

Abstract
Herpes simplex virus encephalitis (HSE) is the most common fatal sporadic encephalitis in humans. HSE is primarily caused by herpes simplex virus (HSV)-1 infection of the brain. HSE results in increased levels of oxidative stress, including the production of reactive oxygen species, free radicals, and neuroinflammation. The most biologically active form of vitamin E (VE) is α-tocopherol (α-TOC). In cellular membranes, α-TOC prevents lipid peroxidation by scavenging free radicals and functioning as an antioxidant. Supplementation with VE has been shown to decrease immunosenescence, improve immune function, and may be neuroprotective. To determine how VE deficiency and VE supplementation would alter the pathogenesis of HSE, we placed weanling male BALB/cByJ mice on VE-deficient (VE-D), VE-adequate (VE-A), or 10× VE-supplemented diets for 4 wk, and then infected the mice intranasally with HSV-1. VE-D mice had more severe symptoms of encephalitis than VE-A mice, including weight loss, keratitis, hunched posture, and morbidity. VE-D mice had increased cytokine and chemokine expression in the brain and increased viral titers. In contrast, VE supplementation failed to decrease cytokine production and had no effect on viral titer. We demonstrated that adequate levels of VE are important in limiting HSE pathology and that 10× supplementation does not enhance protection.
Introduction
Herpes simplex virus encephalitis (HSE)3 is the most common fatal sporadic encephalitis in humans (1–3). Ninety percent of all HSE cases are caused by herpes simplex virus (HSV)-1 (4). Untreated, HSE has a 70% mortality rate. Treatment with antiviral medication, such as acyclovir, decreases HSE associated mortality to 20%; however, only 38% of HSE patients recover to normal function (4,5). HSE is a substantial problem for the immunosuppressed, including people with HIV and those undergoing chemotherapy.
When administered intranasally (i.n.), HSV-1 enters the central nervous system (CNS) along neuronal pathways of the olfactory and trigeminal nerves (6). This route of infection results in an acute necrotizing encephalitis involving the olfactory and limbic systems, including the olfactory bulb, hypothalamus, thalamus, amygdala, hippocampus, and olfactory and entorhinal cortices. In this model, HSV-1 primarily infects neurons and glial cells (7,8). The infection of neurons and glia induce the production of proinflammatory cytokines produced by microglia and infiltrating macrophages, as well as the production of chemokines and antiviral cytokines (9,10). As virus replication continues, both CD4+ and CD8+ T lymphocytes infiltrate the brain (11–13). The intranasal route of infection mimics the hypothesized route of human HSE, where it is believed that the virus enters the CNS via the olfactory pathway or via the trigeminal ganglion (1,14,15). The intranasal model of HSV-1 infection has been well characterized in mice (8,13,16–19).
Vitamin E (VE) is a family of tocopherols and tocotrienols, of which α-tocopherol (α-TOC) is the most biologically active and second most abundant in food (20). These lipid soluble anti-oxidant vitamins are found in cellular membranes and prevent lipid peroxidation by scavenging free radicals (21). Deficiency in VE is associated with increased oxidative stress, central and peripheral neuropathies, and impaired immune function (22–24). VE deficiency increases the parasite load and pathology in mice that are experimentally infected with Heligmosomoides polygyrus (23). VE deficiency also decreases T- and B-cell numbers in rats infected with Trypanosoma cruzi (24).
Supplementation with VE has been shown to decrease immunosenescence, improve immune function, and may be neuroprotective. VE supplementation is capable of modulating T-cell cytokines, including interferon (IFN)-γ (25,26). Short-term, high-dose VE supplementation in colorectal cancer patients increases the production of both IFNγ and interleukin (IL)-2 (25). High dietary VE increases IFNγ and IL-2 production in aged mice after an influenza infection (26). In a recent study, Han et al. (27) determined that VE affects a wide range of immune-related genes in old, but not young, mice. Additionally, VE has been shown to stop the age-related decline in the formation of CD4 T-cell synapses (28). In young restraint-stressed mice, VE has been shown to increase the production of IFNγ and IL-2 in concanavalin A–stimulated splenocytes (29).
Studies from our laboratory have pointed to a key role for antioxidant micronutrients, including VE, in the pathogenesis of infectious diseases (30–34). Specifically, we showed that VE supplementation is capable of decreasing coxsackievirus-induced myocarditis in selenium-deficient mice. In the absence of VE, iron-loaded mice significantly increased coxsackievirus-induced myocarditis compared with iron-loaded, VE-adequate (VE-A) mice. Together, studies from our laboratory and others indicate that VE has the potential to modulate the immune response to a viral pathogen.
Because the brain is rich in lipids, we hypothesized that a deficiency in VE would increase HSE pathology in mice, and furthermore, VE supplementation would reduce the symptoms of HSV-1 encephalitis.
Materials and Methods
Mice, diets, and infection
Weanling BALB/cByJ male mice (Jackson Labs) were fed ad libitum 1 of 3 diets: 1) a VE-deficient (VE-D) diet (TD 88163), 2) a VE-A (dl-α -tocopheryl acetate) diet (38.4 mg/kg), or 3) a VE-supplemented (VE-S) diet (384 mg/kg) (Harlan Teklad) (Table 1). After 4 wk on the diets, the mice were lightly anesthetized with a ketamine (0.6 mg/kg) and xylazine (0.35 mg/kg) solution and infected i.n. with a 1.5 × 106 plaque forming unit (PFU) of HSV-1 in 10 μL total volume. All mice were housed 4 per cage in the University of North Carolina Animal Facility, which is fully accredited by the American Association for Accreditation of Laboratory Animal Care. Animals were maintained under protocols approved by the Institutional Animal Use and Care Committee.
TABLE 1.
Composition of experimental diets1
| Diet
|
|||
|---|---|---|---|
| Deficient | Adequate | Supplemented | |
| g/kg | |||
| Casein, “vitamin-free” | 200.0 | 200.0 | 200.0 |
| DL-methionine | 3.0 | 3.0 | 3.0 |
| Dextrose, monohydrate | 674.3 | 674.3 | 674.3 |
| Corn oil, tocopherol-stripped | 50.0 | 50.0 | 50.0 |
| Cellulose | 30.0 | 30.0 | 30.0 |
| Mineral mix, AIN-76 | 35.0 | 35.0 | 35.0 |
| DL-α-tocopheryl acetate (500 IU/g) | 0 | 0.077 | 0.77 |
Additional minerals and vitamins: calcium carbonate, 3.5; choline, 3.5; vitamin A palmitate (500,000 U/g), 0.04; cholecaliferol (500,000 U/g), 0.0044; vitamin B-12, 0.05; biotin, 0.0004; calcium pantothenate, 0.066; folic acid, 0.002; inositol, l0.11; menadione, 0.05; niacin, 0.1; pyridoxine HCl, 0.022; riboflavin, 0.022; thiamin HCl, 0.022.
