior distributions because it provided the point estimates that were closest to the predicted breeding values obtained by ASReml. To compare the rank, the top 100 individuals from the additive plus dominance models were matched with their rank obtained with the additive models. Correlations between the rank vectors and the average difference in rank were computed using the software R (R Project 2002).
Scots pine data:
Real data from a 26-year-old Scots pine (Pinus sylvestris L.) progeny test in northern (64°18′N) Sweden were analyzed. Fifty-two unrelated parent trees were crossed according to an approximate circulant partial diallel design (Kempthorne and Curnow 1961). In 1971, ∼8000 seedlings were planted out unrestricted, randomly using 2.2-m2 spacing. The plantation was thoroughly mapped and subdivided into 70 nearly square blocks that are used as a fixed effect in our statistical models. The remaining trees (4970 individuals) were measured in 1997 for various traits of breeding interest. In this study we concentrate on total tree height and diameter (at 130 cm above ground). No data were available from the parent trees. Moreover, we did not standardize the data as in Waldmann and Ericsson (2006), and comparison with their results should be done bearing this in mind. For further details of the experiment, see Waldmann and Ericsson (2006). Bivariate analysis was not considered here because of computational limitations.
Simulated data:
A North Carolina design II (Lynch and Walsh 1998) was used to generate the pedigree for the simulated data. We decided to focus on two large data sets instead of many small ones because of identifiability problems with the dominance variance (detected in some earlier test runs). Hence, 70 unrelated parents were crossed so that each of the 35 mothers was mated with all 35 fathers and each mating resulted in 3 offspring (totally 3675 offspring). The A and D matrices were calculated from this design as described in the Model section. The population mean (μ) was set to 100, the random polygenic effects were drawn from 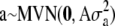 and
and 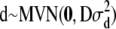 , and the error terms
, and the error terms 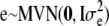 . Data on phenotypic records were generated also for the parents. To evaluate that the posterior predictive loss statistic penalized overfitted models, two data sets were generated with two different levels of the variance components: one data set using
. Data on phenotypic records were generated also for the parents. To evaluate that the posterior predictive loss statistic penalized overfitted models, two data sets were generated with two different levels of the variance components: one data set using 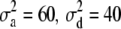 , and
, and 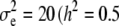 and
and 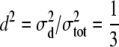 ), and the other data set employing
), and the other data set employing 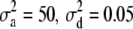 , and
, and 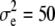 (
(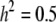 and
and 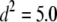 × 10−4). The phenotypic values (y) for each individual were then generated by summing the effects according to
× 10−4). The phenotypic values (y) for each individual were then generated by summing the effects according to 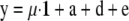 , where 1 is a vector containing only ones and of the size equal to the number of individuals in the simulated design. Both simulated data sets were analyzed with full
, where 1 is a vector containing only ones and of the size equal to the number of individuals in the simulated design. Both simulated data sets were analyzed with full 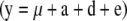 and reduced
and reduced 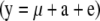 models to test that the
models to test that the 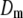 statistic selected the correct model.
statistic selected the correct model.
RESULTS
Scots pine data:
On the basis of visual inspection of some preliminary MCMC chains, we decided to use one chain of 225,000 iterations per trait. We set the burn-in to 25,000 iterations and thinned every 10th iteration, yielding a total sample of 20,000 iterations for both traits. The mode of the posterior densities of the parameters was obtained using a kernel density approach (Hoti et al. 2002; Waldmann and Ericsson 2006). The highest probability density (HPD) intervals were defined throughout this article as 95% of the posterior distributions, and they were estimated by using language R, library boa (Smith 2005).
Summary results of the analyses of both tree diameter and height are shown in Table 1. The posterior distributions were skewed and therefore the mode, median, and mean estimates were different for both the additive and the dominance variance components (Table 1). However, the distribution of the residual variance was closer to normality. Mean, median, and mode estimates of the posterior distributions were larger for the dominance variance than for the additive variance. Moreover, the lower and upper HPD interval estimates were higher for the dominance variance than for the additive variance. For tree diameter, the summary point estimates of the posterior distribution for the dominance proportion were higher than the corresponding estimates of the posterior distribution for the heritability. Also, the lower and upper HPD estimates were larger for the dominance proportion.
TABLE 1.
Point and interval (95% highest posterior density) estimates of the variance components, narrow-sense heritability h2, and dominance proportion d2 of height and diameter in Scots pine from the Gibbs sampling analyses of the full models
| Diameter
|
Height
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Median | Mode | REML | HPD 2.5 | HPD 97.5 | Mean | Median | Mode | REML | HPD 2.5 | HPD 97.5 | |
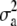 |
62.52 | 59.62 | 54.70 | 55.95 (17.38) | 27.67 | 103.7 | 33.61 | 32.76 | 32.01 | 30.25 (7.20) | 18.89 | 50.56 |
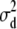 |
88.41 | 85.71 | 82.88 | 83.06 (25.17) | 39.70 | 142.2 | 16.78 | 16.44 | 16.27 | 16.10 (4.41) | 8.632 | 26.46 |
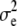 |
721.7 | 722.6 | 722.2 | 728.5 (26.97) | 665.3 | 776.8 | 104.2 | 103.0 | 103.8 | 104.8 (5.57) | 90.01 | 114.0 |
| h2 | 0.0714 | 0.0685 | 0.0630 | 0.0645 | 0.0327 | 0.1170 | 0.2183 | 0.2144 | 0.2103 | 0.2002 | 0.1299 | 0.3143 |
| d2 | 0.1014 | 0.0983 | 0.0939 | 0.0957 | 0.0457 | 0.1616 | 0.1095 | 0.1075 | 0.1052 | 0.1065 | 0.0549 | 0.1696 |
REML estimates were obtained with ASReml. REML standard errors are presented in parentheses.
