

Abstract
Human papillomaviruses (HPVs) are maintained latently in dividing epithelial cells as nuclear plasmids. Two virally encoded proteins, E1, a helicase, and E2, a transcription factor, are important players in replication and stable plasmid maintenance in host cells. Recent experiments in yeast have demonstrated that viral genomes retain replication and maintenance function independently of E1 and E2 (Angeletti et al., 2002; Kim et al., 2005). Flow-cytometry studies of EGFP-reporter vectors containing subgenomic HPV fragments with or without a human ARS (hARS), revealed that six fragments located in E6–E7, E1–E2, L1, and L2 regions showed a capacity for plasmid stabilization in the absence of E1 and E2 proteins. Interestingly, four fragments within; E7, the 3’ end of L2, and the 5’ end of L1 exhibited stability in plasmids that lacked an hARS, indicating that they possess both replication and maintenance functions. Two fragments lying in E1–E2 and the 3’ region of L1 were stable only in the presence of hARS, that they contained only maintenance function. Mutational analyses of HPV16-GFP reporter constructs provided evidence that genomes lacking E1 and E2 could replicate to an extent similar to wildtype HPV16. Together these results support the concept that cellular factors influence HPV replication and maintenance, independently, and perhaps in conjunction with E1 and E2, suggesting a role in the persistent phase of the viral lifecycle.
Keywords: extrachromosal DNA, persistent infection, human papillomavirus
INTRODUCTION
Human papillomaviruses (HPVs) are small circular double-stranded DNA viruses that establish long-term persistent infections in squamous epithelial cells. The viral life cycle is tightly linked with the differentiation state of the host cells. It is thought that an early amplificational event occurs following virus entry into basal layer cells. The viral DNA persists as a nuclear episome in infected cells. In the non-productive stage of infection, HPVs replicate at low copy number in mitotically active basal layer cells within stratified epithelia (Howley, 1996).
In the suprabasal keratinocyte layers, the viral genome undergoes amplification, which yields from 100 to 1000 viral copies per cell. The percentage of differentiated cells that amplify viral DNA is often very low (Flores et al., 2000). Papillomaviruses utilize two viral factors, E1, a helicase, and E2, a transcriptional activator and auxiliary replication factor (Rabson et al., 1986; Ustav and Stenlund, 1991; Ustav et al., 1991). The E1 protein forms hexamers and unwinds viral DNA similarly to cellular helicases, such as; Werner’s (WRN) and Bloom’s (BLM) syndrome helicases and minichromosome maintenance (MCM) proteins (Ellis et al., 1995; Fouts et al., 1999; Kass, Landsberger, and Wolffe, 1997; Mohaghegh et al., 2001; Patel and Picha, 2000). E1 has homology to SV40 DNA helicase, TAg, and both proteins direct polα activity to their respective origins (Clertant and Seif, 1984; Mansky, Batiza, and Lambert, 1997; Park et al., 1994). In the final phase of HPV infection, the virus amplifies differentiating suprabasal cells, and finally viral capsid are produced (Howley, 1996).
HPV genomes can persist in proliferating keratinocytes for years or decades. This persistent phase of the viral lifecycle is characterized by detectable levels of viral genome but the absence of virus production (Stubenrauch and Laimins, 1999), likely as a strategy to evade immune surveillance. Maintenance of viral episomes requires that newly synthesized DNAs be faithfully partitioned to daughter cells during mitosis. A similar strategy is employed by Epstein-Barr virus (EBV) and Koposi’s sarcoma-associated Herpesvirus (KSHV). These viruses maintain their genomes in dividing cells through the function of a virally encoded anchoring protein, which links the viral DNAs, via repeat binding sites, to host mitotic chromosomes (Ballestas, Chatis, and Kaye, 1999; Ballestas and Kaye, 2001; Harrison, Fisenne, and Hearing, 1994; Lupton and Levine, 1985; Sears et al., 2004; Yates et al., 1984; Yates, Warren, and Sugden, 1985). The same scenario has also been observed in bovine papillomavirus type 1 (BPV1). Several studies in BPV1 have shown that the major players, E1 and E2, are key players in replication along with multiple E2 binding sites located in the long control region (LCR) (Abroi et al., 2004; Bastien and McBride, 2000; Ilves, Kivi, and Ustav, 1999; Piirsoo et al., 1996; Ustav et al., 1991; Wu, Ceccarelli, and Frappier, 2000). However, some studies suggest that BPV-1 E1 contributes to viral establishment, but is not required for the maintenance stage of viral life cycle (Kim and Lambert, 2002).
The E2 viral protein contributes to genome persistence by linking viral DNAs, through specific DNA binding sites (ACCGN4CGGT), to mitotic chromosomal proteins via an interaction with the E2 N-terminus (Abroi et al., 2004; Bastien and McBride, 2000; Ilves, Kivi, and Ustav, 1999; McBride, Oliveira, and McPhillips, 2006). This mechanism was first observed in BPV1 studies, in which E2 was found to stably bind to mitotic chromosomes via a cellular protein, bromodomain protein 4 (Brd4) (Baxter and McBride, 2005; Baxter et al., 2005; McPhillips, Ozato, and McBride, 2005; You et al., 2004). However, recent observations of mitotic chromosome interactions with E2 protein from multiple papillomaviruses has revealed that there is significant variation in the timing of binding and the specificity to the binding partner, Brd4 (McBride, Oliveira, and McPhillips, 2006). The E2 proteins from α papillomaviruses (HPV11, HPV16, HPV31 and HPV57) are able to bind to mitotic chromosomes but only in telophase of mitotic process (Oliveira, Colf, and McBride, 2006). Further studies have suggested that the chromosome attachment of HPV E2s is independent of Brd4 since the Brd4-binding affinities of the E2s are much lower than that of BPV1. Additionally, point mutations in E2 that eliminate the Brd4 binding do not affect the mitotic chromosomal localization of these proteins (McPhillips et al., 2006). This indicates that HPVs, particularly in the alpha genus, have evolved a different strategy to maintain and equally distribute their genomes to daughter cells.
In the case of BPV1, a total of 17 E2 binding site (E2BS) have been identified; 12 of them are located within the LCR region (Li et al., 1989). For efficient partitioning and long-term persistence of episomal BPV DNA, an element containing at least 10 E2BSs, known as the minichromosome maintenance element (MME) was needed (Piirsoo et al., 1996). This minimal number of E2 binding sites sufficient to provide the minichromosome maintenance function exceeds the number of sites generally found in LCR of many HPV types. Taken together, E2 and its binding sites, may provide maintenance/partitioning function in HPVs with the accompaniment of cellular maintenance factors. Thus, we hypothesized that stable maintenance of the HPV genomes in proliferating cells requires additional cellular factors for efficient retention and faithful segregation of their genomes.
In previous experiments in Saccharomyces cerevasiae, we demonstrated that HPV16 genomes can be replicated and stably maintained in the absence of any particular viral protein, notably, E1 and E2 (Angeletti et al., 2002; Kim et al., 2005). The existence of an E1 and E2-independent mode of replication and maintenance of HPV is supported by mapping studies in yeast (Kim et al., 2005; Kim and Lambert, 2002). In those studies, elements, referred to as mtc elements, present outside LCR sequence that could substitute for autonomously replicating sequence (ARS) and centromere (CEN) function in yeast. The results suggest that remaining cis-elements are recognized by cellular factors and confer both replication and maintenance functions to the viral genome.
In the present study, we confirm an E1- and E2-independent mode of viral replication in human cells by using a series of HPV16 deletion mutants. Those mutants that lack E1, E2, and LCR sequences showed a comparable level of replication to the wild type genome in short-term replication assays. This experiment provided firm evidence that E1, E2 and E2BSs were not entirely essential for viral replication and that the requirement for viral factors could be supplanted by cellular replication machinery, under certain conditions. We hypothesized that undefined cis-elements outside the LCR, and recognized by cellular factors, could confer a degree of maintenance activity. In order to determine which regions of the genome had intrinsic maintenance function, we performed a complementation assay in mammalian cells using plasmids containing HPV16 subgenomic fragments, an EGFP cassette, with or without a previously defined human ARS (hARS) (Masukata et al., 1993). We then tested each plasmid for short and long-term extrachromosomal replication and we measured their mitotic stability. The results described here support the concept that HPVs make use of E1 and E2-independent cis-acting replication and maintenance signals, which likely allows greater regulation on copy number and segregation.
MATERIALS AND METHODS
Mammalian cells and transfection methods
Human embryonic kidney (HEK) 293, HeLa and HaCaT cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum in a 5% CO2 incubator at 37°C. All transfections were performed using Dreamfect reagent (Ozbiosciences), 2 µg of DNA and 4 × 105 cells in 6-cm plates. Cells were seeded in plates for 16 to 24 h prior to transfection.
Plasmids and library constructions
To identify cis-elements in HPV16 that are responsible for stability of viral genomes, we first created a vector that carries a human autonomously replicating sequence (hARS) and an enhanced green fluorescent protein (EGFP) cassette. The pGFP vector was fused to a 4980-bp segment of hARS (from clone P1W1) (Masukata et al., 1993) resulting in an hARS+ pGFP vector (pGAB1) which is able to replicate autonomously in human cells. HPV16 library constructs were then made by cloning HPV16 subgenomic fragments into pGAB1 vector (Fig 3A). The HPV16 genome was digested with Sau3AI into multiple fragments ranging in size from 2.6 Kb to 18 bp which were the cloned into the pGab1 vector using linkers (top, 5’-CGAGCTAAGCTTGCAA-3’; bottom, 5’-GATCTTGCAAGCTTAGCTCG-3’; HindIII site (underlined)). Individual plasmids containing each fragment of the HPV16 genome were isolated.