HSV-1 virus stocks and virus inactivation
HSV-1 McIntyre (ATCC) stocks were propagated in Vero cells (ATCC), collected, centrifuged(750 × g; 5 min), and stored at −80°C. Vero cells were maintained in DMEM supplemented with 2 mmol/L glutamine and adjusted with 1.5 g/L sodium bicarbonate, 0.1 mmol/L nonessential amino acids, 1.0 mmol/L sodium pyruvate, and 10% fetal bovine serum.
HSV-1 was inactivated by placing 1 mL aliquots in 30 mm tissue culture dishes (Becton-Dickinson) 2.5 cm from a germicidal UV light source for 6 min. Inactivation was confirmed by adding the inactivated virus to Vero cells to verify lack of viral replication.
Pathology and tissue collection
Following infection, mice were weighed, examined daily, and scored on the following scale: 0, no symptoms; 1, ruffled fur, ataxia; 2, hind-limb paralysis/forelimb clasping; 3, hind-limb paralysis with forelimb weakness; 4, moribund; 5, dead. For PCR and viral titer experiments, uninfected (UNI, d0), d 3 and 7 postinfection (p.i.) mice were killed by rapid cervical dislocation, and the brain was removed and quickly dissected on ice and flash frozen.
Liver and brain α-TOC measurements
α-TOC levels were measured by HPLC following standard methods (35).
Brain cytokine measures
Levels of mRNA were determined by isolating total RNA from the forebrain region (thalamus and hypothalamus) using the TRIzol method (Invitrogen). Reverse transcription was carried out using Superscript II First Strand Synthesis kit (Invitrogen) with oligo (dT) primers. Expression of cytokine and chemokine mRNA was determined by quantitative RT-PCR (34). The levels of mRNA for glyceraldehyde-3-phosphate dehydrogenase were determined for all samples and used to normalize gene expression.
Brain stems (BS) were collected into 0.5 mL of ice-cold DMEM and homogenized, clarified by centrifugation (2500 × g; 3 min.), and stored at −80°C until assayed for regulated upon activation, normal T-cell expressed and secreted (RANTES) and IFNγ inducible protein-10 with a Luminex-based multiplex ELISA kit (Biosource) and IL-1β and tumor necrosis factor (TNF)-α ELISAs (BD Pharmingen) following the manufacturer’s instructions.
HSV-1 titers in brain
HSV-1 viral titers from the whole brain were determined from homogenized brain tissue by standard plaque assay on Vero cells. For viral titers from the olfactory bulb and brain stem genomic DNA, PCR was performed as previously described (36). DNA from UNI tissue was extracted in parallel and served as a negative control.
Statistics
Mortality data were analyzed by Kaplan-Meier survival analysis. All other data were analyzed by the nonparametric Kruskal-Wallis test. Kaplan-Meier survival analysis was performed using Prism 4 (GraphPad). All other statistical analyses were performed with JMP 6 software (SAS Institute). Values are the mean ± SEM. Data were considered statistically significant if P < 0.05.
Results
Brain and liver α-TOC levels
After 4 wk on the diet, mice were killed and their brains and livers removed. Altering α-tocopheryl acetate levels in the diets was effective in changing peripheral α-TOC levels. Liver levels of α-TOC were significantly decreased in VE-D mice compared with VE-A and VE-S mice (Fig. 1). The VE-S mice had nearly 7.5 times as much α-TOC as the VE-A mice. In the CNS, the temporal lobes of VE-D mice had significantly less α-TOC compared with the VE-A and VE-S mice (Fig. 1). However, in contrast to the liver, the high α-tocopheryl acetate diet was not effective in increasing brain α-TOC levels.
FIGURE 1.
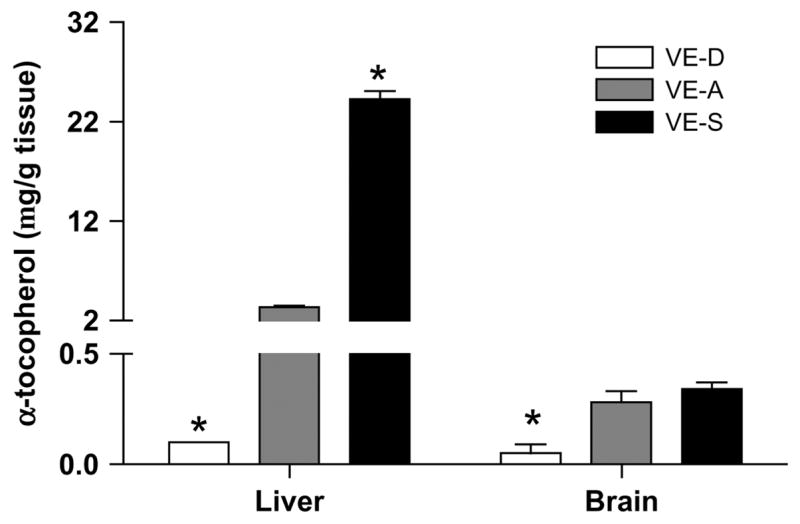
α-TOC concentrations in the liver and brain of mice fed VE-D, VE-A, and VE-S diets for 4 wk. Data are the mean ± SEM, n = 4. *Different from VE-A at that time point, P < 0.05. α-TOC MW = 430.7.
Mice on VE-D diets have earlier onset of HSE symptoms compared with mice on the VE-A diet
After 4 wk, mice were infected with 1.5 × 106 PFU of HSV-1 and followed for symptoms of HSE. VE-D mice had increased HSE symptoms as well as a 28.6% mortality by d 7 p.i., whereas no VE-A mice died by d 7 p.i. (P = 0.03). VE-A mice had fewer symptoms of encephalitis (and no mortality) compared with VE-D mice (Table 2).
TABLE 2.
Increased mortality and HSE symptoms in VE-D mice1
| Group | Mortality2 | Total symptom score3 |
|---|---|---|
| % | ||
| VE-D | 28.60 | 2.00 ± 0.44 |
| VE-A | 0.00 | 1.00 ± 0.26 |
| VE-S | 0.00 | 1.58 ± 0.45 |
Mice were examined daily for symptoms of HSE.
Percent survival through d 9 p.i. (P = 0.03); VE-D vs. VE-A, n = 14 mice/diet.
Values are means ± SEM; n = 6–10 mice (P < 0.01); VE-D vs. VE-A on d 7 p.i.