The difference in estimates of mode, median, and mean for additive variance of height suggests that the additive variance posterior distribution was somewhat skewed, whereas the densities of both dominance and residual variance were close to normality (Table 1). The heritability was considerably higher for height than for diameter. This result has been found in several studies in conifer species (e.g., Yanchuk 1996; Fries and Ericsson 1998; Waldmann and Ericsson 2006). The REML estimates agreed well with the Bayesian point estimates, but were generally closer to the mode and median than to the mean (Table 1).
The results of the model comparison analysis for the Scots pine data using the posterior predictive loss criterion are presented in Table 2. As expected, inclusion of both additive and dominance terms yielded a lower Dm compared to the simpler models for both traits. The difference is relatively small between the additive and the full model for diameter. For height on the other hand, the difference in Dm is larger between the models and the full model is clearly the preferred one. The fixed-effect model gave the largest predictive variance and the worst fit (highest level of Pm and Gm, respectively) for both traits. Also AIC was clearly lowest for the full models for both traits (Table 2). Hence, it seems as if the dominance term improves the statistical models and gives a better fit to the data compared to simpler models.
TABLE 2.
Model choice parameters for the Scots pine data
| Diameter (×105)
|
Height (×105)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Model for y | Gm | Pm | Dm | AIC | Gm | Pm | Dm | AIC |
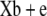 |
46.45 | 47.75 | 94.20 | 0.18492 | 84.01 | 86.59 | 170.6 | 0.09909 |
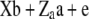 |
41.67 | 46.95 | 88.63 | 0.18378 | 61.10 | 79.86 | 140.9 | 0.09498 |
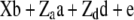 |
41.61 | 46.07 | 87.68 | 0.18361 | 49.04 | 78.95 | 128.0 | 0.09473 |
A lower posterior predictive loss statistic 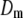 indicates a better model.
indicates a better model. 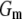 is a goodness-of-fit term, whereas
is a goodness-of-fit term, whereas 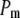 is a penalty term. AIC is the standard Akaike's information criterion (calculated as number of fixed-effect classes plus number of variance components).
is a penalty term. AIC is the standard Akaike's information criterion (calculated as number of fixed-effect classes plus number of variance components).
The difference in ranks of the additive effects (mode estimates) of the top 100 individuals from the additive and additive plus dominance models is plotted in Figures 1 (diameter) and 2 (height). The correlation between the rank positions of the 100 highest-ranked individuals was 0.668 for diameter and 0.696 for height. Thirteen of the top 100 individuals from the additive plus dominance model were not among the top 100 individuals in the additive model for diameter. The corresponding value for height was 21 of 100.
Figure 1.—

Rank of the additive genetic effects for the additive (A) and additive plus dominance (A + D) models of Scots pine diameter. The y-axis plots the position in the A model of the 100 highest ranked individuals in the A + D model. The straight line indicates no discrepancy in rank between the two models.
Figure 2.—

Rank of the additive genetic effects for the additive (A) and additive plus dominance (A + D) models of Scots pine height. The y-axis plots the position in the A model of the 100 highest ranked individuals in the A + D model. The straight line indicates no discrepancy in rank between the two models.
Simulated data:
The hybrid Gibbs sampler with block update every 50th iteration was almost 100 times faster than the pure blocked Gibbs sampler (0.035 sec/iteration compared to 3.0 sec/iteration) when applied to a simulated data set containing 850 individuals. Moreover, the nonnull autocorrelations extended over considerably less iterations for both the additive and the dominance variances in the hybrid sampler than in the single-site sampler (implemented following Sorensen and Gianola 2002). For example, the lag-10 autocorrelation decreased from 0.61 (single site) to 0.22 (hybrid) and from 0.95 (single site) to 0.64 (hybrid) for the additive and dominance variance, respectively. Note that these numbers reflect the MCMC efficiency similarly as the effective sample size, because the latter can be estimated as a direct function of autocorrelation.
The flat priors worked fine with the simulated data with high dominance variance. Once again, the mode and REML estimates agreed best (Table 3). That the additive variance was slightly higher than the expected value of 60 could be attributed to the stochastic generation of the simulated data. Evaluation of a large number of large simulated data sets is currently not computationally feasible for the Gibbs sampler. For the simulated data with low dominance variance, the uninformative prior with degree of belief parameter (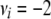 ) resulted in considerable overestimation of the dominance variance (Table 3). Therefore, we also chose to use an informative prior with a degree of belief of 1% on the dominance variance, which was obtained by setting
) resulted in considerable overestimation of the dominance variance (Table 3). Therefore, we also chose to use an informative prior with a degree of belief of 1% on the dominance variance, which was obtained by setting 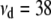 (Wang et al. 1994). With this prior the posterior point estimates of
(Wang et al. 1994). With this prior the posterior point estimates of 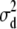 from the Gibbs sampler matched the REML estimate much better.
from the Gibbs sampler matched the REML estimate much better.
TABLE 3.