FIGURE 3. Plasmid maps of pGAB1 and pGFP.
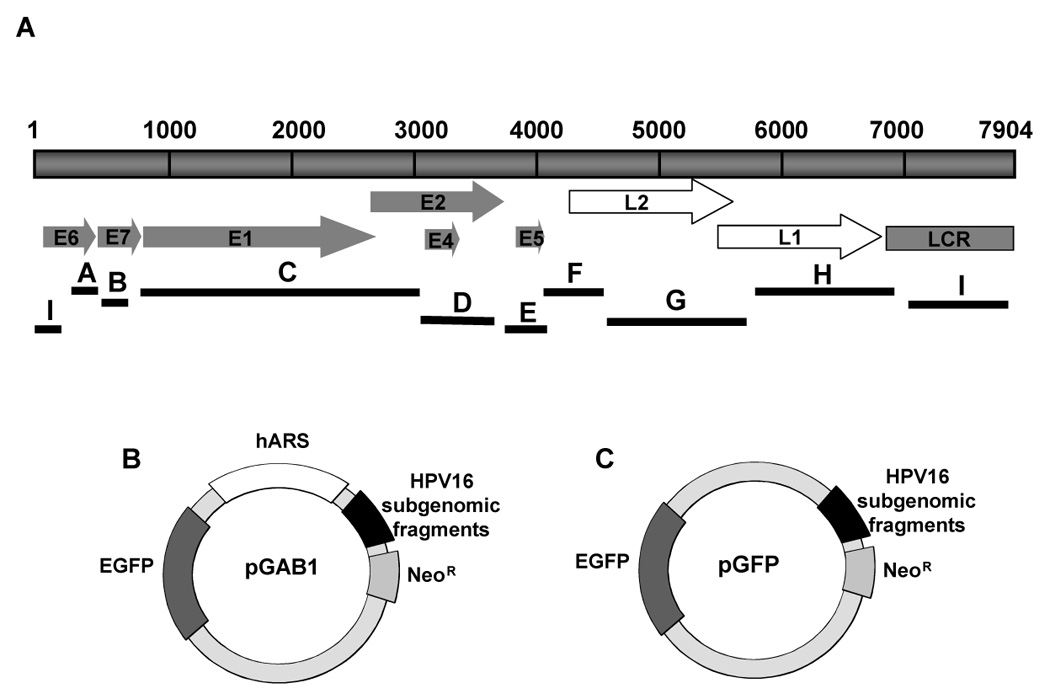
(A) A representation of HPV16 subgenomic fragments derived from Sau3AI-treated HPV16 genome. The top bar represents a linear map of the HPV16 genome with each of eight ORFs indicated by arrows and the control region, LCR shown as a gray bar. The lines designated A to I represent the HPV16 Sau3AI subgenomic fragments (B) A map of pGAB1 used in identification of maintenance elements in HPV16 genome. pGAB1 contains an EGFP gene, a neomycin-resistance gene and an hARS element obtained from clone P1W1 (Masukata et al., 1993). To obtain HPV16 library constructs in the pGAB1 backbone, HPV16 subgenomic fragments were cloned into the HindIII site of pGAB1. (C) The map of pGFP used in identification of replication elements within the HPV16 genome. pGFP is an hARS-negative version of pGAB1. HPV16 library constructs in pGFP backbone derived from removal of the hARS portion from pGAB1plasmids.
To map regions in the viral genome that conferred both replication and maintenance functions, a second set of HPV16 library was constructed by removing the hARS from pGab1 library plasmids by EcoRI.
To rule out any bias effects due to the SV40 promoter that exists in the previous sets of HPV16 library, the third HPV16 library was created in pUC19 carrying an expression cassette of EGFP gene. To generate an EGFP+ pUC19 vector, an entire EGFP gene including CMV promoter was amplified from pEGFP-C1 (Clontech) using 5’CMV (5’-GCATCCTCGAGGTAATCAATTACGGG-3’) and 3’GFP (5’-CGAAGCTTGAGCTCGAGATCTGAG-3’) primers containing XhoI site (underlined). The PCR products was trimmed with XhoI and directly inserted into pUC19 vector at the XhoI site. The resultant vector was named pUCGFP. Then, the full-length HPV16 genome that was excised from pEF399 with a BamHI digest was ligated into pUCGFP at the BamHI site, resulting in the plasmid referred to as HPV16GFP. All mutants derived from HPV16GFP, shown in Figure 1, were made by multiple restriction enzyme digestion to remove certain regions of HPV16 genome and the remaining DNA was religated with T4 ligase. The ΔMSC mutant was generated by deleting the fragment between 2 MscI sites of HPV16GFP that contains entire sequence of L2 coding region and part of the E2 gene 5’ region. ΔE1–E2 was generated by deleting the StuI-BsaBI fragment of HPV16GFP that contains E1 and E2 coding regions. ΔLCR-E2 was generated by deleting the StuI-PmlI fragment of HPV16GFP that contains the E2 coding region through LCR. ΔL1 was generated by deleting the PmlI-XbaI fragment of HPV16GFP that contains 5’ end of L1 coding region. The last mutant, L1, was created by deleting the SmaI-PmlI fragment of HPV16GFP and left fragment in it.
FIGURE 1. Map of the pUCGFP plasmid.
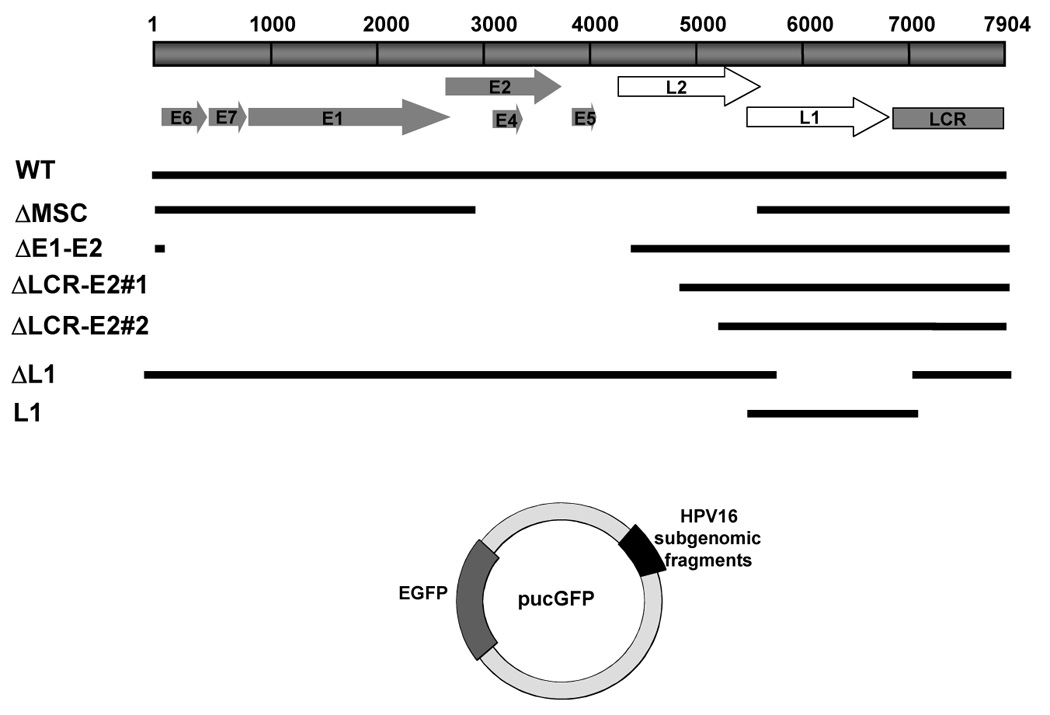
pUCGFP was created from the plasmid pUC19. An expression cassette containing the EGFP gene, driven by a CMV promoter, was cloned into the multiple cloning site (MCS). The wild type HPV16 and various mutants, with respective deletions, shown in diagram, were inserted into pUCGFP at BamHI site.
Plasmid maintenance assay
The plasmid maintenance experiment was modified from the protocol previously described by Vogel et.al. (Vogel et al., 1998). HPV16 library plasmids were transfected into human cell lines using Dreamfect. The media was changed 24 h after transfection to remove excess DNAs and residual transfection reagent. On the next day, the cells were treated with 0.5 mg/mg G418 to favor the growth of transfected cells containing the plasmids that confer G418 resistance. The EGFP-expressing cells were left under selective conditions for 4 days to allow untransfected cells to die. The surviving cells were released into nonselective media for 10–14 days and periodically subjected to analysis by flow cytometry for GFP and Southern blot analysis. In order to determine the stability of the plasmids, the relative percentages of EGFP positive cells were calculated and plotted versus the period of time after cells were released from the drug. To obtain the relative percentages of EGFP positive cells, the final percentages of EGFP positive cells, at several time points of sampling, were divided by the initial percentage of EGFP positive cells at day 0 which is the first day cells were released from selection media. We calculated the loss rate of GFP-positive cells per cell generation and normalized with that of the most stable plasmid to obtain the loss rates per cell generation.
