

Abstract
Background
The S1103Y-SCN5A polymorphism has been implicated as a pro-arrhythmic, sudden death predisposing risk factor in African Americans including one postmortem investigation of African American infants with sudden infant death syndrome (SIDS).
Objective
This study sought to assess whether or not the relatively African American-specific common polymorphism, S1103Y, in the SCN5A-encoded cardiac sodium channel is overrepresented in SIDS in African Americans.
Methods
Seventy-one cases from a population-based cohort of unexplained infant deaths among African Americans (37 females, 34 males, average age = 3 ± 2 months, range from birth to 11 months) were submitted to the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory for postmortem genetic testing. Polymerase chain reaction and a restriction digest assay was performed to genotype this cohort for S1103Y.
Results
Targeted mutational analysis of exon 18 in SCN5A of the African American SIDS cohort (n = 71) revealed the S1103Y polymorphism in 16/71 African American cases of SIDS (22.5%) compared to 135/1161 (11.6%) ostensibly healthy adult African Americans (p = 0.01).
Conclusion
This study provides an independent assessment of the prevalence of S1103Y-SCN5A among African American infants with a sudden, unexpected and unexplained death prior to their first birthday. Further scrutiny and quantification of the risk apparently associated with S1103Y seems warranted.
Keywords: cardiac sodium channel gene SCN5A, long QT syndrome, arrhythmia, sudden infant death syndrome, sudden death
INTRODUCTION
Sudden Infant Death Syndrome (SIDS) is defined as the sudden death of an infant under 1 year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of clinical history.1 Despite the success of the national “Back-to-Sleep” campaigns, SIDS claims the lives of over 2500 infants in the United States each year, remaining a leading cause of death in this vulnerable population.2–5
There is a significant disparity in SIDS rates among racial and ethnic groups. Specifically, rates are highest among African American infants and more than 2 times greater than white infants, thus suggesting a potential role for ethnic-specific genetic predisposition.6 While the pathophysiological mechanisms underlying most of these tragic deaths remain elusive, heritable cardiac arrhythmia syndromes such as long QT syndrome (LQTS), Brugada syndrome (BrS), and catecholoaminergic polymorphic ventricular tachycardia (CPVT) appear to account for 10–15% of SIDS, with the majority of SIDS related mutations being identified in the SCN5A-encoded cardiac sodium channel alpha subunit (Nav1.5) or its interacting proteins.7–15
In 2002, Splawski and colleagues reported on the common African American-specific polymorphism S1103Y-SCN5A (denoted previously as S1102Y) with a prevalence of 13% among African Americans and associated with an increased risk for arrhythmia susceptibility, particularly in the context of other acquired risk factors such as medications, hypokalemia or structural heart disease.16 Subsequently, Burke and colleagues observed an over-representation of S1103Y among African American adolescents and adults whose deaths were classified as autopsy negative sudden unexplained death (SUD).17
Plant and colleagues recently reported an over-representation of S1103Y homozygotes (YY) in a large cohort of African American SIDS (n = 133), suggesting a 24-fold risk for SIDS in those infants who are homozygous for the Y1103-encoding allele (YY genotype).18 In addition, elegant heterologous expression of Y1103-containing sodium channels yielded significant late sodium current when environmentally stressed with acidosis fulfilling the triple-risk hypothesis of SIDS, namely the vulnerable host (S1103Y positive), an exogenous stressor (acidosis), and a critical development period.18
As is the case for any association study, population stratification can create false positive associations, and Plant and colleagues stated unequivocally that their findings “need to be replicated in a separate SIDS cohort of African Americans.”18 Here, we assess the prevalence of S1103Y-SCN5A in a smaller African American SIDS cohort.
Population-Based Cohort of SIDS
Between January 1996 and December 2000, 71 cases of unexplained sudden deaths among African American infants (37 females, 34 males, average age = 3 ± 2 months, range from birth to 11 months) derived from three population-based cohorts of SIDS were submitted to the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory for postmortem genetic testing. To be rendered SIDS, the death of the child under age one year had to be sudden, unexpected, and unexplained following a comprehensive medico-legal autopsy.1 Infants whose death was due to asphyxia or specific disease were excluded.