Point and interval estimate summary of the variance components, narrow-sense heritability h2, and dominance proportion d2 of the simulated data with high and low dominance variance
| Low dominance
|
High dominance
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Median | Mode | REML | HPD 2.5 | HPD 97.5 | Mean | Median | Mode | REML | HPD 2.5 | HPD 97.5 | |
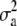 |
50.18 | 49.74 | 47.92 | 48.95 (7.45) | 36.58 | 63.90 | 69.74 | 68.70 | 65.46 | 66.57 (11.34) | 47.42 | 93.80 |
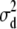 |
4.242 | 3.657 | 1.990 | 2.00 × 10−6 (0.0) | 0.05494 | 9.906 | 40.13 | 40.26 | 39.78 | 39.05 (6.92) | 26.37 | 53.37 |
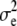 |
47.77 | 48.09 | 48.94 | 52.36 (3.98) | 38.26 | 56.10 | 17.66 | 17.94 | 18.83 | 20.14 (8.68) | 0.0175 | 32.75 |
| h2 | 0.4893 | 0.4878 | 0.4837 | 0.4832 | 0.3868 | 0.5865 | 0.5436 | 0.5414 | 0.5284 | 0.5293 | 0.4170 | 0.6784 |
| d2 | 4.15 × 10−2 | 3.58 × 10−2 | 1.97 × 10−2 | 1.97 × 10−8 | 5.63 × 10−4 | 9.72 × 10−2 | 0.3152 | 0.3158 | 0.3134 | 0.3105 | 0.2059 | 0.4223 |
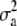 a a
|
50.20 | 49.82 | 49.39 | — | 36.38 | 64.20 | — | — | — | — | — | — |
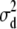 a a
|
1.03 × 10−5 | 1.00 × 10−5 | 1.00 × 10−5 | — | 1.00 × 10−5 | 1.10 × 10−5 | — | — | — | — | — | — |
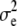 a a
|
51.89 | 51.98 | 52.76 | — | 44.66 | 59.50 | — | — | — | — | — | — |
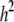 a a
|
0.4899 | 0.4893 | 0.4874 | — | 0.3855 | 0.5900 | — | — | — | — | — | — |
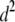 a a
|
1.02 × 10−7 | 1.00 × 10−7 | 9.85 × 10−8 | — | 8.99 × 10−8 | 1.17 × 10−7 | — | — | — | — | — | — |
Simulated values were 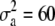 ,
, 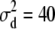 , and
, and 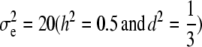 for the high dominance data and
for the high dominance data and 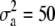 ,
, 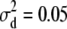 , and
, and 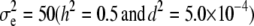 for the low dominance. The Gibbs sampling results are based on MCMC chains with uninformative priors, except for those listed in footnote a. REML standard errors are presented in parentheses.
for the low dominance. The Gibbs sampling results are based on MCMC chains with uninformative priors, except for those listed in footnote a. REML standard errors are presented in parentheses.
A degree of belief of 1% on 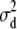 .
.
We also calculated correlations between the simulated random effects and the estimated posterior modes of all individuals of both the additive and dominance effects from the full additive plus dominance model with high genetic variances. The correlation between the additive effects was 0.804 and between the dominance effects 0.662. The corresponding correlations for the REML estimates were 0.805 and 0.667, respectively.
The results of the model comparison analysis for the simulated data are presented in Table 2. When the analyses of the data set with high dominance variance were performed with uninformative priors, Dm was lowest for the full model (Table 4). Once the uninformative priors were used for all parameters on the data with low dominance, Dm turned out to be smallest for the full model. The reason for this is the overestimation of 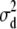 . On the other hand, with the informative prior on
. On the other hand, with the informative prior on 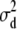 , Dm could rightly select the additive model as best fit. AIC correctly picked up the additive model for the low dominance data and the full model for the high dominance data (Table 4).
, Dm could rightly select the additive model as best fit. AIC correctly picked up the additive model for the low dominance data and the full model for the high dominance data (Table 4).
TABLE 4.
The values of model choice parameters for simulated data
| Low dominance (×105)
|
High dominance (×105)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Model for y | Gm | Pm | Dm | AIC | Gm | Pm | Dm | AIC |
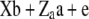 |
1.291 | 2.637 | 3.927 | 0.201854 | 1.469 | 3.126 | 4.596 | 0.20871 |
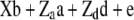 |
1.097 | 2.658 | 3.756 | 0.201874 | 0.1279 | 2.401 | 2.530 | 0.20835 |
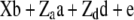 a a
|
1.296 | 2.640 | 3.936 | — | — | — | — | — |
A lower posterior predictive loss statistic 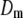 indicates a better model.
indicates a better model. 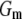 is a goodness-of-fit term, whereas
is a goodness-of-fit term, whereas 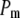 is a penalty term. The posterior predictive losses are based on MCMC chains with uninformative priors, except for that listed in footnote a. AIC is the standard Akaike's information criterion (calculated as number of fixed-effect classes plus number of variance components).
is a penalty term. The posterior predictive losses are based on MCMC chains with uninformative priors, except for that listed in footnote a. AIC is the standard Akaike's information criterion (calculated as number of fixed-effect classes plus number of variance components).
A degree of belief of 1% on 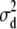 .
.
DISCUSSION
In this study, we have developed an efficient strategy for estimation of genetic parameters including dominance variance by using Bayesian inference and variable transformation. The method performs well when evaluated on both real and simulated data. Moreover, the narrow-sense heritability in this study was lower than what was found by Waldmann and Ericsson (2006) for a pure additive model on the same data. Hence, depending on the experimental design and trait data, it is sometimes important to estimate both additive and nonadditive genetic components in populations. Otherwise, ranking of breeding values may be nonoptimal and heritability estimates can be overestimated, sometimes considerably.
Impact of nonadditive genetic effects on breeding:
In dairy cattle, Wall et al. (2005) investigated the effects of inbreeding, heterosis, recombination loss, and migration on fertility (fitness) traits and milk production. One of the purposes of the study was to examine if nonadditive effects changed the estimates of breeding values and the ranking of bulls. They reported that nonadditive effects had an effect on fertility traits and milk production, although not very dramatic. In addition, there was a difference in ranking of bulls if nonadditive effects were included, which resulted in considerable changes in rank for some individuals. In our study, the ranking of the additive genetic effects differs considerably between the additive and additive plus dominance models for both Scots pine traits. For the top 100 individuals, the risk of selecting a nonoptimal candidate is as high as 21% (height) and 13% (diameter). This shows that it could be important to include nonadditive effects in ranking and selection of individuals in breeding programs, to the extent that they exist.
Moreover, in a simulation study (Varona and Misztal 1999), it was shown that inclusion of the dominance component could contribute an increase in genetic response to selection of ∼10% compared to selection based on a pure additive model (in pedigree designs with specific combining abilities). The highest increase in selection response occurred when the additive heritability was low, the dominance heritability high, selection intensity low, and proportion of full sibs high (Varona and Misztal 1999). The design and results from the Scots pine analysis coincide well with these findings and we could therefore expect that inclusion of the dominance component will increase the selection response in future generations of this population, especially if the breeding target is diameter.