Assay for plasmid DNA replication
Cells were plated in 100 mm dishes and transfected with 5 µg of DNAs, while the cells were 50 to 70% confluent. The cells were incubated with the DNAs for 24 h, then the media was changed. For the short-term replication assay, plasmid DNA was isolated using the Hirt method (Hirt, 1967) at 4 days after transfection. For the long-term replication assay, the transfected cells were grown in media containing 0.5 mg/ml G418 for 4 days after transfection then released to media without the drug for 10–14 days. DNA was isolated using the Hirt method. The plasmids were double-digested for 16–24 h with DpnI and XhoI for HPV16 library in pUCGFP, or with DpnI and XhoI for HPV16 library in pGAB1 or pGFP. DNA samples were electrophoresed in a 0.8% agarose gel then analyzed by Southern blot using EGFP as a probe.
Analysis of the HPV16 genome for maintenance protein binding sites
The HPV16 genome was analyzed for perfect matches with the high-mobility-group (HMG) protein consensus binding site (WWWWWWS) using the Fuzznuc program (http://bioweb.pasteur.fr/seqanal/interfaces/fuzznuc.html). The frequency of predicted HMG sites was plotted against the HPV16 genome with the threshold setting of >10 sites per 100 nucleotides. Similarly, topoisomerase II (topo II) consensus sites (RNYNNCNNGYNGKTNYNY) were plotted with a threshold frequency of >7 sites per 100 nucleotides. The presence of single binding sites of centromere binding protein B (CENP-B; YTTCGTTGGAARCGGGA) and telomere-repeat factor (TRF; TTAGGTTA) were plotted against the HPV16 genome. Matrix or scaffolding attachment regions (MARS/SARS) were located in the HPV16 genome using the Marscan program (http://bioweb.pasteur.fr/seqanal/interfaces/marscan.html). MARS are made up of bipartite sequence elements of 8 bp (AATAAYAA) and 16 bp (AWWRTAANNWWGNNNC) within a 200 bp distance from each other. The can be on either strand of the DNA and can overlap (van Drunen et al., 1999). In addition, we included previously reported MAR locations mapped in HPV16 (Tan et al., 1998).
RESULTS
E1 and E2-independent replication of HPV16
Recent studies in yeast showed that, under certain conditions, HPV16 could replicate in the absence of functional E1 and E2 proteins (Kim et al., 2005). In order to confirm the E1 and E2-independent mode of replication of HPV16 in human cells, we generated a series of HPV16 deletion mutants cloned into a modified pUC19 vector, referred to pUCGFP that carries an expression cassette of EGFP. A panel of HPV16 deletion constructs was transiently transfected in HEK 293 cells in parallel with a parental wild-type HPV16 and vector alone. Low molecular weight DNAs were isolated by the Hirt method at 4 days after transfection and then subjected to overnight digestion with DpnI and HindIII prior to Southern blot analysis using pUCGFP as a probe. By these means, the input DNA originated from bacteria that is sensitive to DpnI was completely degraded. Only newly synthesized, DpnI-resistant, plasmids were still intact and observed as linear DNAs on the Southern blot when they were cleaved by HindIII (FIG. 2A). In this experiment, a similar level of replication was observed in most mutants compared to the wild-type HPV16, even though they lacked E1, E2 coding regions and the LCR sequence. This result is in agreement with the data from the pervious studies in yeast that give us the evidence of E1-and E2-independent mode of replication. The same experiments were performed in HaCaT and HeLa cells and simlar results were achieved, but since transfection efficiency was superior in 293 cells we chose to do the analyses in 293 cells. Many other studies have used 293 cells to identify replication and maintenance elements in EBV and KSHV (Skalsky, Hu, and Renne, 2007; Yates, Camiolo, and Bashaw, 2000).
FIGURE 2. E1 and E2-independent replication and episomal maintenance of HPV16 and its mutants.
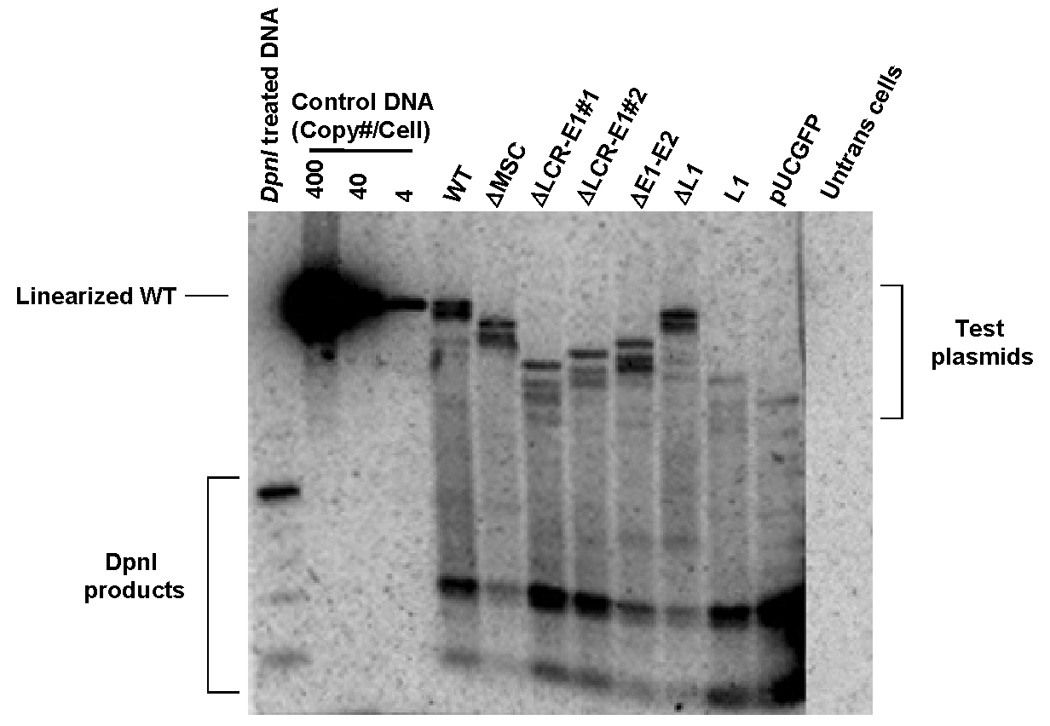
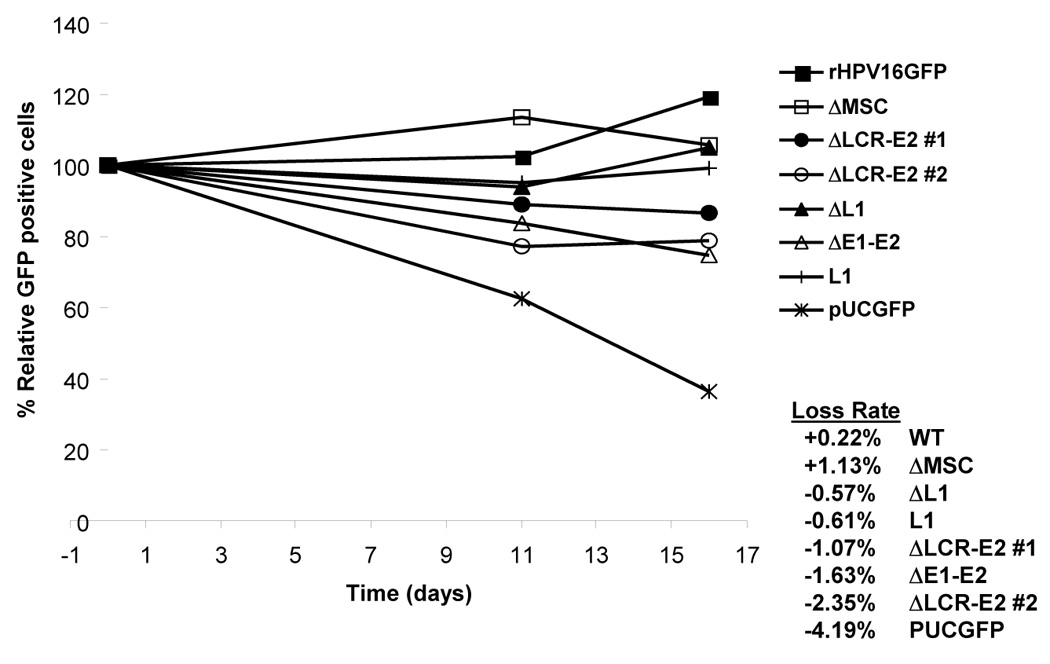
(A) Short-term replication of HPV1 and deletion mutants. 293 cells were transfected with pUCGFP containing entire HPV16 genome or its mutant derivatives. Plasmids were harvested 4 days after transfection. DNA samples were double digested overnight with DpnI and HindIII prior to southern analysis. The left lane contains a control for the completeness of DpnI digestion of DNA that was extracted from bacteria. (B) Plasmid stability of HPV16 genome and mutants. 293 cells were cotransfected with full-length HPV16 or its mutants and pCDNA3.1 (+) that contains a neomycin-resistant gene. Cells were grown in the presence of the drug, G418, for 2 weeks. After the drug was removed, the cells were cultured under nonseletive conditions for another 16 days. Meanwhile, the GFP-positive cells were monitered by flow cytometry at 0, 6, and 11 days after release from drug. The Relative percent GFP-positive cells was calculated and plotted against time. The loss rate of each plasmid was determined as percent change of GFP-positive cell per cell generation.