This study was approved by Mayo Clinic’s Institutional Review Board as an anonymous study. As such, only limited medical information was available, including sex, ethnicity, and age at death. Time of day, medication use, and position at death were not available. By definition, the infant’s medical history and family history were negative.
S1103Y-SCN5A Genotyping
DNA was extracted from autopsy blood using the Puregene DNA Isolation kit (Gentra, Minneapolis, MN) or from frozen necropsy tissue using the Qiagen DNeasy Tissue Kit (Qiagen, Inc, Valencia, CA). Genomic DNA derived from 100 healthy African American subjects was obtained from the Human Genetic Cell Repository sponsored by the National Institute of General Medical Sciences and the Coriell Institute for Medical Research (Camden, NJ) and served as ethnic matched controls.
The S1103Y genotype was determined by PCR amplification of SCN5A exon 18, restriction enzyme analysis and gel electrophoresis. PCR reactions were performed in 20 µL volumes using 50 ng DNA, 16 pmol of each primer19 (sense primer 5’AGGGTCTGAAACCCCCAGGGTCA3’ and antisense primer 5’CCAGCTGGCTTCAGGGACAAA 3’), 200 µM of each dNTP, 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 2.0 mM MgCl2, and 1.0 U Amplitaq Gold (Applied Biosystems, Foster City, CA). Restriction enzyme analysis was performed using 1 µL of PCR product, 1 µL of enzyme digestion buffer (10 mM Tris-HCl, 10 mM MgCl2, 50 mM NaCl, 1 mM dithiothreitol (pH 7.9 at 25°C), 2 U BseRI (New England Biolabs, Ipswich, MA), and 7.5 µL of deionized water. The reaction mixture was incubated at 37° C for 2hr, followed by 65° C for 20 min. Digested samples were separated on a 3% agarose gel and visualized with a UV transiluminator (Figure 1). Direct sequencing of samples showing representative gel profiles confirmed the presence of S1103Y as described previously.20
Figure 1. Molecular characterization of S1103Y polymorphism.
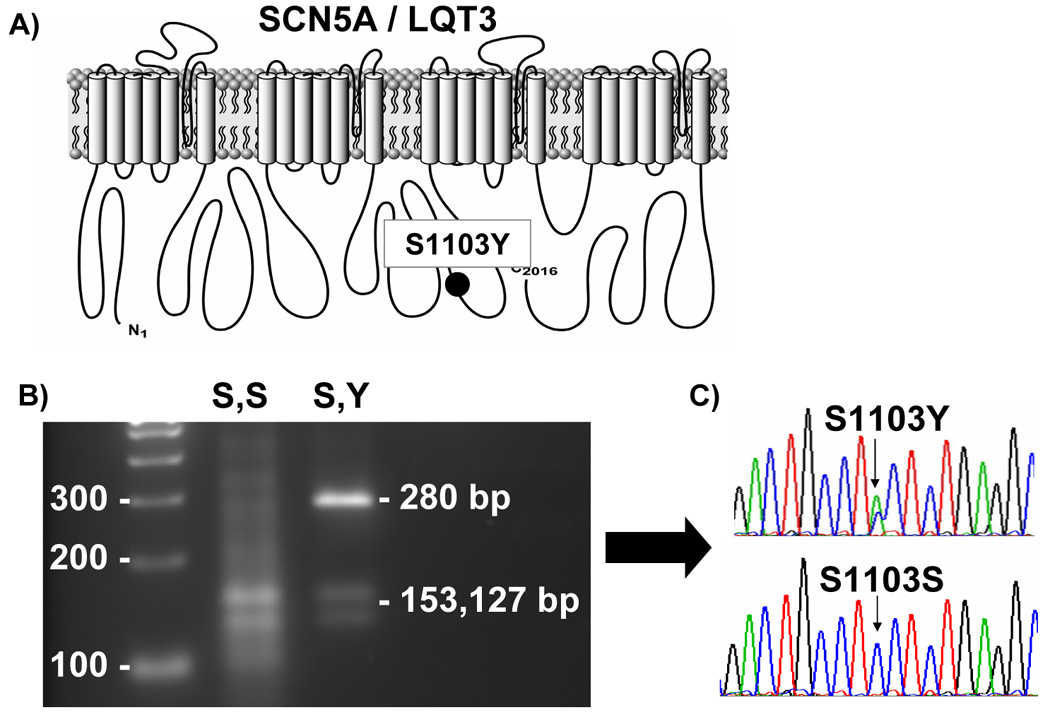
(A) depicts the S1103Y polymorphism within the SCN5A sodium channel. (B) depicts the restriction digest, yielding the associated fragments and subsequent DNA sequencing (C).