Empirical estimates of nonadditive genetic effects:
The association between dominance variance and selection was examined in a comprehensive review by Crnokrak and Roff (1995). Their compilation of studies showed that the level of dominance variance varied between different trait categories for wild species. Dominance was highest for life-history traits, slightly lower for physiological traits, and lowest for morphological traits (VD/VA = 1.17, VD/VA = 1.06, and VD/VA = 0.19, respectively). Moreover, they found that there was no general trend in the level of dominance variance between different trait categories for domestic species, but VD/VA was relatively high (between 0.79 and 0.91).
In a recent review, Roff and Emerson (2006) compiled studies of both dominance and epistasis from line-cross experiments. They found that dominance interactions existed in almost all studies of both life-history and morphological traits (96.5 and 97.4%, respectively). But, the ratio of dominance to additive effects in life-history traits was twice as high as for morphological traits. Epistatic interactions were less common than dominance, but still found in 79.4 and 67.1% for life-history and morphological traits, respectively. Similar to the case of dominance, the ratio of epistatic to additive effects was higher in life-history than in morphological traits.
Nonadditive parameters in forest trees:
Historically, attention to nonadditive variance components has been limited in forest tree breeding. Unfortunately, many studies on nonadditive variance components in forest trees are based on far too small data sets and therefore we restrict our discussion to a few well-designed experiments. Fries and Ericsson (1998) estimated the dominance variance in six different traits in Scots pine and found high dominance values, especially for tree diameter (VD/VA = 2.80). However, they could not estimate the dominance variation in tree height. In loblolly pine, Jansson and Li (2004) estimated the variance ratio of specific combining ability to general combining ability 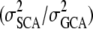 in growth volume at 0.36, while Balocchi et al. (1993) investigated age trends in height and reported that VD/VA varied between 0.20 and 4.42 over time. In radiata pine, Wu and Matheson (2004, 2005) estimated
in growth volume at 0.36, while Balocchi et al. (1993) investigated age trends in height and reported that VD/VA varied between 0.20 and 4.42 over time. In radiata pine, Wu and Matheson (2004, 2005) estimated 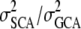 for tree height at 0.90 and 0.95, respectively. Wu and Matheson (2004) suggested that special mating designs should be applied to utilize nonadditive variance in breeding purposes while, on the other hand, Jansson and Li (2004) argued that only additive variance should be taken into consideration in breeding programs. Also in Douglas fir, Yanchuk (1996) used a large data set to estimate nonadditive variance. They concluded that the level of additive variance was on average three times greater compared to nonadditive variance and that diameter had greater levels of nonadditive variance compared to tree height. In conclusion, there seems to be no general trend about the level of dominance compared to additive variance, but it often seems as if at least some dominance is present.
for tree height at 0.90 and 0.95, respectively. Wu and Matheson (2004) suggested that special mating designs should be applied to utilize nonadditive variance in breeding purposes while, on the other hand, Jansson and Li (2004) argued that only additive variance should be taken into consideration in breeding programs. Also in Douglas fir, Yanchuk (1996) used a large data set to estimate nonadditive variance. They concluded that the level of additive variance was on average three times greater compared to nonadditive variance and that diameter had greater levels of nonadditive variance compared to tree height. In conclusion, there seems to be no general trend about the level of dominance compared to additive variance, but it often seems as if at least some dominance is present.
Statistical issues:
Use of family-based models for estimation of dominance variance (via the interaction factor) may bias both the additive and the dominance variance estimates because the information in the pedigree is not used simultaneously. Pedigree-based approaches (i.e., animal models) have been used several times in animal breeding for estimation of dominance variances (e.g., Hoeschele and Vanraden 1991; Misztal 1997). Typically, genetic evaluations are performed in populations of nonrandom mating (e.g., breeding populations), where the animal model accounts for selection in multiple generations unlike family-based approaches (Kennedy et al. 1988). Furthermore, estimates of general and specific combining abilities using a family-based model will include a portion of higher-order epistatic terms (Lynch and Walsh 1998) that generally are ignored. As a result, overestimations of both additive and dominance variances may occur.
In the animal model, the number of random effects to be estimated is typically large compared to the number of observations, resulting in an overparameterized system of equations that may result in nonidentifiability of the parameters. This is especially challenging if several genetic factors are fitted in the model. One can improve the problem of identifiability by adding extra generations without records to the pedigree. In other words, using complicated pedigrees to separate the nonadditive variance components from the additive components is more efficient because the nonadditive identical-by-descent matrices tend to have more nonzero elements in complicated pedigrees than in simple pedigrees (Mao and Xu 2005).
Informative priors can be used to make parameters more identifiable in Bayesian models (Gelfand and Sahu 1999; Sorensen and Gianola 2002). It is also well known in statistics that repeated and correlated trait measurements will alleviate the problem of identifiability. The use of a more economical parameterization could also help, for example, the sire or reduced animal model where nonparental breeding values are expressed in terms of parental breeding values (Quaas and Pollak 1980). Alternatively, the number of equations can be reduced by manipulating the coefficient matrix of animal models, which leads to asymmetric coefficient matrices (see Henderson 1984; Lynch and Walsh 1998). Finally, it is also possible to use a finite-locus approximation to infinite-locus models for estimation of nonadditive parameters (e.g., Du and Hoeschele 2000). A general drawback, however, is that the estimates depend on the number of loci used.
We used a posterior predictive loss criterion to compare models including different numbers of random genetic components. In frequentist statistics it is common to perform hypothesis testing of genetic variance components using LRTs, which evaluate whether a reduced model gives the same fit to the data as a full model. In general, this test performs well for two-sided hypotheses if the sample size is large because the distribution of LRTs then follows a chi-square distribution, asymptotically. On the other hand, when the amount of data increases, all hypotheses eventually become statistically significant (Sillanpää and Auranen 2004) and, for bounded parameters like variance components, the null distribution might be difficult to estimate in general (Crainiceanu and Ruppert 2004). Consequently, in these situations, approximating the distribution of LRTs using a chi-square distribution can give incorrect results. Another commonly used statistic is AIC that uses both likelihood (goodness of fit) and a penalty terms that corresponds to the number of parameters (K) in the model. K is typically calculated as the rank of the design matrix of the fixed effects plus the number of variances. Vaida and Blanchard (2005) argued that when the focus of inference is on the random effects in mixed models, K needs to be substituted with a parameter referred to as the effective number of parameters (which takes correlation between parameters into account). Estimation of the effective number of parameters is not straightforward and was not performed in our analysis.