Plasmid stability of HPV16 deletion mutants that lack E1 and E2 coding regions
In order to assess the capability of wild type HPV16 and its mutant derivatives to be stably maintained in mammalian cells, the plasmids were individually transfected into 293 cells along with pCNDA3.1 (+) that confers G418 resistance. The drug-resistance property of the transfected cells allows them to outgrow untransfected cells under selective conditions. After transfection, the cells were grown under selective conditions for 4 days to enrich cells containing the plasmids and to allow plasmids to become established. After removal of selection, the stability of plasmids was evaluated by monitoring the remaining GFP-positive cells in the total cell population over the course of at least 2 weeks. Using flow cytometry, we could measure percent of GFP-positive cells at different time points over several cell generations and the percent loss of GFP-positive cells per cell generation was calculated (Fig. 2B). The change of GFP-positive cells over time, which indicates the stability of plasmid, depends on two main processes; replication and partitioning. If both replication and partitioning of the GFP-containing plasmid occurs in dividing cells, the number of GFP-positive cells remains essentially unchanged. If only one mechanism takes place (either replication or partitioning), the number of GFP-positive cells should be decreasing. In the case of partitioning plasmids that lack replicative activity, the number of GFP-positive cells should remain constant over the course of only the first cell divisions. After each cell division, even if partitioning is functional, the copy number of the nonreplicating plasmids will fall below a detectable level. As shown in figure 2B, the vector itself that did not contain any segments of the viral genome was lost at a rate of 4% per cell generation. In contrast, all derivative mutants of HPV16 showed similar pattern of plasmid stability to that observed in wild-type with the lower loss rate compare to the vector alone. Again, the mutants that lack E1, E2, and LCR regions were retained in the cells for 16 days. Interestingly, the vector that carries only L1 sequence was efficiently maintained in mammalian in the absence of selection for at least 16 cell generations. This suggests that the maintenance of HPV16 genome is independent of the viral proteins E1 and E2 and therefore the E2 binding sites as well as the viral origin of replication that are located within the LCR regions are not required for the retention of plasmid in the cells. It is likely that undefined cis-acting elements residing outside the LCR region would be recognized by cellular factors and that confers the stability of the plasmid.
Mapping of E2-independent maintenance function
In previous studies, it was discovered that PV genomes could be stably maintained in human cells in the absence of functional E1 and E2 proteins (Angeletti et al., 2002; Kim et al., 2005; Kim and Lambert, 2002). To identify such elements, we created a library of plasmids in which subgenomic fragments of the HPV16 genome, generated by digestion with Sau3AI, were inserted into pGAB1, a vector containing a human ARS element, a neomycin resistant gene, and an expression cassette for EGFP under the strong constitutive CMV promoter. The vector backbone carries the human ARS element to provide the replication function in mammalian cells to ensure that loss of plasmids over the course of the experiment was not due to lack of such activity and to allow the identification of maintenance function in regions where no replication function resides. To examine whether this HPV16 library plasmids were able to replicate in human cells, we performed a short-term replication assay with DpnI treatment at 4 days after transfection. All HPV16 subgenomic containing plasmids replicated in 293 cells as efficiently as vector itself that does not harbor any segment of HPV16 DNA (Fig 4A). Similar observed replication efficiencies rules out the possibility of interference of replication function in long-term maintenance assays. Therefore, the HPV16 subgenomic fragments present in the Gab1 vector, that provides replication function, would be considered as maintenance elements and this stability did not result from the advantage of replication. We noted that there was a steady loss of replication function after 10 days post-release from G418, yet plasmids persisted as evidenced by Southern analysis and by the presence of EGFP signal (Fig 4B). The gradual loss of replication function and continued DNA persistence was witnessed with all constructs to varying extents. To ensure that the persisting non-replicating DNAs were not simply extracellular, we washed transfect cells with PBS and subcultured them every 3 days. Cells were then externally treated with 1 unit of DnaseI prior to Southern analsis. In these experiments, persisting plasmids were indeed intracellular and episomal for greater than 14 days.
FIGURE 4. Short- and long-term replication of HPV16 subgenomic DNA in the pGAB1 vector.
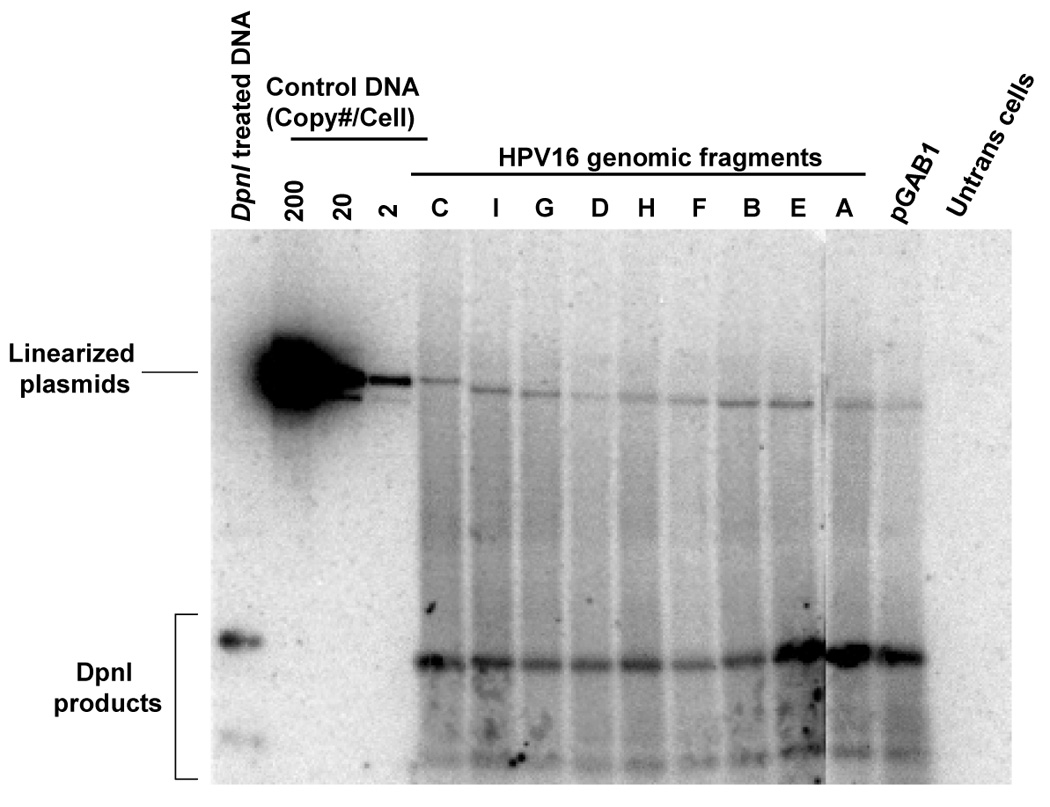
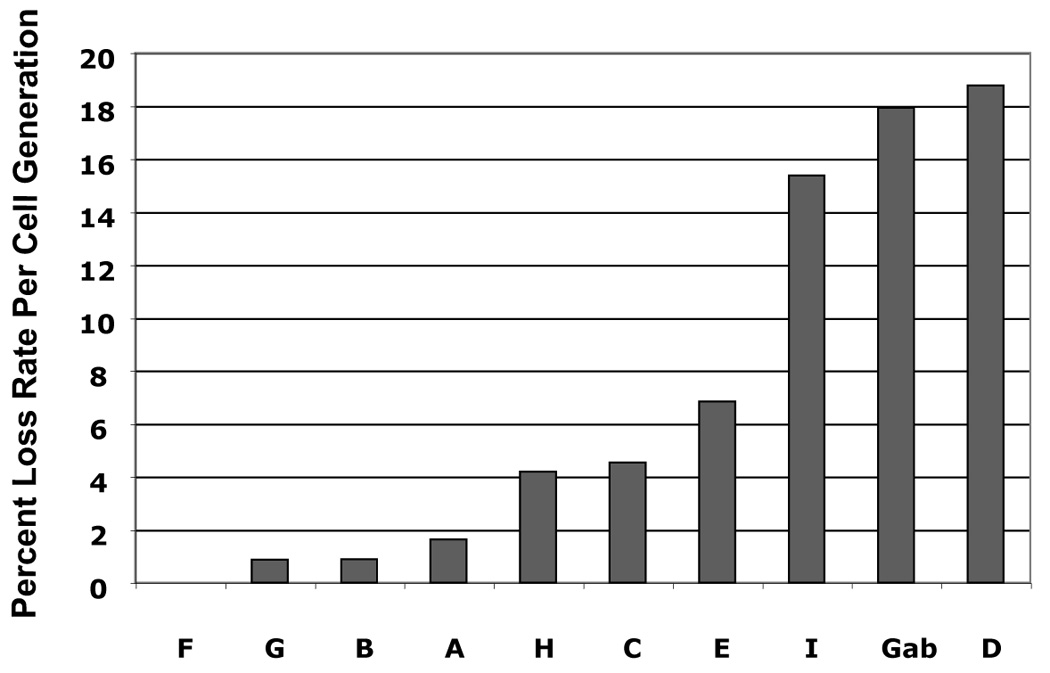
(A) DpnI-resistant newly synthesized DNAs were linearized with XhoI and detected by Southern blot at 4 days posttransfection. XhoI–treated DNAs indicated copy number were electrophoresed alongside. At the far left, DpnI-treated control DNA showed the completeness of DpnI cutting of DpnI-sensitive DNA from bacteria. (B) Southern analysis of Hirt extracted DNAs at 11 days after transfection. (C) The stability of plasmids containing HPV16 subgenomic fragments was demonstrated as the relative loss rate per cell generation. The percent of GFP-positive cells was determined by flow cytometry at different time points. The change in percent GFP-positive cells per cell generation (loss rate) was computed relative to the first time point of each transfection. The loss rates of each plasmid were normalized with that of the most stable plasmid to obtain the relative loss rate per cell generation.