Following HSV-1 infection, VE deficiency increased cytokine and chemokine expression in the forebrain
Microglial cells are a key producer of proinflammatory and antiviral cytokines and chemokines in the brain during HSV infection (37). We examined the expression of cytokine and chemokines in the forebrain of VE-D, VE-A, and VE-S HSV-1 infected mice on d 0, 3, and 7 p.i. On d 7 p.i., the VE-D mice had significantly higher expressions of IL-6, TNFα, and IL-1β. IL-10 was significantly increased in VE-D mice on both d 3 and 7 p.i. (Fig. 2). As with the pathology scores, proinflammatory cytokine expression in VE-S mice did not differ from VE-A mice.
FIGURE 2.

Forebrain IL-6 (A), TNFα (B), IL-1β (C), and IL-10 (D) gene expression in VE-D, VE-A and VE-S mice. Data are the mean ± SEM, n = 6 or 7, and are expressed as the fold of the mean of the UNI VE-A group (d 0). *Different from VE-A at that time point, P < 0.05.
Gene expression for the antiviral cytokine IFNβ was significantly increased in VE-D mice compared with the VE-A mice on d 7 p.i. Gene expression of IFNγ was increased in VE-D mice on both d 3 and 7 p.i. Inducible nitric oxide synthase (iNOS) was significantly increased on d 7 p.i. in VE-D mice compared with VE-A mice, whereas VE-S mice had a decrease in iNOS on d 3 p.i. compared with VE-A mice (Fig. 3).
FIGURE 3.
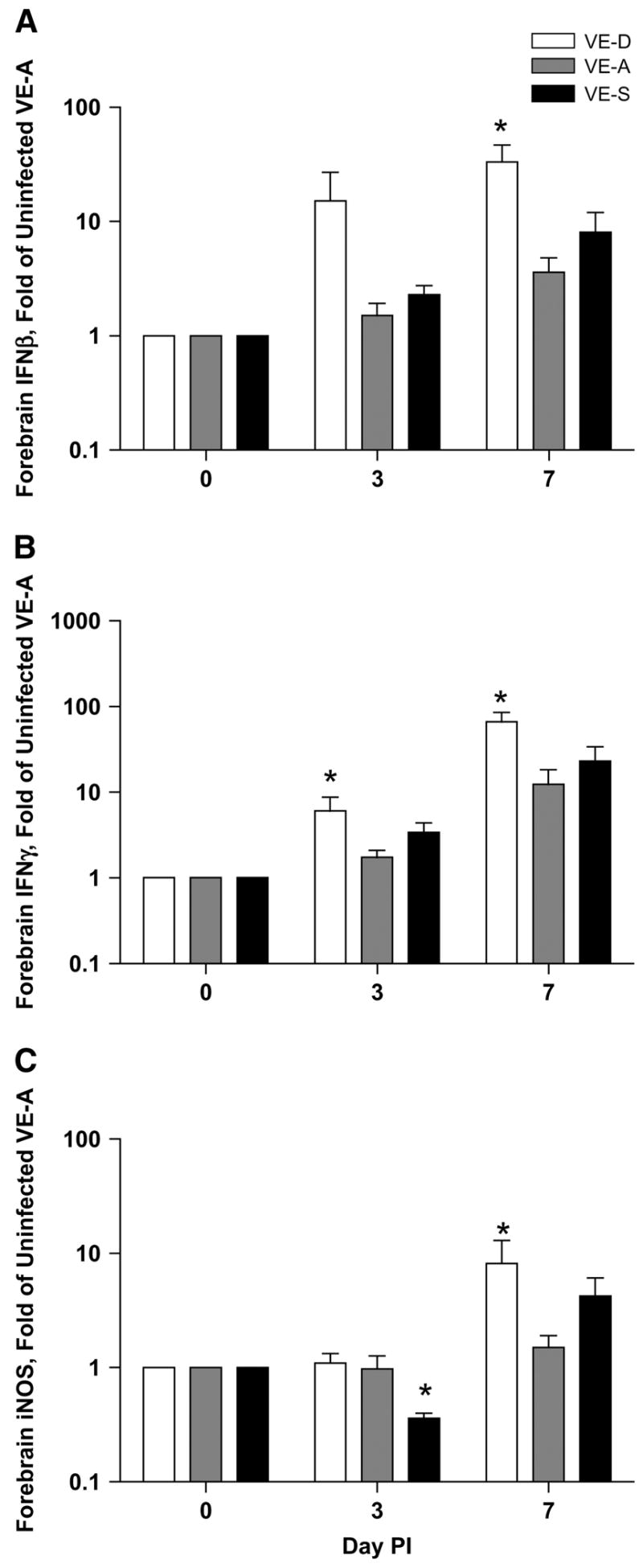
Forebrain IFNβ (A), IFNγ (B), and iNOS (C) gene expression in VE-D, VE-A and VE-S mice. Data are the mean ± SEM, n = 6 or 7, and are expressed as the fold of the mean of the UNI VE-A group (d 0). *Different from VE-A at that time point, P < 0.05.
The expression of chemokines, which preceded the infiltration of large numbers of lymphocytes, and the upregulation of adhesion molecules were essential for T cells to enter the brain to clear HSV-1. Monocyte chemotactic protein-1, RANTES, macrophage inflammatory protein-1α, and intercellular adhesion molecule-1 were significantly higher on d 7 p.i. in the forebrain of VE-D mice compared with VE-A mice (Fig. 4).
FIGURE 4.
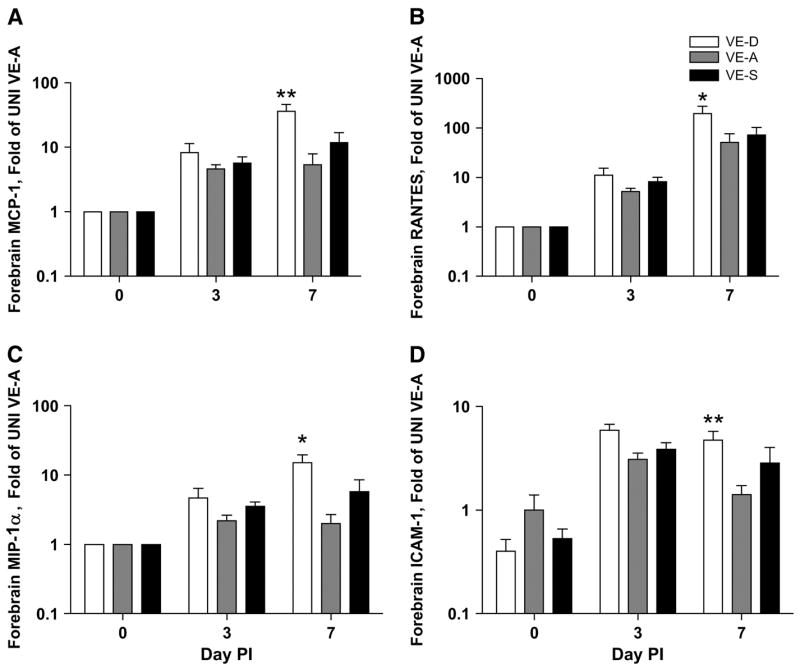
Forebrain monocyte chemotactic protein (MPC)-1 (A), RANTES (B), macrophage inflammatory protein (MIP)-1α (C), and intercellular adhesion molecule (ICAM)-1 (D) gene expression in VE-D, VE-A and VE-S mice. Data are the mean ± SEM, n = 6 or 7, and are expressed as the fold of the mean of the UNI VE-A group (d 0). *Different from VE-A at that time point, P < 0.05.