In Bayesian analysis, the Bayes factor has been used for testing polygenic genetic parameters in animal models both with and without molecular markers (García-Cortés et al. 2001). However, the Bayes factor is suitable only for models with fully proper priors and is sensitive to the parameterization of the model. The DIC was recently proposed as a general model choice criterion and has been used for evaluation in animal models (e.g., Rekaya et al. 2003). Sorensen and Waagepetersen (2003) provide an extensive discussion of different Bayesian model comparison criteria for animal models. In this study, after disappointing initial experiments with the DIC, we have chosen to use the posterior predictive loss approach. The results of the model comparison of the Scots pine data showed that models including both additive and dominance effects should be favored over the reduced models. The analyses of the simulated data also showed that Dm worked, but was sensitive to the levels of the variance components and we recommend estimation of this statistic from several runs with different priors in the case of small variances.
Damgaard (2007) used a transformation for breeding values based on the ideas from Mrode and Thompson (1989) to obtain a priori uncorrelated breeding values for application of single-site Gibbs sampling. During development of our method, we tried a similar approach for large pedigrees considered here but encountered mixing problems due to too high a posteriori correlation of breeding values (results not shown). To overcome this mixing problem here, we propose the hybrid sampler. Moreover, we do not rescale variance components to unit variance in our transformation and therefore we are not required to apply a back-transformation. Two other Gibbs sampling approaches for additive and dominance models were presented by Chalh and El Gazzah (2004). The first approach obtains estimates from an animal model with only an additive (A) component and then calculates dominance effects as a direct function of additive effects. The second approach is based on modifying mixed model equation residuals from the animal model with only additive (A) components. The speedup compared to the traditional additive plus dominance single-site Gibbs sampler was considerable, but further comparative analyses are required regarding mixing properties.
Acknowledgments
This work was supported by The Research School in Forest Genetics and Breeding, Swedish Agricultural University (SLU), Umeå, Sweden, and by a research grant (202324) from the Academy of Finland.
APPENDIX
Construction of diagonalizing transformations for the covariance matrices:
Let A be a symmetric matrix (this holds for the genetic relationship matrices). The singular value decomposition of A is USU′, where U is an orthonormal matrix; i.e., 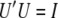 and
and 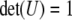 , and S is a diagonal matrix with nonnegative elements on the diagonal; i.e.,
, and S is a diagonal matrix with nonnegative elements on the diagonal; i.e., 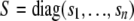 . Now, the required diagonalizing transformations, the square-root matrix and the inverse of the square-root matrix, are defined as
. Now, the required diagonalizing transformations, the square-root matrix and the inverse of the square-root matrix, are defined as 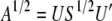 and
and 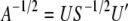 , respectively.
, respectively.
Hybrid Gibbs sampler with block sampling on transformed relationship matrices:
The single-site Gibbs sampler (Sorensen and Gianola 2002) updates each parameter in 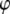 consecutively and therefore often suffers from slow mixing properties in the Markov chain because of strong autocorrelations between iterations. In some cases, a sampler can even get stuck within some range of values for one variable because other correlated variables will practically prevent it from moving to other parts of the parameter space. The autocorrelations tend to reach over more iterations the more individuals there are in the animal model and therefore sometimes require MCMC chains of several million iterations. However, the single-site sampler is fast because there is no need for matrix inversions. On the other hand, the blocked Gibbs sampler suggested by García-Cortés and Sorensen (1996) has good mixing properties, but entails huge computational demands, and is therefore very time consuming. As a compromise, we have implemented a hybrid Gibbs sampler that combines the single-site Gibbs sampler and the blocked Gibbs sampler. The main idea is that the hybrid sampler is less time consuming than a sampler based only on block updating, but has better mixing properties than the single-site sampler.
consecutively and therefore often suffers from slow mixing properties in the Markov chain because of strong autocorrelations between iterations. In some cases, a sampler can even get stuck within some range of values for one variable because other correlated variables will practically prevent it from moving to other parts of the parameter space. The autocorrelations tend to reach over more iterations the more individuals there are in the animal model and therefore sometimes require MCMC chains of several million iterations. However, the single-site sampler is fast because there is no need for matrix inversions. On the other hand, the blocked Gibbs sampler suggested by García-Cortés and Sorensen (1996) has good mixing properties, but entails huge computational demands, and is therefore very time consuming. As a compromise, we have implemented a hybrid Gibbs sampler that combines the single-site Gibbs sampler and the blocked Gibbs sampler. The main idea is that the hybrid sampler is less time consuming than a sampler based only on block updating, but has better mixing properties than the single-site sampler.
The fixed and random genetic effects are collected in the location parameter vector 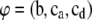 for the transformed variables.
for the transformed variables. 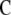 is the coefficient matrix of the left-hand side of the mixed-model equation as described in (9). Let
is the coefficient matrix of the left-hand side of the mixed-model equation as described in (9). Let 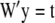 represent the right-hand side of the transformed mixed-model equation. Moreover, denote
represent the right-hand side of the transformed mixed-model equation. Moreover, denote 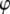 without the ith component as
without the ith component as 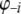 and C without element i of the ith row as
and C without element i of the ith row as 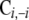 . The steps of the hybrid Gibbs sampler are as follows:
. The steps of the hybrid Gibbs sampler are as follows:
Initialize the parameters
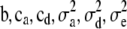 .
.- Single-site sampling:
- Draw
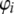 from
from 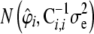 , where
, where 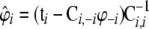 .