To track the maintenance function imparted by elements in HPV16 DNA, we conducted flow cytometry to quantify the EGFP signal over the course of 10–14 days after cells were released from the G418 selection. Thus, the loss of EGFP signal represents plasmid the loss and indicates differences in relative intrinsic stability of the plasmids in human cells. To analyze plasmid stability, we calculated the loss of GFP-positive cells per cell generation and normalized with that of the most stable plasmid to obtain the relative loss rates. As shown in Fig.4C, the plasmids that carried certain parts of HPV16 genome were more stably maintained than was pGAB1. Among the HPV16 library plasmids, six of the nine Sau3AI subgenomic fragments (fragments A (nt 525–621), B(nt 622–870), C (nt 871–3,478), F (nt 5072–5233), G (nt 5234–6150), and H (nt 6151–6950) were identified to support long term maintenance as they exhibited lower relative loss rates comparing to vector alone. Those fragments are located within two distinct regions in the HPV16 genome; the first region resides in the early genes lying from the 3’portion of E6 ORF to the 5’segment of E2 ORF (fragments A, B, and C) and the other region is in the late genes that covers the 3’end of L2 and the entire L1 ORF (fragment F, G, and H) (Fig.4C). Interestingly, these maintenance elements mapped in human cells are overlap the regions of HPV16 that were previously identified as maintenance elements in yeast system (Fig. 6).
FIGURE 6. Comparison of replication and maintenance elements mapped in yeast and human cells.
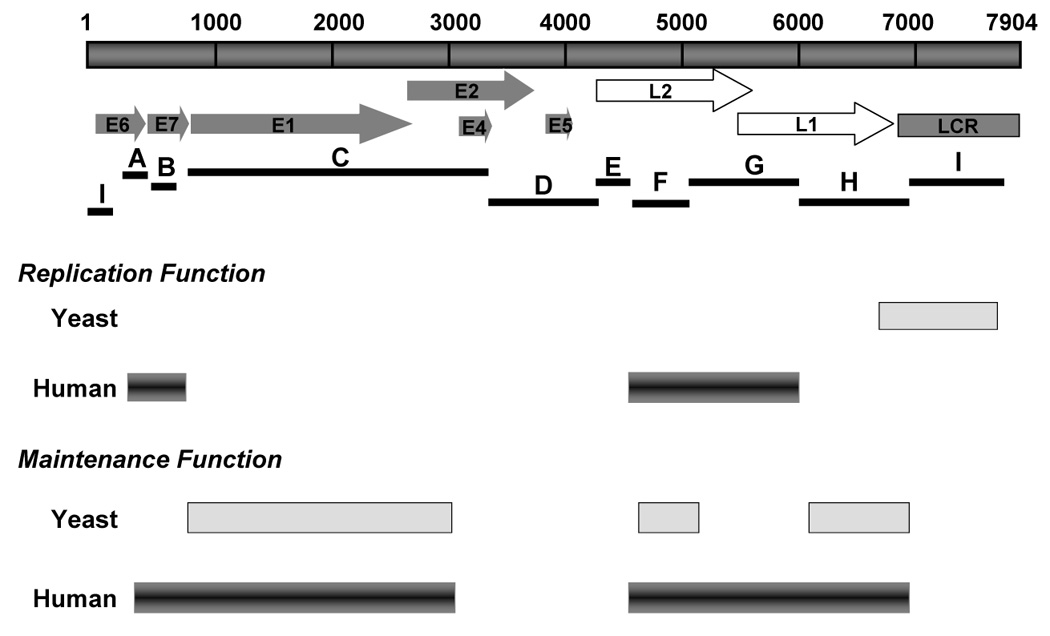
The replication and maintenance elements mapped in yeast are shown as light gray bars. The dark beveled bars show significant replication and maintenance functions mapped in human cells in this study.
Four fragments within maintenance elements contain both replication and maintenance functions
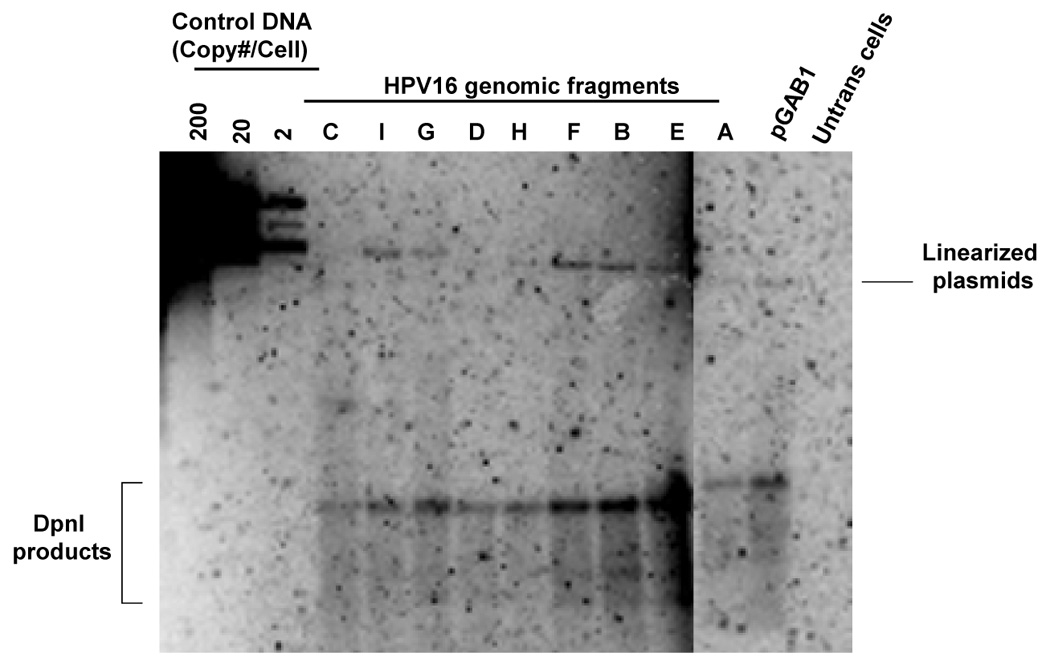
The plasmid library in pGAB1 backbone that is mentioned above was used to map region of the HPV16 genome that provides plasmid stability. Although all plasmids exhibited similar replication efficiency, we cannot rule out the possibility that some of these fragments would contain replication activity since such elements have been mapped in HPV16 genome using a yeast system. In order to identify replication elements in the viral genome, we generated another plasmid library in the absence of hARS by removing the hARS element from each HPV16 library constructs that were made in pGAB1. These plasmids allow us to identify the elements that support DNA replication based on their ability to substitute for the function of hARS. We tested whether these plasmids were able to replicate in 293 cells using a short-term replication assay and found that most of them replicated to differing extents (Fig 5A). The short-term replication assay showed the strongest initially detectable levels of DpnI-resistant DNA in fragments A (nt 525–621), B (nt 622–870), C (nt 871–3478), E (nt 4538–5071), F (nt 5072–5233), G (nt 5234–6150), and I (nt 7014-524) (Fig 5A). However, in long-term maintenance assays, using flow cytometry, we found plasmids harboring 4 subgenomic fragments were lost at the slower rate compared to vector alone, pEGFP, whereas those baring other fragments lost rapidly. The fragments that contained both replication and moderate to significant levels of maintenance function, included fragments A (nt 525–621), B (nt 622–870), F (nt 5072–5233), and G (nt 5234–6150) (Fig 5B). Interestingly, these fragments are located within two regions in the viral genome that we identified as maintenance elements using the pGab1 vector (Fig. 4B). The first region is in partial portion of E6 and E7 whereas the other one is in L2 and 5’ part of L1. We noted that the vector itself (pUCGFP) had some replication activity and was maintained to an extent over 10 days, however the regions A, B, F and G were maintained to a greater degree than the vector alone.
FIGURE 5. Identification of regions in HPV16 that is able to substitute for replication element.