VE deficiency increases proinflammatory cytokine and chemokine protein levels in the brain stem after HSV-1 infection
HSV-1 infected VE-A mice had an increase in protein levels for IL-1β, TNFα, and RANTES in the brain stem. VE-D mice had significantly more IL-1β and TNFα on d 7 p.i. Additionally, RANTES was significantly higher in the BS of VE-D mice on d 7 p.i. compared with VE-A mice (Fig. 5). Inducible protein-10 was increased with infection; however, no significant differences among the diet groups were found (data not shown). Similar to the mRNA levels in the forebrain, cytokine and chemokine production in the BS of VE-S mice did not differ from VE-A mice.
FIGURE 5.
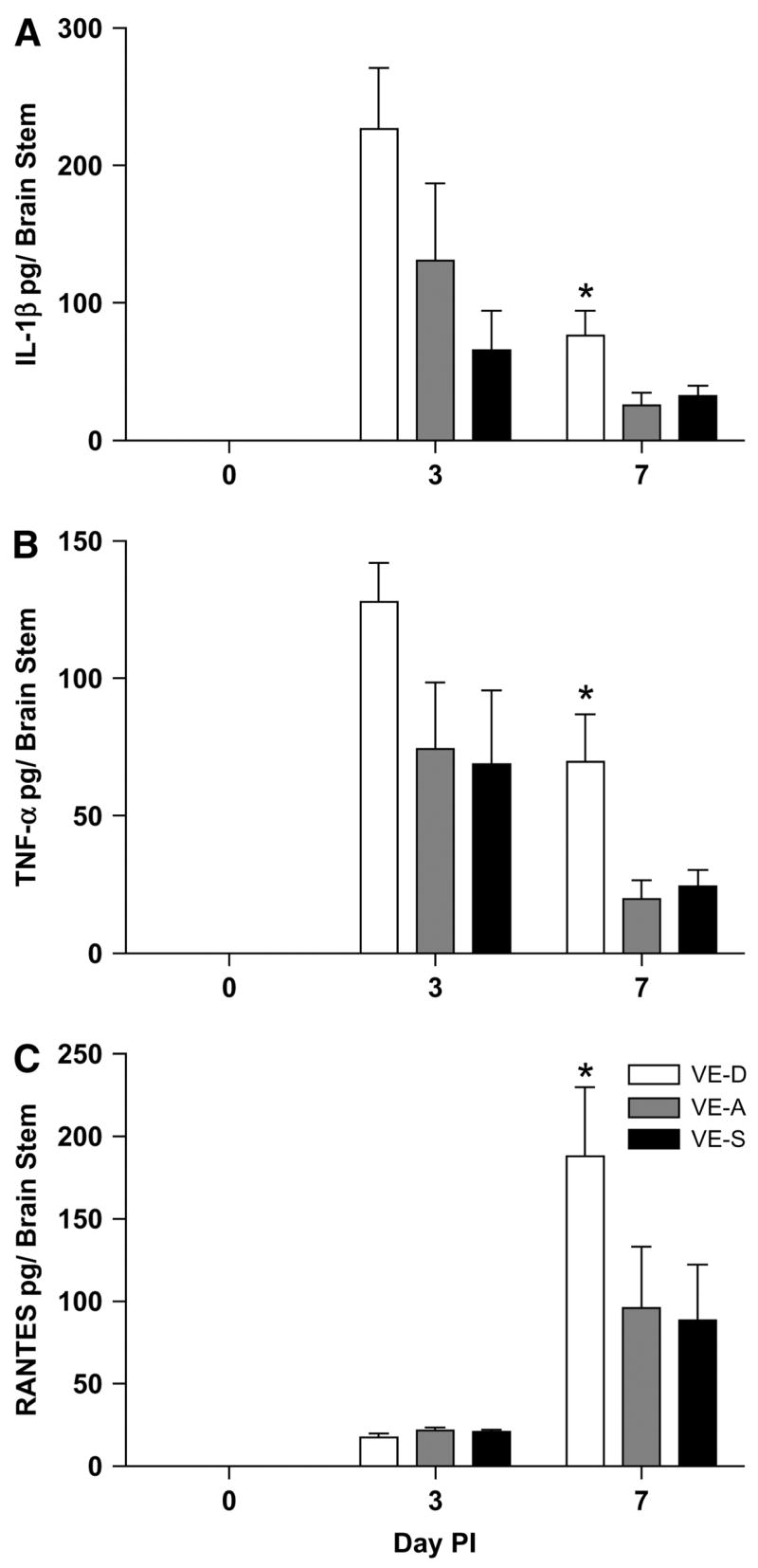
Brain stem production of IL-1β (A), TNFα (B), and RANTES (C) in VE-D, VE-A and VE-S mice. Data are the mean ± SEM, n = 6 or 7, and are expressed as pg/brain stem. *Different from VE-A at that time point, P < 0.05.
Increased HSV-1 viral load in VE-D mouse brains
HSV-1 viral replication in the brain leads to neuronal damage (38). To determine whether a deficiency in VE would lead to increased viral replication and, conversely, if increasing VE would lead to decreased viral replication, we measured HSV-1 titers in the brains of the mice. On d 7 p.i., both VE-D and VE-S mice showed a trend toward increased viral titers in the brain (Fig. 6A).
FIGURE 6.