. - Calculate
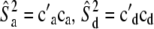 , and
, and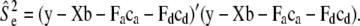
- Sample
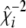 from
from 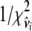 , where
, where 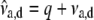 and
and 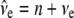 .
. - Calculate
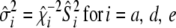 .
. - If
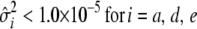 ,
, 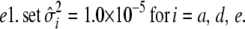
- Update
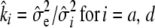 .
.
- Block sampling (every 50th iteration):
- Generate
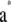 from
from 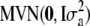 and
and 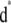 from
from 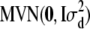 .
. - Generate adjustment factors
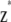 from
from 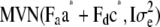 .
. - Denote the adjustment
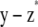 with f.
with f. - Compute a sample of the location effects as
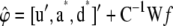 , where u is zero vector of length nfixed.
, where u is zero vector of length nfixed. - Calculate
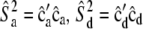 , and
, and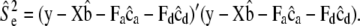
- Sample
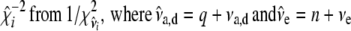 .
. - Calculate
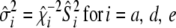 .
. - If
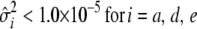 ,
, 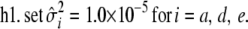
- Update
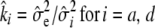 .
. - Go to 2a.
It should be emphasized that having bounds in the prior of the variance components in the single-site sampler would have been another way to guarantee that the variance components do not become too small (step 2e) during the iteration process. To solve the equation system 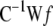 , several matrix decomposition and iterative methods are available (see Golub and Van Loan 1989). In this study, we have used a conjugate gradient iterative method (Barrett et al. 1994) in the numerical algorithm group (NAG) C library for a real symmetric linear system. Matrix C contains many zero entries and was therefore stored in sparse symmetric coordinate storage (SCS) format. The code is freely available from the authors.
, several matrix decomposition and iterative methods are available (see Golub and Van Loan 1989). In this study, we have used a conjugate gradient iterative method (Barrett et al. 1994) in the numerical algorithm group (NAG) C library for a real symmetric linear system. Matrix C contains many zero entries and was therefore stored in sparse symmetric coordinate storage (SCS) format. The code is freely available from the authors.
Modifications resulting from the use of transformed genetic effects:
The largest effect is at steps 3a and 3b of the above algorithm, where samples are drawn from the prior distributions of the genetic effects. When transformed genetic effects are used the sampling is done from a multivariate normal with uncorrelated components, which is a much simpler task than sampling from a multivariate normal with correlated components. Also in step 2b and 3e the calculations of 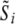 simplify from
simplify from 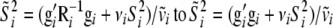 .
.
Computation of model comparison parameters:
We use the posterior predictive loss approach (Laud and Ibrahim 1995; Gelfand and Gosh 1998) to perform model comparison analysis. In this method, a MCMC chain of simulated/predicted phenotypes for each individual 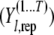 conditional on the observed phenotype
conditional on the observed phenotype 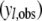 is derived by randomly sampling elements from the MCMC chains of
is derived by randomly sampling elements from the MCMC chains of 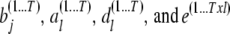 , respectively. The joint residual
, respectively. The joint residual 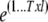 is obtained by first computing
is obtained by first computing
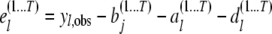 |
and then merging the error term of each individual 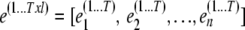 . Here,
. Here, 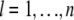 is the number of individuals with observations,
is the number of individuals with observations, 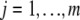 is the number of levels of the fixed effects, and T is the number of MCMC samples. The posterior predictive loss criteria are computed as
is the number of levels of the fixed effects, and T is the number of MCMC samples. The posterior predictive loss criteria are computed as
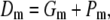 |
where Gm is the goodness of fit and Pm is the summation of all individual simulated/predicted phenotypic variance and provides a penalty term when computing Dm. These terms can be obtained by using
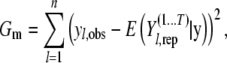 |
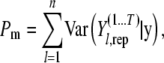 |
where
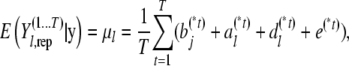 |
and
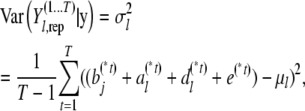 |
with * denoting a random sample from the posterior MCMC chains.
References
- Balocchi, C. E., F. E. Bridgewater, B. J. Zobel and S. Jahromi, 1993. Age trends in genetic parameters for tree height in a nonselected population of loblolly pine. For. Sci. 39 231–251. [Google Scholar]
- Barrett, R., M. Berry, T. F. Chan, J. Demmel, J. M. Donato et al., 1994. Templates for the Solution of Linear Systems: Building Blocks for Iterative Methods, Ed. 2. Society for Industrial and Applied Mathematics, Philadelphia.
- Barton, N. H., and M. Turelli, 2004. Effects of genetic drift on variance components under a general model of epistasis. Evolution 58 2111–2132. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and B. Rannala, 2004. The Bayesian revolution in genetics. Nat. Rev. Genet. 5 251–261. [DOI] [PubMed] [Google Scholar]
- Blasco, A., 2001. The Bayesian controversy in animal breeding. J. Anim. Sci. 79 2023–2046. [DOI] [PubMed] [Google Scholar]
- Chalh, A., and M. El Gazzah, 2004. Bayesian estimation of dominance merits in noninbred populations by using Gibbs sampling with two reduced sets of mixed model equations. J. Appl. Genet. 45 331–339. [PubMed] [Google Scholar]
- Cockerham, C. C., 1954. An extension of the concept of partitioning hereditary variances for analysis of covariances among relatives when epistasis is present. Genetics 39 859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham, C. C., and B. S. Weir, 1984. Covariances of relatives stemming from a population undergoing mixed self and random mating. Biometrics 40 157–164. [PubMed] [Google Scholar]
- Crainiceanu, C. M., and D. Ruppert, 2004. Likelihood ratio tests in linear mixed models with one variance component. J. R. Stat. Soc. B 66 165–185. [Google Scholar]
- Crnokrak, P., and D. A. Roff, 1995. Dominance variance: association with selection and fitness. Heredity 75 530–540. [Google Scholar]
- Damgaard, L. H., 2007. Technical note: how to use WinBUGS to draw inferences in animal models. J. Anim. Sci. 85 1363–1368. [DOI] [PubMed] [Google Scholar]
- de Boer, I. J. M., and I. Hoeschele, 1993. Genetic evaluation methods for populations with dominance and inbreeding. Theor. Appl. Genet. 86 245–258. [DOI] [PubMed] [Google Scholar]
- Du, F.-X., and I. Hoeschele, 2000. Estimation of additive, dominance and epistatic variance components using finite locus models implemented with a single-site Gibbs and a descent graph sampler. Genet. Res. 76 187–198. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longman, New York.