Statistical analysis
In addition to the genotype data derived here from 100 additional African American control subjects, S1103Y genotype data reported for 1061 healthy, ethnic-matched adult controls from previous studies were included for analysis16, 18. Chi squared analysis was used to analyze the significance of any observed variation in the genotype distribution between the groups. Statistical significance was determined by a p value < 0.05.
RESULTS
Our cohort of 71 African American SIDS cases was comprised of 34 males and 37 females with an average age at death of 3 ± 2 months. Consistent with prior epidemiologic studies of SIDS, the majority of infant deaths occurred prior to 6 months. Only 4/71 infants (5.6%) died ≥ 6 months of age.
Depicted in Figure 1 is the channel location of S1103Y and the methodology by which S1103Y genotyping was performed. Briefly, the S1103Y polymorphism is a result of a single nucleotide substitution of a cytosine (c) for an alanine (a) in the second position of codon 1103 (n. 3308 c>a). The restriction enzyme BseRI recognizes the S1103 “wildtype” allele and cleaves the PCR product DNA into two distinct bands (153 base pairs and 127 base pairs) when separated by gel electrophoresis. The Y1103 “mutant” allele abolishes this naturally occurring restriction enzyme recognition site (CTCCTC > CTACTC), resulting in a 280 base pair size band. Hence, S1103Y heterozygotes will yield three DNA bands, whereas S1103 homozygotes will yield two bands following gel electrophoresis.
Overall, genotyping analysis revealed that 16/71 (22.5%) African American SIDS cases were heterozygous for the pro-arrhythmic S1103Y-SCN5A polymorphism compared to 135/1161 (11.6%) healthy African American control subjects (Figure 2, p = 0.01). None of the SIDS victims assessed were homozygous for Y1103. However, our study was not powered sufficiently to draw any significant conclusions from the absence of YY genotypes. There was no significant difference in the presence of S1103Y genotype between male (7/34, 20.6%) and female SIDS victims (9/37, 24.3%, p = 0.8). While SIDS cases with S1103Y genotype appeared older (average age = 3.2 ± 1.5 months) than SIDS cases wildtype for S1103 (average age = 2.89 ± 2.2 months), this was not significant.
Figure 2. Frequency of S1103Y Polymorphism Between Cases and Controls.