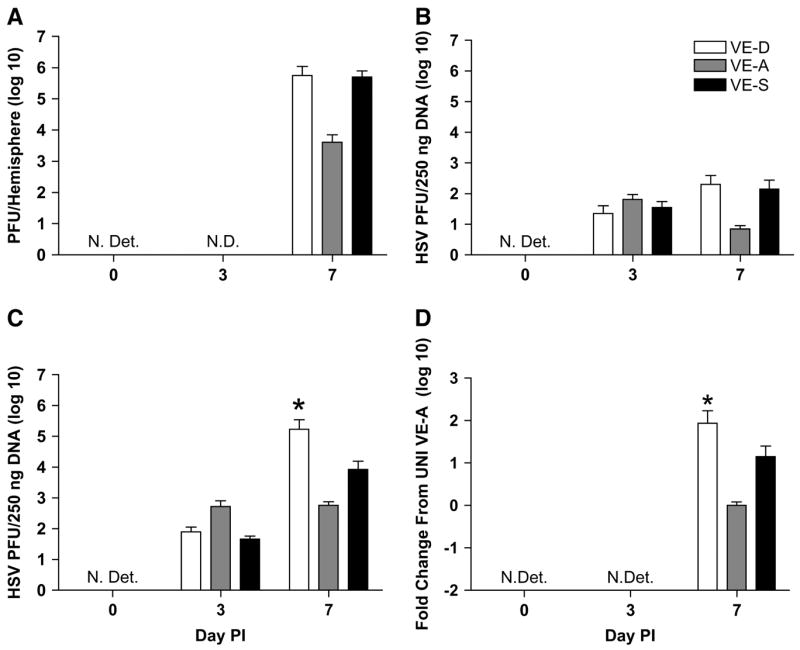
Brain HSV titer is expressed as means ± SEM, n = 5, of PFU per hemisphere of brain in VE-D, VE-A and VE-S mice (A). HSV-1 genomic DNA and mRNA in olfactory bulb (B), brain stem (C), and forebrain (D), respectively. Data are expressed as means ± SEM, n = 6 or 7. Abbreviations: N.Det., not detected; N.D., not done. *Different from VE-A at that time point, P < 0.05.
The i.n. route of HSV-1 infection does not lead to the entire brain becoming infected, but rather HSV-1 spreads along the olfactory and trigeminal nerves to distinct regions (8); therefore, we examined individual regions of the brain for the presence of HSV-1. The HSV-1 genome was measured by DNA or mRNA in the olfactory bulb, brain stem (midbrain, pons, and medulla), and forebrain, respectively. On d 3 p.i., HSV-1 DNA was found in the olfactory bulb of all groups of mice, but it was not significantly higher in the VE-D or VE-S mice (Fig. 6B). However, the HSV-1 viral load was significantly higher in the brain stem and forebrain of VE-D mice on d 7 p.i. (Fig. 6C,D). No HSV-1 DNA or mRNA was detected in the brains of UNI mice.
Discussion
VE has been suggested as a treatment for HSV infections (39). However, there are few studies that have examined the effect of VE on HSV infections (40–42). Whereas supplementation with VE has become controversial (43,44), 93% of men and 96% of women in the United States do not consume the recommended daily allowance of VE (45,46), and data from NHANES III indicate that many have low serum levels of α-TOC(47).
In this study, VE deficiency increased HSE pathology, but VE supplementation did not improve symptoms compared with VE-A mice. As previously demonstrated, HSV-1 infection in VE-A mice increased the expression of pro- and anti-inflammatory cytokines, antiviral cytokines, and chemokines in the brain (37,38). VE deficiency significantly increased the expression of all of these mediators on d 7 p.i. compared with VE-A mice. Interestingly, VE supplementation failed to decrease inflammation. Although VE supplementation clearly enhanced liver α-TOC levels, 10× supplementation failed to enhance brain α-TOC levels, suggesting that the brain tightly controls cellular membrane composition. The fact that VE supplementation failed to increase brain levels of VE likely is the reason for the lack of effect of VE supplementation on brain cytokine and chemokine levels.
Nitric oxide may act as an immune mediator that leads to neuronal damage (48). iNOS, the enzyme that produces nitric oxide, is upregulated during HSV-1 infection. Its production plays a dual role in the response to HSV because iNOS is important for clearing infection (49), but too much is deleterious. iNOS is upregulated during HSV-1 infection in a temporal and spatial pattern that follows viral replication (50). iNOS inhibitors administered to mice infected i.n. with HSV-1 were demonstrated to significantly reduce paralysis and mortality (50). This suggests that iNOS plays a critical role in the pathogenesis of HSV-1 and that increased levels in VE-D mice may be a contributing factor that leads to the mortality in these mice.
Microglial cells are identified as a source of proinflammatory cytokine production during HSV-1 infection in both humans and mice (37,51,52). During HSV-1 infection, microglia from BALB/c mice produce a vigorous, but not protective response to HSV-1 (37). In the VE-D mice, the proinflammatory response was even more robust than in the VE-A mice. In light of the neurotoxic nature of these cytokines (53,54), it is likely that increased pathogenesis in VE-D mice is linked to this overly robust response.
Glutamate is released by microglia after activation by pro-inflammatory stimuli, including cytokines (55). An excessive glutamate release is neurotoxic, resulting in neuronal damage and neuroinflammation. In vitro, HSV-1–infected microglial cells release neurotoxic factors that result in neuronal death when the supernatants are transferred to neuronal cultures. The neurotoxic effects of these substances are partially blocked by iNOS inhibitors and N-methyl-D-aspartate (NMDA) receptor antagonists (54). Therefore, iNOS and glutamate-induced neurotoxicity via NMDA receptors may be partially responsible for HSV-1–associated neuronal damage. In vivo, administration of an NMDA-receptor antagonist to restraint-stressed HSV-1 mice decreases HSE pathology and mortality (19). In the brain, VE deficiency results in increased glutamate production (56). Together with the increase in glutamate, the high levels of proinflammatory cytokines produced in VE-D mice likely led to neurotoxicity, which might have been amplified by an increase in activated microglia.
Chemokines and adhesion molecules are upregulated in VE-A mice following the HSV-1 infection (10,37,57,58). This response was even more pronounced in VE-D mice. Chemokines and adhesion molecule expression are needed for T cells to cross the blood-brain barrier and enter the brain during HSV-1 infection (57,59). Future studies will examine the impact of increased chemokine and adhesion molecules on T-cell trafficking in VE-D mice. Additionally, the high α-TOC concentration in the periphery may alter T-cell function or trafficking in the VE-S mice.
CNS infection with HSV-1 results in oxidative stress and lipid peroxidation (60,61). Because VE-D alone increases oxidative stress and lipid peroxidation (22,62), and VE-D mice in this study had increased HSV-1 viral replication, it was not surprising that they had increased cytokine/chemokine production p.i. Previous studies demonstrate that VE is effective at controlling both peripheral and central inflammation, as well as reducing sickness behavior in LPS-treated mice (63–65). VE was considered a very good candidate for decreasing the symptoms of HSE. However, a 10× VE supplementation was unable to increase α-TOC levels in the brain over VE-A levels. Therefore, the lack of effect on cytokine and chemokine levels in the brains of VE-S mice was not unexpected. It is possible that a longer supplementation with 10× VE might increase brain α-TOC levels enough to be protective, this will require further study.