- Fernandez, A., M. A. Toro and C. Lopez-Fanjul, 1995. The effect of inbreeding on the redistribution of genetic variance of fecundity and viability in Tribolium castaneum. Heredity 75 376–381. [Google Scholar]
- Fries, A., and T. Ericsson, 1998. Genetic parameters in diallel-crossed Scots pine favor heartwood formation breeding objectives. Can. J. For. Res. 28 937–941. [Google Scholar]
- Fuerst, C., J. W. James, J. Sölkner and A. Essl, 1997. Impact of dominance and epistasis on the genetic make-up of simulated populations under selection: a model development. J. Anim. Breed. Genet. 114 163–175. [DOI] [PubMed] [Google Scholar]
- García, N., C. Lopez-Fanjul and A. García-Dorado, 1994. The genetics of viability in Drosophila melanogaster: effects of inbreeding and artificial selection. Evolution 48 1277–1285. [DOI] [PubMed] [Google Scholar]
- García-Cortés, L. A., and D. Sorensen, 1996. On a multivariate implementation of the Gibbs sampler. Genet. Sel. Evol. 28 121–126. [Google Scholar]
- García-Cortés, L. A., C. Cabrillo, C. Moreno and L. Varona, 2001. Hypothesis testing for the genetic background of quantitative traits. Genet. Sel. Evol. 33 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman, A., 2006. Prior distributions for variance parameters in hierarchical models. Bayesian Anal. 1 515–533. [Google Scholar]
- Gelfand, A. E., and S. K. Gosh, 1998. Model choice: a minimum posterior predictive loss approach. Biometrika 95 1–11. [Google Scholar]
- Gelfand, A. E., and S. K. Sahu, 1999. Identifiability, improper priors, and Gibbs sampling for generalized linear models. J. Am. Stat. Assoc. 94 247–253. [Google Scholar]
- Gelfand, A. E., and A. F. M. Smith, 1990. Sampling-based approaches to calculating marginal densities. J. Am. Stat. Assoc. 85 398–409. [Google Scholar]
- Geman, S., and D. Geman, 1984. Stochastic relaxation, Gibbs distributions and Bayesian restoration of images. IEEE Trans. Pattern Anal. Mach. Intell. 6 721–741. [DOI] [PubMed] [Google Scholar]
- Gianola, D., and R. L. Fernando, 1986. Bayesian methods in animal breeding theory. J. Anim. Sci. 63 217–244. [Google Scholar]
- Gilmour, A. R., B. J. Gogel, B. R. Cullis and R. Thompson, 2006. ASReml User Guide, Release 2.0. VSN International, Hemel Hempstead, UK.
- Golub, G. H., and C. F. Van Loan, 1989. Matrix Computations, Ed. 2. Johns Hopkins University Press, Baltimore.
- Hallander, J., and P. Waldmann, 2007. The effect of non-additive genetic interactions on selection in multi-locus genetic models. Heredity 98 349–359. [DOI] [PubMed] [Google Scholar]
- Henderson, C. R., 1976. A simple method for computing the inverse of a numerator relationship matrix used in prediction of breeding values. Biometrics 32 69–83. [Google Scholar]
- Henderson, C. R., 1984. Applications of Linear Models in Animal Breeding. University of Guelph, Guelph, ON, Canada.
- Henderson, C. R., 1985. a Best linear unbiased prediction of nonadditive genetic merits in noninbred populations. J. Anim. Sci. 60 111–117. [Google Scholar]
- Henderson, C. R., 1985. b MIVQUE and REML estimation of additive and nonadditive genetic variances. J. Anim. Sci. 61 113–121. [Google Scholar]
- Hoeschele, I., and P. M. VanRaden, 1991. Rapid inversion of dominance relationship matrices for noninbred populations by including sire by dam subclass effects. J. Dairy Sci. 74 557–569. [DOI] [PubMed] [Google Scholar]
- Hoti, F. J., M. J. Sillanpää and L. Holmström, 2002. A note on estimating the posterior density of a quantitative trait locus from a Markov chain Monte Carlo sample. Genet. Epidemiol. 22 369–376. [DOI] [PubMed] [Google Scholar]
- Jansson, G., and B. Li, 2004. Genetic gains of full-sib families from disconnected diallels in loblolly pine. Silvae Genet. 53 60–64. [Google Scholar]
- Kearsey, M. J., and H. S. Pooni, 1996. The Genetical Analysis of Quantitative Traits. Chapman & Hall, London.