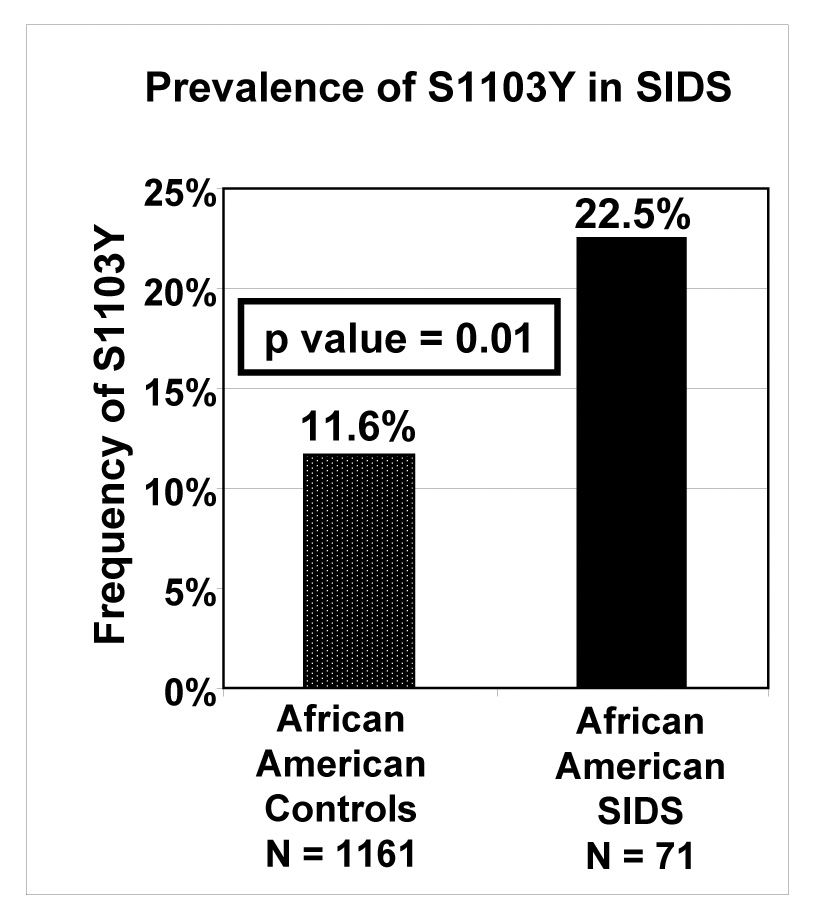
The graph depicts the overall prevalence of S1103Y in ostensibly healthy African American adults (controls) and African American cases of SIDS (p = 0.01).
DISCUSSION
A significant substrate for “infant vulnerability” to SIDS that has emerged over the last decade involves pathogenic defects in the genes that encode critical ion channel proteins and channel interacting proteins (i.e. cardiac channelopathies) in the heart.7–11, 13–15 Such perturbations may account for up to 15% of SIDS9, 11, 12, 14 with mutations in the SCN5A-encoded cardiac sodium channel being the most abundant.21 Besides pathogenic mutations, some rare SCN5A genetic variants observed among cases and healthy individuals may be functionally relevant and confer risk for SIDS under certain conditions.21 However, common polymorphisms, such as the African American-specific S1103Y-SCN5A channel variant present in approximately 10–13% of healthy African Americans, may also confer increased risk for sudden cardiac arrest.16–18
In the original discovery that linked S1103Y with cardiac risk in African Americans, Splawski and colleagues reported an over-representation of S1103Y in unrelated African American adults with arrhythmias (47.8% versus 13.0% of controls) and further showed that Y1103 was linked with QTc prolongation and/or a history of syncope in a single large African-American family in which nearly 50% of the phenotypically affected individuals were heterozygous for S1103Y.16 Burke and colleagues subsequently noted an 8-fold increased risk for sudden unexplained arrhythmic death in young African Americans (age > 10 years), where 28% were identified as heterozygous for S1103Y.17 In our own small cohort of young African Americans with autopsy negative SUD over the age of 1 year, 57% (4 of 7) hosted S1103Y (unpublished data).
Recently, Plant and colleagues demonstrated that S1103Y was overrepresented among deceased African American infants whose death was classified as SIDS.18 Specifically, they reported that infants homozygous for Y1103 had a 24-fold increased risk for SIDS due to an LQT3-like increase in late sodium current elicited by an exogenous stressor of acidosis, a known risk factor for SIDS. In their examination of African American SIDS victims (n = 133), Plant and colleagues reported that 3 (2%) of the decedents were homozygous for the minor Y1103-encoding allele which was statistically significant. Unlike the prior observations of S1103Y over-representation among adult African Americans with premature, unexplained sudden cardiac arrest, S1103Y heterozygotes (9.8%) were observed with a similar frequency as seen among the ethnic-matched reference alleles that were derived from healthy subjects.