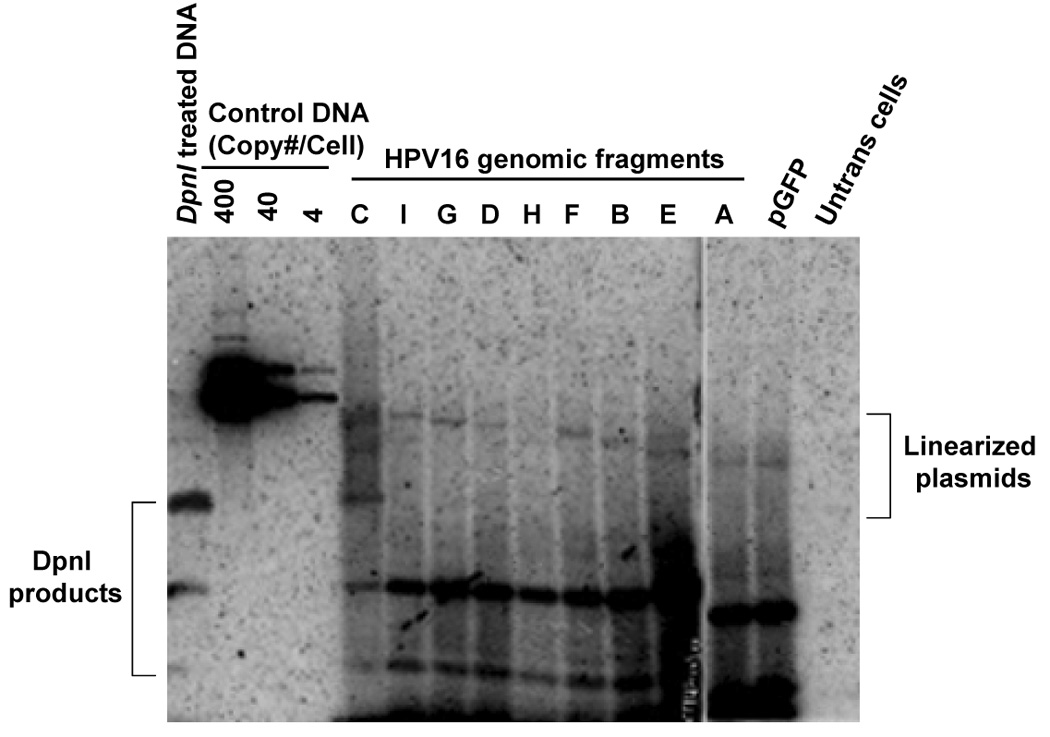
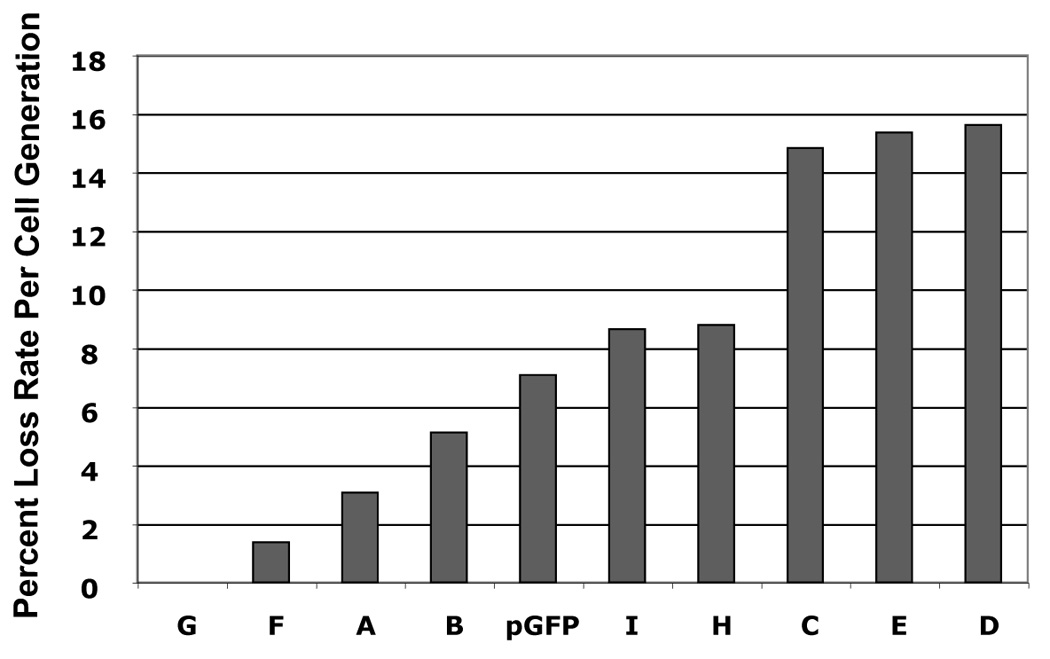
(A) Short-term replication of HPV16 library constructs in pGFP vector. Hirt-extracted DNA at 4 days posttransfection was analyzed by Southern blot after overnight digestion with DpnI and XhoI. (B) The loss rates of each plasmid was determined as the relative loss rate per cell generation. The loss rates of each plasmid was compared to that of the most stable constrtuct.
Analysis of the HPV16 genome for maintenance factor binding sites
We reasoned that factors that function in cellular chromosome maintenance are most likely to contribute to HPV maintenance, either independently, or in conjunction with E2. Therefore, we mapped the HPV16 genome, for the presence of HMG, CENP-B, TRF, MAR/SAR and Topo II binding sites, which others previously showed influence EBV maintenance (Deng et al., 2002; Hung, Kang, and Kieff, 2001; Masumoto et al., 1989). We discovered that the HPV16 genome contains high concentrations of Topo II sites in the LCR, L1, L2 and E7 ORFs, as well as HMG sites overlapping in L1 and E1. In addition, at least eight CENP-B binding sites were detected in the early and late regions of the genome. Of particular interest are the locations of MARS in L2, overlapping flanked by TRF binding sites. MAR elements are known to provide stability to replicating episomes in human cells (Cossons et al., 1997; Papapetrou et al., 2006) and mapping studies have identified MARS in HPV16 in E6, E1, E5, L2, and L1-LCR regions (Tan et al., 1998).
DISCUSSION
To establish a persistent infection, HPVs gains access to mitotically active basal-layer keratinocytes where low-copy replication begins. Viruses that replicate their DNA extrachromosomally, such as HPVs, EBV and KSHV, possess high fidelity mechanisms to maintain their genomes. The primary threats to stability of viral genomes are; loss by diffusion through the nuclear pores, loss during mitosis, or by nuclease-mediated decay, and integration into the host genome (Calos and Sclimenti, 1998; Deng et al., 2002). Viruses combat these problems by: (i) Nuclear retention and compartmentalization of genomes, thus protecting them from loss and degradation (Deng et al., 2002; Hung, Kang, and Kieff, 2001; Jankelevich et al., 1992; Nielsen et al., 2000; Swindle et al., 1999; van Brabant, Fangman, and Brewer, 1999). (ii) Improving segregation efficiency by attaching genomes to mitotic machinery (Ikeno et al., 1998; Kanda, Otter, and Wahl, 2001; Lehman and Botchan, 1998; Longtine et al., 1992).
During the maintenance phase, it is thought that low levels of E1 and E2 proteins are expressed in the infected basal cells. Given this and the fact that HPV genomes can persist for decades, this raises the question of whether cellular factors have a more prominent role in genome maintenance of HPVs than previously expected.
BPV1 E2 has been shown to interact with a chromatin-binding bromodomain protein (Brd4), thus implicating it as the mitotic chromosome anchor point for viral genomes (McBride, McPhillips, and Oliveira, 2004; You et al., 2004). The resulting model proposed for all PVs, is that chromosome anchoring allows equal partitioning of newly synthesized DNAs to daughter cells (McBride, McPhillips, and Oliveira, 2004). E2 from BPV and many HPVs (types 1, 4, and 8) have been shown to bind tightly to mitotic chromosomes. However, α-HPVs (types 11, 16, 31 and 57) appear to bind mitotic chromosomes to a much weaker extent (Oliveira, Colf, and McBride, 2006). In specific, α-HPV E2 proteins could be detected near chromosomes in prophase and telophase, but not in metaphase or anaphase (Oliveira, Colf, and McBride, 2006). A potential explanation for this disparity could be that partitioning observed among the α-HPVs could be dependent to a greater extent on cellular factors than HPVs for which E2 binds mitotic chromosomes tightly. Recent studies have shown that the interaction of E2 and Brd4 is not required for genome partitioning of all papillomaviruses, given by the fact that several HPV E2 proteins are capable of associating with mitotic chromosomes in the absence of Brd4 binding. Mutations in E2 have been described that disrupt the Brd4 interaction but do not affect the association of E2 with mitotic chromosomes (McPhillips et al., 2006). Thus, segregation of HPVs by interaction with chromosomes may occur through different cellular anchoring partners and the involvement of E2 may differ between genotypes. Ultimately, the role Brd4 in HPV segregation remains in question since recent work has shown that Brd4 functions as a chromatin binding transcriptional regulator and can function as a silencer (McPhillips et al., 2006; Wu and Chiang, 2007; Wu et al., 2006). Further studies have demonstrated that though Brd4 has a role in transcription of viral genes, it is not required for maintenance for all PVs.
In addition to Brd4, E2 has been shown to interact with a cellular helicase (ChlR1), which was initially discovered by an interaction with the yeast homologue of ChlR1 (Parish et al., 2006). In further studies, siRNA downregulation of ChlR1 resulted in a lack of E2 association with mitotic chromosomes, indicating a potential role for ChlR1 in loading of E2 onto chromosomal anchoring sites. Furthermore, a mutant of BPV E2 (W130R), which does not associate with mitotic chromosomes also fails to bind ChlR1 (Parish et al., 2006). Dao and colleagues described an HPV11 E2 interaction with mitotic spindles leading to an alternative model for E2-dependent genome segregation (Dao et al., 2006; Van Tine et al., 2004). Although these models disagree on the anchoring target for segregation, the role of E2 as the tethering factor is well accepted.
Motivating our studies, was the dramatic disparity in the number of E2BSs in PV genomes. For example, BPV-1 has at least 16 E2BSs, of which, at least 10 E2BSs are required for stable replication, per a study by Piirsoo et al. (Piirsoo et al., 1996). In contrast, α-HPVs usually have 4 E2BSs, only 3 of which are required for stable replication (Stubenrauch, Lim, and Laimins, 1998). In addition, certain regions of HPV16 genome have been shown to interact with nuclear matrix that might be involved in nuclear retention of the viral genome (Tan et al., 1998) These observations reinforce the concept that α-HPVs may rely to a lesser extent on E2 than BPV-1 for stable replication. We therefore, hypothesized that unknown cis-elements, together with their cellular binding partners, contribute to α-HPV maintenance. In our ongoing studies of HPV replication in yeast, we have found that 3 E2BSs are insufficient to restore stability to an unstable yeast plasmid (ARS+ CEN−) (unpublished data). Even in the presence of E2, E2BS-containing plasmids lacking a CEN become integrated into the yeast chromosome. In contrast, the HPV genome is stably maintained in the absence of E2 in yeast, yet the addition of E2 increases the copy number and improves genome stability. This indicates that E2 requires additional cellular factors for stabilization of HPV DNA and E2BSs alone, are not sufficient.