- Kempthorne, O., and R. N. Curnow, 1961. The partial diallel cross. Biometrics 17 229–250. [Google Scholar]
- Kennedy, B. W., L. R Schaeffer and D. A. Sorensen, 1988. Genetic properties of animal models. J. Dairy Sci. 71(Suppl. 2): 17–26. [Google Scholar]
- Laud, P. W., and J. G. Ibrahim, 1995. Predictive model selection. J. R. Stat. Soc. B 57 247–262. [Google Scholar]
- Lee, S. H., and J. H. J. Van der Werf, 2006. Using dominance relationship coefficients based on linkage disequilibrium and linkage with a general complex pedigree to increase mapping resolution. Genetics 174 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Fanjul, C., A. Fernandez and M. A. Toro, 2002. The effect of epistasis on the excess of the additive and nonadditive variances after population bottlenecks. Evolution 56 865–876. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Mao, Y., and S. Xu, 2005. A Monte Carlo algorithm for computing the IBD matrices using incomplete marker information. Heredity 94 305–315. [DOI] [PubMed] [Google Scholar]
- Misztal, I., 1997. Estimation of variance components with large-scale dominance models. J. Dairy Sci. 80 965–974. [Google Scholar]
- Mrode, R., and R. Thompson, 1989. An alternative algorithm for incorporating the relationships between animals in estimating variance components. J. Anim. Breed. Genet. 106 89–95. [Google Scholar]
- Ovaskainen, O., J. M. Cano and J. Merilä, 2007. A Bayesian framework for comparative quantitative genetics. Proc. R. Soc. Lond. Ser. B 275 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaas, R. L., and E. J. Pollak, 1980. Mixed model methodology for farm and ranch beef cattle testing programs. J. Anim. Sci. 51 1277. [Google Scholar]
- R Project, 2002. The R project for statistical computing. www.r-project.org.
- Rekaya, R., K. A. Weigel and D. Gianola, 2003. Bayesian estimation of parameters of a structural model for genetic covariances between milk yield in five regions of the United States. J. Dairy Sci. 86 1837–1844. [DOI] [PubMed] [Google Scholar]
- Roff, D. A., and K. Emerson, 2006. Epistasis and dominance: evidence for differential effects in life-history versus morphological traits. Evolution 60 1981–1990. [PubMed] [Google Scholar]
- Searle, S. R., G. Casella and C. E. McCulloch, 1992. Variance Components. Wiley, New York.
- Shoemaker, J. S., I. S. Painter and B. S. Weir, 1999. Bayesian statistics in genetics: a guide for the uninitiated. Trends Genet. 15 354–358. [DOI] [PubMed] [Google Scholar]
- Sillanpää, M. J., and K. Auranen, 2004. Replication in genetic studies of complex traits. Ann. Hum. Genet. 68 646–657. [DOI] [PubMed] [Google Scholar]
- Smith, B. J., 2005. Bayesian Output Analysis Program (BOA), Version 1.1.5. University of Iowa, Iowa City, IA. http://www.public-health.uiowa.edu/boa.
- Sorensen, D., and D. Gianola, 2002. Likelihood, Bayesian and MCMC Methods in Quantitative Genetics. Springer-Verlag, New York.
- Sorensen, D., and R. Waagepetersen, 2003. Normal linear models with genetically structured residual variance heterogeneity: a case study. Genet. Res. 82 207–222. [DOI] [PubMed] [Google Scholar]
- Sorensen, D. A., C. S. Wang, J. Jensen and D. Gianola, 1994. Bayesian analysis of genetic change due to selection using Gibbs sampling. Genet. Sel. Evol. 26 333–360. [Google Scholar]
- Vaida, F., and S. Blanchard, 2005. Conditional Akaike information for mixed-effects models. Biometrika 92 351–370. [Google Scholar]
- Varona, L., and I. Misztal, 1999. Prediction of parental dominance combinations for planned matings, methodology, and simulation results. J. Dairy Sci. 82 2186–2191. [DOI] [PubMed] [Google Scholar]
- Waldmann, P., and T. Ericsson, 2006. Comparison of REML and Gibbs sampling estimates of multi-trait genetic parameters in Scots pine. Theor. Appl. Genet. 112 1441–1451. [DOI] [PubMed] [Google Scholar]
- Wall, E., S. Brotherstone, J. F. Kearney, J. A. Woolliams and M. P. Coffey, 2005. Impact of nonadditive genetic effects in the estimation of breeding values for fertility and correlated traits. J. Dairy Sci. 88 376–385. [DOI] [PubMed] [Google Scholar]
- Walsh, B., 2001. Quantitative genetics in the age of genomics. Theor. Popul. Biol. 59 175–184. [DOI] [PubMed] [Google Scholar]
- Wang, C. S., J. J. Rutledge and D. Gianola, 1993. Marginal inference about variance components in a mixed linear model using Gibbs sampling. Genet. Sel. Evol. 21 41–62. [Google Scholar]
- Wang, C. S., D. Gianola, D. A. Sorensen, J. Jensen, A. Christensen et al., 1994. Response to selection for litter size in Danish Landrace pigs: a Bayesian analysis. Theor. Appl. Genet. 88 220–230. [DOI] [PubMed] [Google Scholar]
- Wang, J., A. Caballero, P. D. Keightley and W. G. Hill, 1998. Bottleneck effect on genetic variance: theoretical investigation of the role of dominance. Genetics 150 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C., and K. Fowler, 1999. The changes in genetic and environmental variance with inbreeding in Drosophila melanogaster. Genetics 152 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, J. H., and H. A. Orr, 1993. Increased heritable variation following population bottlenecks: the role of dominance. Evolution 47 949–957. [DOI] [PubMed] [Google Scholar]
- Wu, H. X., and A. C. Matheson, 2004. General and specific combining ability from partial diallels of radiata pine: implications for utility of SCA in breeding and deployment populations. Theor. Appl. Genet. 108 1503–1512. [DOI] [PubMed] [Google Scholar]
- Wu, H. X., and A. C. Matheson, 2005. Genotype by environment interactions in an Australia-wide radiata pine diallel mating experiment: implications for regionalized breeding. For. Sci. 51 29–40. [Google Scholar]
- Xu, S., 2003. Advanced statistical methods for estimating genetic variances in plants. Plant Breed. Rev. 22 113–163. [Google Scholar]
- Yanchuk, A. D., 1996. General and specific combining ability from disconnected partial diallels of coastal Douglas-fir. Silvae Genet. 45 37–45. [Google Scholar]
- Yi, N., and S. Xu, 2000. Bayesian mapping of quantitative trait loci under the identity-by-descent-based variance component model. Genetics 156 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]