In this study, however, the S1103Y common polymorphism was over-represented among African American SIDS cases when compared to the heterozygous frequency observed and replicated in multiple independent cohorts of ostensibly healthy African Americans. These data provide the fourth data set overall and the second independent data set among SIDS in African American infants that demonstrates and confirms that the Y1103-encoding allele confers increased susceptibility for premature sudden death. We did not discover any SIDS infants homozygous for Y1103, but our sample size was too small to draw any conclusions from this observation.
S1103Y-SCN5A should now be viewed as a relatively African American-specific common polymorphism that 1) functionally alters the properties of the NaV1.5 cardiac sodium channel in a manner that yields a pro-arrhythmic molecular phenotype in the setting of concomitant pharmacological inhibition of the KCNH2-encoded IKr potassium channel (i.e. HERG block16) and acidosis18 and 2) clinically has been shown to be overrepresented among i) living African American adults with a significant arrhythmic phenotype,16 ii) previously healthy deceased African American adults with a postmortem diagnosis of autopsy negative SUD,17 and iii) previously healthy, deceased African American infants with a postmortem diagnosis of SIDS.18
Taken together, these studies suggest that S1103Y may confer increased susceptibility for a premature sudden cardiac arrest among African Americans at any age. Obviously, this could have tremendous implications among the African American community where 10–15% of individuals are positive for this pro-arrhythmic, sudden death predisposing risk factor. With reported odds ratios/relative risks on the order of 8 – 24 fold increased risk of premature sudden cardiac arrest, the relative strength of this genetic risk factor certainly equals or exceeds that of traditional risk factors for coronary artery disease that have prompted lifestyle modifications and initiation of pharmacotherapy intended to attenuate risk. Nevertheless, given that the absolute risk for SCA secondary to S1103Y remains extremely low, it is difficult to know what interventions are appropriate and necessary to provide meaningful risk reduction for the 10–15% of African Americans who will host this at-risk biomarker.
In theory, the ability to perform routine genetic testing for S1103Y in newborn African American infants would be quite simple and straightforward. Routine testing for many metabolic, endocrine, and hematologic disorders, among others, are already being performed on newborns as part of a universal newborn screen. Approximately 616,000 African American infants are born each year, with an estimated 60,000 – 90,000 hosting the S1103Y polymorphism,6 and Splawski and colleagues suggested that overall, 4.6 million African Americans of any age carry S1103Y.16 However, despite an impressive odds ratio for increased sudden death susceptibility, the overwhelming majority will not die suddenly secondary to S1103Y and although SIDS has an increased incidence in African Americans,6 its overall incidence with respect to the African American population is still quite low (< 1,000 African American SIDS cases per year).6
This raises a provocative dilemma as to the necessity, nature, and extent of a possible intervention for the identified host who possesses this sudden death risk factor. Perhaps a prudent initial approach may be the cautious use of the 50 FDA-approved medications that are known to prolong the QT interval as an unwanted effect. Clearly, large prospective studies are needed with respect to genotyping for this pro-arrhythmic polymorphism in the African American subpopulation and evaluating appropriate interventions for the S1103Y-positive host.
CONCLUSION
This study provides an independent assessment of the prevalence of S1103Y-SCN5A among African American infants with a sudden, unexpected and unexplained death prior to their first birthday. Further scrutiny and quantification of the risk apparently associated with S1103Y seems warranted.
ACKNOWLEDGEMENTS
We gratefully acknowledge the medical examiners, coroners, and forensic pathologists from across the United States for their referrals of SIDS cases.
Dr. Michael J. Ackerman’s research program is supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program (Rochester, MN), the CJ Foundation for SIDS (Hackensack, NJ), the Dr. Scholl Foundation (Northbrook, IL), the Hannah Wernke Memorial Foundation (Strongsville, OH), the American Heart Association (Established Investigator Award – Dallas, TX), and the National Institutes of Health (HD042569-05A1 – Bethesda, MD). The project described was supported by Grant Number R01HD042569 from the National Institute Of Child Health And Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute Of Child Health And Human Development or the National Institutes of Health.