Our studies have defined cis-acting elements that provide a measurable degree of stability to HPV genomes independently of E1 and E2 functions. We created vectors that had either a complementing replication element (hARS) or not, each containing Sau3AI fragments of the HPV16 genome (Fig. 3A–C).
The pGAB1 plasmid, mentioned above, was used to map the regions of the HPV16 genome that provides plasmid stability. Although all plasmids exhibited similar replication efficiency but we cannot rule out the possibility that some of these fragments would contain replication activity since such elements have been mapped in HPV16 genome using a yeast system. In order to identify replication elements in the viral genome, we generated another plasmid library in the absence of hARS by introducing individual Sau3AI-HPV16 subgenomic fragments in pGFP, a derivative vector of pGAB1 that lacks hARS portion (Fig. 3C). This library plasmid allowed us to identify the elements that support DNA replication based on their ability to substitute the function of hARS. We tested whether these plasmids were able to replicate in 293 cells using a short-term replication assay and found that most of them replicated to varying extents. In a long-term maintenance assay using flow cytometry, the results revealed that the vector itself was maintained somewhat over the course of 10 days. However, four subgenomic fragments in HPV16 genome showed more stability with increasing of percent GFP-positive cells over time. Interestingly, these fragments are located within two regions in the viral genome that have been identified as maintenance elements. The first region is in partial portion of E6 and E7 whereas the other one is in L2 and 5’ part of L1.
Our mapping studies revealed that we mapped the HPV16 genome for the presence of cis-acting functions that could complement replication and maintenance functions, normally provided by the ARS and CEN elements in yeast plasmids (Kim et al., 2005). In that study, we discovered that three regions within HPV16 (nucleotides 871 to 3480, nucleotides 4538 to 5072, and nucleotides 6151 to 6951) were capable of providing maintenance function to complement an ARS+ /CEN− plasmid vector (pPA94).
In the current study, we found that in the absence of the hARS fragment, replication activity in HPV DNA segments could be detected, above the level of the vector (pGFP), in fragments A (nt 525–621), B (nt 622–870), C (nt 871–3478), E (nt 4538–5071), F (nt 5072–5233), G (nt 5234–6150), and I (nt 7014-524) (Fig. , 5B, 6). Perhaps the strongest replication activity was from fragments C and E. The fact that there is low-level replication activity not driven by E1 or E2, may be consistent with an alternate mode of replication. We discovered that at least four fragments within the genome contain both replication and maintenance functions in fragments A (nt 525–621), B (nt 622–870), F (nt 5072–5233), and G (nt 5234–6150) (Fig. 5B, 6). These data indicates that intrinsic low-level replication function could provide a wider range of copy control for HPV, depending on the level of quiescence required to evade cell-mediated immune responses present in the stratified epithelium. Furthermore, the assistance of cellular factors in viral DNA maintenance is has already been established in other viral systems.
Several studies using EBV OriP, have described multiple cellular factors that influence maintenance of OriP plasmids. Examples of recently described maintenance factors which influence EBV OriP stability are telomere repeat binding factor 2 (TRF-2), hRap1 and poly-ADP-ribose polymerase (tankyrase) (Deng et al., 2002). It has also been shown that the histone-related proteins, HMG and H1, when fused to EBNA-1 can complement its N-terminal chromatin binding function (Hung, Kang, and Kieff, 2001). A nucleolar protein known as EBP2, which interacts with EBNA-1, appears to be required for EBNA-1-dependent maintenance of OriP plasmids (Kapoor, Shire, and Frappier, 2001). Furthermore, the cellular kinesin-like protein, Kid, which competes with EBNA-1 for its binding sites and is known to bind the metaphase plate, has also been implicated in OriP maintenance (Zhang and Nonoyama, 1994). Finally, OriP artificial chromosomes are known to be stabilized by binding CENP-B (Masumoto et al., 1989).
The concept that evolves from our studies is that cellular factors may impart a greater influence on HPV replication and maintenance than previously expected. The fact that most HPVs contain only 4 E2BSs, while BPV1 contains at least 17 E2BS, suggests HPVs have evolved in a direction, to take greater advantage of cellular factors for genome persistence. There is evidence that extrachromosomal replicating DNA viruses make use of well-conserved chromosomal maintenance proteins. For example, EBV is now known to interact with multiple chromosomal maintenance proteins (TRF, H1 histones, Kid, metaphase plate proteins; SMC, and CENP-B (Deng et al., 2002; Hung, Kang, and Kieff, 2001; Masumoto et al., 1989). The detection of multiple MARS, HMG, Topo II, TRF and CENP-B binding sites and the clustering of multiple types of binding sites in the late region in HPV16 provide some potentially important candidates to consider as cellular maintenance factors. Ultimately, these observations suggest that there is likely to be more than mechanism by which cellular factors influence HPV maintenance.
ACKNOWLEDGEMENTS
We thank the Angeletti lab members for valuable discussion and critique of this work. We thank Paul Lambert (UW-Madison) for his helpful discussions about this work. We thank Kevin Lesh for creating the pGAB1 vector. This work was supported by NIH Grant: K01CA100736 to P.C.A. and by a COBRE grant (5P20RR015635) through the Nebraska Center for Virology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abroi A, Ilves I, Kivi S, Ustav M. Analysis of chromatin attachment and partitioning functions of bovine papillomavirus type 1 E2 protein. J Virol. 2004;78(4):2100–2113. doi: 10.1128/JVI.78.4.2100-2113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti PC, Kim K, Fernandes FJ, Lambert PF. Stable replication of papillomavirus genomes in Saccharomyces cerevisiae. J Virol. 2002;76(7):3350–3358. doi: 10.1128/JVI.76.7.3350-3358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency- associated nuclear antigen. Science. 1999;284(5414):641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- Ballestas ME, Kaye KM. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J Virol. 2001;75(7):3250–3258. doi: 10.1128/JVI.75.7.3250-3258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien N, McBride AA. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology. 2000;270(1):124–134. doi: 10.1006/viro.2000.0265. [DOI] [PubMed] [Google Scholar]
- Baxter MK, McBride AA. An acidic amphipathic helix in the Bovine Papillomavirus E2 protein is critical for DNA replication and interaction with the E1 protein. Virology. 2005;332(1):78–88. doi: 10.1016/j.virol.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Baxter MK, McPhillips MG, Ozato K, McBride AA. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J Virol. 2005;79(8):4806–4818. doi: 10.1128/JVI.79.8.4806-4818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calos MP, Sclimenti CR. Assaying extrachromosomal gene therapy vectors that carry replication/persistence elements. Adv Drug Deliv Rev. 1998;30(1–3):13–21. doi: 10.1016/s0169-409x(97)00103-8. [DOI] [PubMed] [Google Scholar]
- Clertant P, Seif I. A common function for polyoma virus large-T and papillomavirus E1 proteins? Nature. 1984;311(5983):276–279. doi: 10.1038/311276a0. [DOI] [PubMed] [Google Scholar]
- Cossons N, Nielsen TO, Dini C, Tomilin N, Young DB, Riabowol KT, Rattner JB, Johnston RN, Zannis-Hadjopoulos M, Price GB. Circular YAC vectors containing a small mammalian origin sequence can associate with the nuclear matrix. J Cell Biochem. 1997;67(4):439–450. doi: 10.1002/(sici)1097-4644(19971215)67:4<439::aid-jcb3>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Dao LD, Duffy A, Van Tine BA, Wu SY, Chiang CM, Broker TR, Chow LT. Dynamic localization of the human papillomavirus type 11 origin binding protein E2 through mitosis while in association with the spindle apparatus. J Virol. 2006;80(10):4792–4800. doi: 10.1128/JVI.80.10.4792-4800.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Lezina L, Chen CJ, Shtivelband S, So W, Lieberman PM. Telomeric proteins regulate episomal maintenance of epstein-barr virus origin of plasmid replication. Mol Cell. 2002;9(3):493–503. doi: 10.1016/s1097-2765(02)00476-8. [DOI] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83(4):655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000;74(14):6622–6631. doi: 10.1128/jvi.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouts ET, Yu X, Egelman EH, Botchan MR. Biochemical and electron microscopic image analysis of the hexameric E1 helicase. J Biol Chem. 1999;274(7):4447–4458. doi: 10.1074/jbc.274.7.4447. [DOI] [PubMed] [Google Scholar]
- Harrison S, Fisenne K, Hearing J. Sequence requirements of the Epstein-Barr virus latent origin of DNA replication. J Virol. 1994;68(3):1913–1925. doi: 10.1128/jvi.68.3.1913-1925.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Howley PM. Virology. 2nd edition ed. Philadelphia, Pa.: Lippincott-Raven Publishers; 1996. Papillomavirinae: the viruses and their replication; pp. 2045–2076. [Google Scholar]
- Hung SC, Kang MS, Kieff E. Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc Natl Acad Sci U S A. 2001;98(4):1865–1870. doi: 10.1073/pnas.031584698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeno M, Grimes B, Okazaki T, Nakano M, Saitoh K, Hoshino H, McGill NI, Cooke H, Masumoto H. Construction of YAC-based mammalian artificial chromosomes. Nat Biotechnol. 1998;16(5):431–439. doi: 10.1038/nbt0598-431. [DOI] [PubMed] [Google Scholar]