In addition to cytokine and chemokine production, symptoms of HSE are a result of the viral load in the various brain regions. Few studies examine the impact of antioxidant deficiency on viral replication. Of the studies conducted, selenium deficiency results in increased coxsackievirus replication; however, it does not impact the replication of influenza A/PR8 (66,67). Both resveratrol, an antioxidant, and topically applied VE were shown to decrease HSV-1 replication (40,68). In this study, VE-D mice had a significantly higher viral load in the forebrain and brain stem compared with VE-A or VE-S mice. This is important because these regions are vital to maintaining whole-body homeostasis. The hypothalamus (part of the fore-brain) is responsible for maintaining homeostasis by regulating thirst, hunger, circadian rhythms, and control of the autonomic nervous system. The brain stem controls breathing, heart rate, and blood pressure. High viral titers and inflammatory cytokines in these regions causing neuronal damage would be expected to result in the increased mortality seen in VE-D mice. The finding that VE-A and VE-S mice had similar titers is not a surprise given that 10× supplementation was not effective in altering brain levels of α-TOC.
Taken together, these data indicate a global failure of VE-D mice to mount an appropriate immune response to a central HSV-1 infection and a failure of the 10× VE-S to decrease HSE symptoms. These findings are important because the majority of people in the United States do not consume enough VE in their diets, suggesting that immune protection against HSV encephalitis, and perhaps other viral infections as well, may be suboptimal.
Acknowledgments
The authors thank Dr. Allen Smith, USDA, for performing the VE analysis and Dr. Orville Levander and Alexia Smith for insightful discussion.
Footnotes
Supported in part by a grant from the National Institute of Environmental Health Sciences (P30ES10126) and by grants from the NIH to the Clinical Nutrition Research Unit (DK56350) at the University of North Carolina.
Author disclosures: P. A. Sheridan and M. A. Beck, no conflicts of interest.
Abbreviations used: α-TOC, α-tocopherol; BS, brain stem; CNS, central nervous system; HSE, herpes simplex virus encephalitis; HSV-1, herpes simplex virus-1; IFN, interferon; IL, interleukin; i.n., intranasally; iNOS, inducible nitric oxide synthase; NMDA, N-methyl-D-aspartate; PFU, plaque forming unit; p.i., post infection; RANTES, regulated upon activation, normal T-cell expressed and secreted; TNF, tumor necrosis factor; UNI, uninfected; VE, vitamin E; VE-A, VE adequate; VE-D, VE deficient; VE-S, VE supplemented.
Literature Cited
- 1.Johnson M, Valyi-Nagi T. Expanding the clinicopathologic spectrum of herpes simplex encephalitis. Hum Pathol. 1998;29:207–10. doi: 10.1016/s0046-8177(98)90036-3. [DOI] [PubMed] [Google Scholar]
- 2.Whitley R. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Philadelphia, PA: Lippincott Williams & Wilkins; 2001. pp. 2461–509. [Google Scholar]
- 3.Whitley R, Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–8. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 4.Skoldenberg B. Herpes simplex encephalitis. Scand J Infect Dis Suppl. 1996;100:8–13. [PubMed] [Google Scholar]
- 5.Tyler KL. In: Viral meningitis and encephalitis. 15. Braunwald ASFE, Isselbacher KJ, Kasper DL, Hauser SL, Longo DL, Jameson JL, editors. New York: McGraw-Hill; 2002. http://wwwharrisonsonlinecom. [Google Scholar]
- 6.Kennedy PGE, Chaudhuri A. Herpes simplex encephalitis. J Neurol Neurosurg Psychiatry. 2002;73:237–8. doi: 10.1136/jnnp.73.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beers DR, Henkel JS, Kesner RP, Stroop WG. Spatial recognition memory deficits without notable CNS pathology in rats following herpes simplex encephalitis. J Neurol Sci. 1995;131:119–27. doi: 10.1016/0022-510x(95)00099-n. [DOI] [PubMed] [Google Scholar]
- 8.Mori I, Goshima F, Ito H, Koide N, Yoshida T, Yokochi T, Kimura Y, Nishiyama Y. The vomeronasal chemosensory system as a route of neuroinvasion by herpes simplex virus. Virology. 2005;334:51–8. doi: 10.1016/j.virol.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 9.Enquist LW, Husak PJ, Banfield BW, Smith GA. Infection and spread of alphaherpesviruses in the nervous system. Adv Virus Res. 1998;51:237–347. doi: 10.1016/s0065-3527(08)60787-3. [DOI] [PubMed] [Google Scholar]
- 10.Wickham S, Lu B, Ash J, Carr DJJ. Chemokine receptor deficiency is associated with increased chemokine expression in the peripheral and central nervous systems and increased resistance to herpetic encephalitis. J Neuroimmunol. 2005;162:51–9. doi: 10.1016/j.jneuroim.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Hudson SJ, Streilein JW. Functional cytotoxic T cells are associated with focal lesions in the brains of SJL mice with experimental herpes simplex encephalitis. J Immunol. 1994;152:5540–7. [PubMed] [Google Scholar]
- 12.Chan WL, Javanovic T, Lukic ML. Infiltration of immune T cells in the brain of mice with herpes simplex virus-induced encephalitis. J Neuroimmunol. 1989;23:195–201. doi: 10.1016/0165-5728(89)90051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anglen CS, Truckenmiller ME, Schell TD, Bonneau RH. The dual role of CD8+ T lymphocytes in the development of stress-induced herpes simplex encephalitis. J Neuroimmunol. 2003;140:13–27. doi: 10.1016/s0165-5728(03)00159-0. [DOI] [PubMed] [Google Scholar]
- 14.Johnson R. Acute encephalitis. Clin Infect Dis. 1996;23:219–24. doi: 10.1093/clinids/23.2.219. [DOI] [PubMed] [Google Scholar]
- 15.Esiri MM. Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J Neurol Sci. 1982;54:209–26. doi: 10.1016/0022-510x(82)90183-6. [DOI] [PubMed] [Google Scholar]
- 16.Ikemoto K, Pollard R, Fukumoto T, Morimatsu M, Suzuki F. Small amounts of exogenous IL-4 increase the severity of encephalitis induced in mice by the intranasal infection of herpes simplex virus type 1. J Immunol. 1995;155:1326–33. [PubMed] [Google Scholar]
- 17.Esiri MM, Drummond CWE, Morris CS. Macrophages and microglia in HSV-1 infected mouse brain. J Neuroimmunol. 1995;62:201–5. doi: 10.1016/0165-5728(95)00123-8. [DOI] [PubMed] [Google Scholar]