Abbreviations
- SIDS
sudden infant death syndrome
- SCN5A
voltage-gated sodium channel
- LQTS
Long QT syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Krous HF, Beckwith JB, Byard RW, et al. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114:234–238. doi: 10.1542/peds.114.1.234. [DOI] [PubMed] [Google Scholar]
- 2.Willinger M, Hoffman HJ, Hartford RB. Infant sleep position and risk for sudden infant death syndrome: report of meeting held January 13 and 14, 1994, National Institutes of Health, Bethesda, MD. Pediatrics. 1994;93:814–819. [PubMed] [Google Scholar]
- 3.Gibson E, Cullen JA, Spinner S, et al. Infant sleep position following new AAP guidelines. American Academy of Pediatrics. Pediatrics. 1995;96:69–72. [PubMed] [Google Scholar]
- 4.Dwyer T, Ponsonby AL, Blizzard L, et al. The contribution of changes in the prevalence of prone sleeping position to the decline in sudden infant death syndrome in Tasmania. JAMA. 1995;273:783–789. [PubMed] [Google Scholar]
- 5.American Academy of Pediatrics AAP Task Force on Infant Positioning and SIDS: Positioning and SIDS. Pediatrics. 1992;89:1120–1126. [PubMed] [Google Scholar]
- 6.Mathews TJ, MacDorman MF. Infant mortality statistics from the 2004 period linked birth/infant death data set. Natl Vital Stat Rep. 2007;55:1–32. [PubMed] [Google Scholar]
- 7.Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long- QT syndrome. N Engl J Med. 2000;343:262–267. doi: 10.1056/NEJM200007273430405. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz PJ, Priori SG, R. B, et al. Molecular diagnosis in a child with sudden infant death syndrome. Lancet. 2001;358:1342–1343. doi: 10.1016/S0140-6736(01)06450-9. [DOI] [PubMed] [Google Scholar]
- 9.Ackerman MJ, Siu BL, Sturner WQ, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286:2264–2269. doi: 10.1001/jama.286.18.2264. [DOI] [PubMed] [Google Scholar]
- 10.Skinner JR, Chung S-K, Montgomery D, et al. Near-miss SIDS due to Brugada syndrome. Arch Dis Child. 2005;90:528–529. doi: 10.1136/adc.2004.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tester DJ, Ackerman MJ. Sudden infant death syndrome: How significant are the cardiac channelopathies? Cardiovasc Res. 2005;67:388–396. doi: 10.1016/j.cardiores.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Arnestad M, Crotti L, Rognum TO, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 13.Cronk LB, Ye B, Kaku T, et al. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4:161–166. doi: 10.1016/j.hrthm.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tester DJ, Dura M, Carturan E, et al. A mechanism for sudden infant death syndrome (SIDS): Stress-induced leak via ryanodine receptors. Heart Rhythm. 2007;4:733–739. doi: 10.1016/j.hrthm.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Norstrand DW, Valdivia CR, Tester DJ, et al. Molecular and Functional Characterization of Novel GPD1-L Mutations in Sudden Infant Death Syndrome. Circulation. 2007;116:2253–2259. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–1336. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 17.Burke A, Creighton W, Mont E, et al. Role of SCN5A Y1102 polymorphism in sudden cardiac death in blacks. Circulation. 2005;112:798–802. doi: 10.1161/CIRCULATIONAHA.104.482760. [DOI] [PubMed] [Google Scholar]
- 18.Plant LD, Bowers PN, Liu QY, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. Journal of Clinical Investigation. 2006;116:430–435. doi: 10.1172/JCI25618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Li Z, Shen J, et al. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics. 1996;34:9–16. doi: 10.1006/geno.1996.0236. [DOI] [PubMed] [Google Scholar]
- 20.Ackerman MJ, Tester DJ, Jones G, et al. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479–1487. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- 21.Wang DW, Desai RR, Crotti L, et al. Cardiac Sodium Channel Dysfunction in Sudden Infant Death Syndrome. Circulation. 2007;115:368–376. doi: 10.1161/CIRCULATIONAHA.106.646513. [DOI] [PubMed] [Google Scholar]