- Ilves I, Kivi S, Ustav M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which Is mediated by the viral E2 protein and its binding sites. J Virol. 1999;73(5):4404–4412. doi: 10.1128/jvi.73.5.4404-4412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankelevich S, Kolman JL, Bodnar JW, Miller G. A nuclear matrix attachment region organizes the Epstein-Barr viral plasmid in Raji cells into a single DNA domain. Embo J. 1992;11(3):1165–1176. doi: 10.1002/j.1460-2075.1992.tb05157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Otter M, Wahl GM. Coupling of mitotic chromosome tethering and replication competence in epstein-barr virus-based plasmids. Mol Cell Biol. 2001;21(10):3576–3588. doi: 10.1128/MCB.21.10.3576-3588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P, Shire K, Frappier L. Reconstitution of Epstein-Barr virus-based plasmid partitioning in budding yeast. Embo J. 2001;20(1–2):222–230. doi: 10.1093/emboj/20.1.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass SU, Landsberger N, Wolffe AP. DNA methylation directs a time-dependent repression of transcription initiation. Curr Biol. 1997;7(3):157–165. doi: 10.1016/s0960-9822(97)70086-1. [DOI] [PubMed] [Google Scholar]
- Kim K, Angeletti PC, Hassebroek EC, Lambert PF. Identification of cis-acting elements that mediate the replication and maintenance of human papillomavirus type 16 genomes in Saccharomyces cerevisiae. J Virol. 2005;79(10):5933–5942. doi: 10.1128/JVI.79.10.5933-5942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Lambert PF. E1 protein of bovine papillomavirus 1 is not required for the maintenance of viral plasmid DNA replication. Virology. 2002;293(1):10–14. doi: 10.1006/viro.2001.1305. [DOI] [PubMed] [Google Scholar]
- Lehman CW, Botchan MR. Segregation of viral plasmids depends on tethering to chromosomes and is regulated by phosphorylation. Proc Natl Acad Sci U S A. 1998;95(8):4338–4343. doi: 10.1073/pnas.95.8.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Knight J, Bream G, Stenlund A, Botchan M. Specific recognition nucleotides and their DNA context determine the affinity of E2 protein for 17 binding sites in the BPV-1 genome. Genes Dev. 1989;3(4):510–526. doi: 10.1101/gad.3.4.510. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Enomoto S, Finstad SL, Berman J. Yeast telomere repeat sequence (TRS) improves circular plasmid segregation, and TRS plasmid segregation involves the RAP1 gene product. Mol Cell Biol. 1992;12(5):1997–2009. doi: 10.1128/mcb.12.5.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupton S, Levine AJ. Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol Cell Biol. 1985;5(10):2533–2542. doi: 10.1128/mcb.5.10.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky KC, Batiza A, Lambert PF. Bovine papillomavirus type 1 E1 and simian virus 40 large T antigen share regions of sequence similarity required for multiple functions. J Virol. 1997;71(10):7600–7608. doi: 10.1128/jvi.71.10.7600-7608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masukata H, Satoh H, Obuse C, Okazaki T. Autonomous replication of human chromosomal DNA fragments in human cells. Mol Biol Cell. 1993;4(11):1121–1132. doi: 10.1091/mbc.4.11.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J Cell Biol. 1989;109(5):1963–1973. doi: 10.1083/jcb.109.5.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA, McPhillips MG, Oliveira JG. Brd4: tethering, segregation and beyond. Trends Microbiol. 2004;12(12):527–529. doi: 10.1016/j.tim.2004.10.002. [DOI] [PubMed] [Google Scholar]
- McBride AA, Oliveira JG, McPhillips MG. Partitioning viral genomes in mitosis: same idea, different targets. Cell Cycle. 2006;5(14):1499–1502. doi: 10.4161/cc.5.14.3094. [DOI] [PubMed] [Google Scholar]
- McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol. 2006;80(19):9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips MG, Ozato K, McBride AA. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J Virol. 2005;79(14):8920–8932. doi: 10.1128/JVI.79.14.8920-8932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29(13):2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen TO, Cossons NH, Zannis-Hadjopoulos M, Price GB. Circular YAC vectors containing short mammalian origin sequences are maintained under selection as HeLa episomes. J Cell Biochem. 2000;76(4):674–685. doi: 10.1002/(sici)1097-4644(20000315)76:4<674::aid-jcb15>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Oliveira JG, Colf LA, McBride AA. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc Natl Acad Sci U S A. 2006;103(4):1047–1052. doi: 10.1073/pnas.0507624103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou EP, Ziros PG, Micheva ID, Zoumbos NC, Athanassiadou A. Gene transfer into human hematopoietic progenitor cells with an episomal vector carrying an S/MAR element. Gene Ther. 2006;13(1):40–51. doi: 10.1038/sj.gt.3302593. [DOI] [PubMed] [Google Scholar]
- Parish JL, Bean AM, Park RB, Androphy EJ. ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol Cell. 2006;24(6):867–876. doi: 10.1016/j.molcel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Park P, Copeland W, Yang L, Wang T, Botchan MR, Mohr IJ. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc Natl Acad Sci U S A. 1994;91(18):8700–8704. doi: 10.1073/pnas.91.18.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Picha KM. Structure and function of hexameric helicases. Annu Rev Biochem. 2000;69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- Piirsoo M, Ustav E, Mandel T, Stenlund A, Ustav M. Cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. Embo J. 1996;15(1):1–11. [PMC free article] [PubMed] [Google Scholar]
- Rabson MS, Yee C, Yang YC, Howley PM. Bovine papillomavirus type 1 3′ early region transformation and plasmid maintenance functions. J Virol. 1986;60(2):626–634. doi: 10.1128/jvi.60.2.626-634.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears J, Ujihara M, Wong S, Ott C, Middeldorp J, Aiyar A. The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J Virol. 2004;78(21):11487–11505. doi: 10.1128/JVI.78.21.11487-11505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky RL, Hu J, Renne R. Analysis of Viral cis Elements Conferring Kaposi's Sarcoma-Associated Herpesvirus Episome Partitioning and Maintenance. J Virol. 2007;81(18):9825–9837. doi: 10.1128/JVI.00842-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubenrauch F, Laimins LA. Human papillomavirus life cycle: active and latent phases. Semin Cancer Biol. 1999;9(6):379–386. doi: 10.1006/scbi.1999.0141. [DOI] [PubMed] [Google Scholar]
- Stubenrauch F, Lim HB, Laimins LA. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J Virol. 1998;72(2):1071–1077. doi: 10.1128/jvi.72.2.1071-1077.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. Human papillomavirus DNA replication compartments in a transient DNA replication system. J Virol. 1999;73(2):1001–1009. doi: 10.1128/jvi.73.2.1001-1009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SH, Bartsch D, Schwarz E, Bernard HU. Nuclear matrix attachment regions of human papillomavirus type 16 point toward conservation of these genomic elements in all genital papillomaviruses. J Virol. 1998;72(5):3610–3622. doi: 10.1128/jvi.72.5.3610-3622.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. Embo J. 1991;10(2):449–457. doi: 10.1002/j.1460-2075.1991.tb07967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav M, Ustav E, Szymanski P, Stenlund A. Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. Embo J. 1991;10(13):4321–4329. doi: 10.1002/j.1460-2075.1991.tb05010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Brabant AJ, Fangman WL, Brewer BJ. Active role of a human genomic insert in replication of a yeast artificial chromosome. Mol Cell Biol. 1999;19(6):4231–4240. doi: 10.1128/mcb.19.6.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Drunen CM, Sewalt RG, Oosterling RW, Weisbeek PJ, Smeekens SC, van Driel R. A bipartite sequence element associated with matrix/scaffold attachment regions. Nucleic Acids Res. 1999;27(14):2924–2930. doi: 10.1093/nar/27.14.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tine BA, Dao LD, Wu SY, Sonbuchner TM, Lin BY, Zou N, Chiang CM, Broker TR, Chow LT. Human papillomavirus (HPV) origin-binding protein associates with mitotic spindles to enable viral DNA partitioning. Proc Natl Acad Sci U S A. 2004;101(12):4030–4035. doi: 10.1073/pnas.0306848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M, Wittmann K, Endl E, Glaser G, Knuchel R, Wolf H, Niller HH. Plasmid maintenance assay based on green fluorescent protein and FACS of mammalian cells. Biotechniques. 1998;24(4):540–542. 544. [PubMed] [Google Scholar]
- Wu H, Ceccarelli DF, Frappier L. The DNA segregation mechanism of Epstein-Barr virus nuclear antigen 1. EMBO Rep. 2000;1(2):140–144. doi: 10.1093/embo-reports/kvd026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282(18):13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20(17):2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J, Warren N, Reisman D, Sugden B. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci U S A. 1984;81(12):3806–3810. doi: 10.1073/pnas.81.12.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Camiolo SM, Bashaw JM. The minimal replicator of Epstein-Barr virus oriP. J Virol. 2000;74(10):4512–4522. doi: 10.1128/jvi.74.10.4512-4522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Warren N, Sugden B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature. 1985;313(6005):812–815. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]
- You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117(3):349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- Zhang S, Nonoyama M. The cellular proteins that bind specifically to the Epstein-Barr virus origin of plasmid DNA replication belong to a gene family. Proc Natl Acad Sci U S A. 1994;91(7):2843–2847. doi: 10.1073/pnas.91.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]