- 18.Mansur DS, Kroon EG, Nogueira ML, Arantes RME, Rodrigues SCO, Akira S, Gazzinelli RT, Campos MA. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol. 2005;166:1419–26. doi: 10.1016/S0002-9440(10)62359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair A, Hunzeker J, Bonneau RH. Modulation of microglia and CD8+ T cell activation during the development of stress-induced herpes simplex virus type-1 encephalitis. Brain Behav Immun. 2007;21:791–806. doi: 10.1016/j.bbi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Herrera E, Barbas C. Vitamin E: action, metabolism and perspectives. J Physiol Biochem. 2001;57:43–56. [PubMed] [Google Scholar]
- 21.Singh U, Devaraj S, Jialal I. Vitamin E, oxidative stress, and inflammation. Annu Rev Nutr. 2005;25:151–74. doi: 10.1146/annurev.nutr.24.012003.132446. [DOI] [PubMed] [Google Scholar]
- 22.Yokota T, Igarashi K, Uchihara T, Jishage K, Tomita H, Inaba A, Li Y, Arita M, Suzuki H, et al. Delayed-onset ataxia in mice lacking alpha -tocopherol transfer protein: Model for neuronal degeneration caused by chronic oxidative stress. Proc Natl Acad Sci USA. 2001;98:15185–90. doi: 10.1073/pnas.261456098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith A, Madden KB, Yeung KJA, Zhao A, Elfrey J, Finkelman F, Levander O, Shea-Donohue T, Urban JF., Jr Deficiencies in selenium and/or vitamin E lower the resistance of mice to Heligmosomoides polygyrus infections. J Nutr. 2005;135:830–6. doi: 10.1093/jn/135.4.830. [DOI] [PubMed] [Google Scholar]
- 24.Carvalho LSC, Camargos ERS, Almeida CT, Peluzio MdCG, Alvarez-Leite JI, Chiari E, Reis DdA. Vitamin E deficiency enhances pathology in acute Trypanosoma cruzi-infected rats. Trans R Soc Trop Med Hyg. 2006;100:1025–31. doi: 10.1016/j.trstmh.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 25.Malmberg K-J, Lenkei R, Petersson M, Ohlum T, Ichihara F, Glimelius B, Frodin J-E, Masucci G, Kiessling R. A short-term dietary supplementation of high doses of vitamin E increases T helper 1 cytokine production in patients with advanced colorectal cancer. Clin Cancer Res. 2002;8:1772–8. [PubMed] [Google Scholar]
- 26.Han SN, Wu D, Ha WK, Beharka A, Smith DE, Bender BS, Meydani SN. Vitamin E supplementation increases T helper 1 cytokine production in old mice infected with influenza virus. Immunology. 2000;100:487–93. doi: 10.1046/j.1365-2567.2000.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han SN, Adolfsson O, Lee C-K, Prolla TA, Ordovas J, Meydani SN. Age and vitamin E-induced changes in gene expression profiles of T cells. J Immunol. 2006;177:6052–61. doi: 10.4049/jimmunol.177.9.6052. [DOI] [PubMed] [Google Scholar]
- 28.Marko MG, Ahmed T, Bunnell SC, Wu D, Chung H, Huber BT, Meydani SN. Age-associated decline in effective immune synapse formation of CD4+ T cells is reversed by vitamin E supplementation. J Immunol. 2007;178:1443–9. doi: 10.4049/jimmunol.178.3.1443. [DOI] [PubMed] [Google Scholar]
- 29.Wakikawa A, Utsuyama M, Wakabayashi A, Kitagawa M, Hirokawa K. Vitamin E enhances the immune functions of young but not old mice under restraint stress. Exp Gerontol. 1999;34:853–62. doi: 10.1016/s0531-5565(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 30.Beck M, Shi Q, Morris V, Levander O. Rapid genomic evolution of a non-virulent coxsackievirus B3 in selenium-deficient mice results in selection of identical virulent isolates. Nat Med. 1995;1:433–6. doi: 10.1038/nm0595-433. [DOI] [PubMed] [Google Scholar]
- 31.Beck M, Kolbeck P, Rohr L, Shi Q, Morris V, Levander O. Vitamin E deficiency intensifies the myocardial injury of coxsackievirus B3 infection of mice. J Nutr. 1994;124:345–58. doi: 10.1093/jn/124.3.345. [DOI] [PubMed] [Google Scholar]
- 32.Beck MA, Shi Q, Morris VC, Levander OA. Benign coxsackievirus damages heart muscle in iron-loaded vitamin E-deficient mice. Free Radic Biol Med. 2005;38:112–6. doi: 10.1016/j.freeradbiomed.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Beck MA, Williams-Toone D, Levander OA. Coxsackievirus B3-resistant mice become susceptible in Se/vitamin E deficiency. Free Radic Biol Med. 2003;34:1263–70. doi: 10.1016/s0891-5849(03)00101-1. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Maeda N, Beck MA. Vitamin C deficiency increases the lung pathology of influenza virus-infected gulo −/− mice. J Nutr. 2006;136:2611–6. doi: 10.1093/jn/136.10.2611. [DOI] [PubMed] [Google Scholar]
- 35.Zaspel BJ, Csallany AS. Determination of alpha-tocopherol in tissues and plasma by high-performance liquid chromatography. Anal Biochem. 1983;130:146–50. doi: 10.1016/0003-2697(83)90661-9. [DOI] [PubMed] [Google Scholar]
- 36.Lioy D, Sheridan PA, Hurley SD, Walton JR, Martin AM, Olschowka JA, Moynihan JA. Acute morphine exposure potentiates the development of HSV-1-induced encephalitis. J Neuroimmunol. 2006;172:9–17. doi: 10.1016/j.jneuroim.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Marques CP, Hu S, Sheng W, Lokensgard JR. Microglial cells initiate vigorous yet non-protective immune responses during HSV-1 brain infection. Virus Res. 2006;121:1–10. doi: 10.1016/j.virusres.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 38.Sergerie Y, Boivin G, Gosselin D, Rivest S. Delayed but not early glucocorticoid treatment protects the host during experimental herpes simplex virus encephalitis in mice. J Infect Dis. 2007;195:817–25. doi: 10.1086/511987. [DOI] [PubMed] [Google Scholar]
- 39.Gaby A. Natural remedies for Herpes simplex. Altern Med Rev. 2006;11:93–101. [PubMed] [Google Scholar]
- 40.Sheridan J, Kern E, Martin A, Booth A. Evaluation of antioxidant healing formulations in topical therapy of experimental cutaneous and genital herpes simplex virus infections. Antiviral Res. 1997;36:157–66. doi: 10.1016/s0166-3542(97)00049-1. [DOI] [PubMed] [Google Scholar]
- 41.Thomas SL, Wheeler JG, Hall AJ. Micronutrient intake and the risk of herpes zoster: a case-control study. Int J Epidemiol. 2006;35:307–14. doi: 10.1093/ije/dyi270. [DOI] [PubMed] [Google Scholar]
- 42.Starasoler S, Haber G. Use of vitamin E oil in primary herpes gingivostomatitis in an adult. N Y State Dent J. 1978;44:382–3. [PubMed] [Google Scholar]
- 43.Robinson I, de Serna D, Gutierrez A, Schade D. Vitamin E in humans: an explanation of clinical trial failure. Endocr Pract. 2006;12:576–82. doi: 10.4158/EP.12.5.576. [DOI] [PubMed] [Google Scholar]
- 44.Miller ER, III, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-Analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 45.Maras JE, Bermudez OI, Qiao N, Bakun PJ, Boody-Alter EL, Tucker KL. Intake of [alpha]-tocopherol is limited among US adults. J Am Diet Assoc. 2004;104:567–75. doi: 10.1016/j.jada.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Gao X, Wilde PE, Lichtenstein AH, Bermudez OI, Tucker KL. The maximal amount of dietary {alpha}-tocopherol intake in U.S. adults (NHANES 2001–2002) J Nutr. 2006;136:1021–6. doi: 10.1093/jn/136.4.1021. [DOI] [PubMed] [Google Scholar]
- 47.Ford ES, Sowell A. Serum {alpha}-tocopherol status in the United States population: findings from the third national health and nutrition examination survey. Am J Epidemiol. 1999;150:290–300. doi: 10.1093/oxfordjournals.aje.a010001. [DOI] [PubMed] [Google Scholar]
- 48.Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. Pharmacologic suppression of neuronal oxidative damage and dendritic degeneration following direct activation of glial innate immunity in mouse cerebrum. J Neurochem. 2003;87:1518–26. doi: 10.1046/j.1471-4159.2003.02120.x. [DOI] [PubMed] [Google Scholar]
- 49.MacLean A, Wei X, Huang F, Al Alem U, Chan W, Liew F. Mice lacking inducible nitric-oxide synthase are more susceptible to herpes simplex virus infection despite enhanced Th1 cell responses. J Gen Virol. 1998;79:825–30. doi: 10.1099/0022-1317-79-4-825. [DOI] [PubMed] [Google Scholar]
- 50.Fujii S, Akaike T, Maeda H. Role of nitric oxide in pathogenesis of herpes simplex virus encephalitis in rats. Virology. 1999;256:203–12. doi: 10.1006/viro.1999.9610. [DOI] [PubMed] [Google Scholar]
- 51.Aravalli R, Hu S, Lokensgard J. Toll-like receptor 2 signaling is a mediator of apoptosis in herpes simplex virus-infected microglia. J Neuroinflammation. 2007;4:11. doi: 10.1186/1742-2094-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR. Cutting Edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol. 2005;175:4189–93. doi: 10.4049/jimmunol.175.7.4189. [DOI] [PubMed] [Google Scholar]
- 53.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–44. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 54.Lokensgard JR, Cheeran MC, Hu S, Gekker G, Peterson PK. Glial cell responses to herpesvirus infections: role in defense and immunopathogenesis. J Infect Dis. 2002;186(Suppl 2):S171–9. doi: 10.1086/344272. [DOI] [PubMed] [Google Scholar]
- 55.Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-{alpha} induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–8. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- 56.Steffen V, Vizuete ML, Machado A, Cano J. The effect of a vitamin E-deficient diet on amino acid levels in the substantia nigra, striatum and hippocampus of rats. Life Sci. 1994;54:375–9. doi: 10.1016/0024-3205(94)00794-2. [DOI] [PubMed] [Google Scholar]
- 57.Carr DJJ, Ash J, Lane TE, Kuziel WA. Abnormal immune response of CCR5-deficient mice to ocular infection with herpes simplex virus type 1. J Gen Virol. 2006;87:489–99. doi: 10.1099/vir.0.81339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Melchjorsen J, Pedersen FS, Mogensen SC, Paludan SR. Herpes simplex virus selectively induces expression of the CC chemokine RANTES/CCL5 in macrophages through a mechanism dependent on PKR and ICP0. J Virol. 2002;76:2780–8. doi: 10.1128/JVI.76.6.2780-2788.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorries R. The role of T-cell-mediated mechanisms in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:219–45. doi: 10.1007/978-3-662-10356-2_11. [DOI] [PubMed] [Google Scholar]
- 60.Valyi-Nagy T, Olson SJ, Valyi-Nagy K, Montine TJ, Dermody TS. Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology. 2000;278:309–21. doi: 10.1006/viro.2000.0678. [DOI] [PubMed] [Google Scholar]
- 61.Nucci C, Palamara A, Ciriolo M, Nencioni L, Savini P, D’Agostini C, Rotilio G, Cerulli L, Garaci E. Imbalance in corneal redox state during herpes implex virus 1-induced keratitis in rabbits. Effectiveness of exogenous glutathione supply. Exp Eye Res. 2000;70:215–20. doi: 10.1006/exer.1999.0782. [DOI] [PubMed] [Google Scholar]
- 62.MacEvilly CJ, Muller DPR. Lipid peroxidation in neural tissues and fractions from vitamin E-deficient rats. Free Radic Biol Med. 1996;20:639–48. doi: 10.1016/0891-5849(95)02147-7. [DOI] [PubMed] [Google Scholar]
- 63.Godbout JP, Berg BM, Kelley KW, Johnson RW. [alpha]-Tocopherol reduces lipopolysaccharide-induced peroxide radical formation and interleukin-6 secretion in primary murine microglia and in brain. J Neuroimmunol. 2004;149:101–9. doi: 10.1016/j.jneuroim.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 64.Godbout JP, Berg BM, Krzyszton C, Johnson RW. [alpha]-Tocopherol attenuates NF[kappa]B activation and pro-inflammatory cytokine production in brain and improves recovery from lipopolysaccharide-induced sickness behavior. J Neuroimmunol. 2005;169:97–105. doi: 10.1016/j.jneuroim.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 65.Berg BM, Godbout JP, Chen J, Kelley KW, Johnson RW. {alpha}-Tocopherol and selenium facilitate recovery from lipopolysaccharide-induced sickness in aged mice. J Nutr. 2005;135:1157–63. doi: 10.1093/jn/135.5.1157. [DOI] [PubMed] [Google Scholar]
- 66.Beck MA, Kolbeck PC, Shi Q, Rohr LH, Morris VC, Levander OA. Increased virulence of a human enterovirus (coxsackievirus B3) in selenium-deficient mice. J Infect Dis. 1994;170:351–7. doi: 10.1093/infdis/170.2.351. [DOI] [PubMed] [Google Scholar]
- 67.Li W, Beck MA. Selenium deficiency induced an altered immune response and increased survival following influenza A/Puerto Rico/8/34 infection. Exp Biol Med (Maywood) 2007;232:412–9. [PubMed] [Google Scholar]
- 68.Faith SA, Sweet TJ, Bailey E, Booth T, Docherty JJ. Resveratrol suppresses nuclear factor-[kappa]B in herpes simplex virus infected cells. Antiviral Res. 2006;72:242–51. doi: 10.1016/j.antiviral.2006.06.011. [DOI] [PubMed] [Google Scholar]