

Abstract
Liver X receptor (LXR) α and β are members of the nuclear receptor superfamily of ligand-activated transcription factors. Best known for triggering “reverse cholesterol transport” gene programs upon their activation by endogenous oxysterols, LXRs have recently also been implicated in regulation of innate immunity. In this study, we define a role for LXRs in regulation of pulmonary inflammation and host defense and identify the lung and neutrophil as novel in vivo targets for pharmacologic LXR activation. LXR is expressed in murine alveolar macrophages, alveolar epithelial type II cells, and neutrophils. Treatment of mice with TO-901317, a synthetic LXR agonist, reduces influx of neutrophils to the lung triggered by inhaled LPS, intratracheal KC chemokine, and intratracheal Klebsiella pneumoniae and impairs pulmonary host defense against this bacterium. Pharmacologic LXR activation selectively modulates airspace cytokine expression induced by both LPS and K. pneumoniae. Moreover, we report for the first time that LXR activation impairs neutrophil motility and identify inhibition of chemokine-induced RhoA activation as a putative underlying mechanism. Taken together, these data define a novel role for LXR in lung pathophysiology and neutrophil biology and identify pharmacologic activation of LXR as a potential tool for modulation of innate immunity in the lung.
Liver X receptor α (LXRα,4 also known as NR1H3) and LXRβ (NR1H2) are members of the nuclear receptor (NR) superfamily of ligand-activated transcription factors, a superfamily which includes the perhaps better known glucocorticoid receptor (GR), estrogen receptor, thyroid receptor, and peroxisome proliferator-activated receptors (PPARs). The LXRs are activated by physiologic sterol ligands (e.g., oxysterols, cholesterol intermediates (1, 2)) and by synthetic agonists (e.g., TO-901317 (3-5)). In recent years, our understanding of the importance of LXRs has expanded across several fields of (patho-)physiology. Perhaps best known from a sizeable literature as homeostatic “cholesterol sensors” that drive transcriptional programs promoting cellular cholesterol efflux, “reverse cholesterol transport,” and bile acid synthesis (2, 5-8), more recent roles for LXRs in atherosclerosis (9-11), renin expression (12), glucose homeostasis (13-15), and innate immunity (3, 4, 16) have also been identified.
LXRs are, in fact, reported to fulfill a pivotal role in innate immunity of the macrophage. They inhibit macrophage apoptosis (17) and negatively regulate proinflammatory gene expression (e.g., IL-6, cyclooxgenase 2) induced by LPS and bacteria (4) in macrophages, at least in part through inhibition of NF-κB (3, 4, 18). LXRs and other NRs such as GR repress overlapping yet distinct sets of proinflammatory genes (19). An interesting negative bidirectional cross-talk has been described between LXRs and TLR3/4 agonists (3). Reports that LPS and LPS-induced cytokines attenuate activity of LXR upon its cognate DNA sequence (3, 20), taken together with a report that LXRαβ double knockout mice are hypersensitive to i.p. LPS (4) suggest that endogenous LXR modulation in inflammatory disease states may play a role in pathogenesis. Exploiting these insights, a potential anti-inflammatory therapeutic role for synthetic LXR agonists has recently been described in vivo in a model of dermatitis (4), and data suggest the possibility of therapeutic synergy among NR agonists (19). Nevertheless, a report that LXRαβ double knockout mice are susceptible to infection with the Gram-positive intracellular bacterial pathogen Listeria monocytogenes (16) suggests that therapeutic manipulation of LXRs cannot progress without an appreciation of their effects upon host defense in vivo.
The lung is continuously exposed to inflammatory environmental stimuli and bacterial microbes alike. Although effective host defense is critical, so too is avoidance of dysregulated inflammatory tissue injury. Hence, precise regulation of pulmonary recruitment of the neutrophil (polymorphonuclear neutrophil (PMN)), a pivotal effector cell in both lung infection and inflammation, is critical. Along with the PMN itself, the alveolar macrophage plays a central regulatory role at this crossroads of pulmonary PMN recruitment. We hypothesized that LXR regulates PMN recruitment to the inflamed and infected lung. LXRα and LXRβ gene expression has been described in the lung (21, 22) and pulmonary expression of endogenous LXR agonists (23-25) and LXR gene targets (18, 26) have also independently been described. Nevertheless, we are aware of no previous reports describing a role for the LXRs in lung pathophysiology nor in the biology of the PMN. In this study, we demonstrate protein expression of LXR in murine PMNs, alveolar macrophages, and alveolar epithelial type II cells. We show that treatment with the synthetic LXR agonist TO-901317 attenuates PMN recruitment to the lung triggered by inhaled LPS, and we identify the lung and the PMN as novel in vivo targets for LXR stimulation. LXR agonism is associated with selective attenuation of airspace cytokine expression and with impairment of PMN migration and PMN Rho GTPase activation. Extending our observations to the antimicrobial face of innate immunity, we demonstrate that LXR stimulation attenuates early PMN recruitment to the lung triggered by the clinically relevant extracellular Gram-negative bacterial pathogen Klebsiella pneumoniae and thereby impairs host defense and survival against this microorganism. These observations shed new light upon the complex roles of LXRs in innate immunity and highlight new areas requiring future investigation.
Materials and Methods
Reagents
TO-901317 was purchased from Cayman Chemical. Aprotinin, leupeptin, and AEBSF were obtained from Sigma-Aldrich. Escherichia coli 0111:B4 LPS purified by phenol extraction was purchased from Sigma-Aldrich, the Rho activation assay from Upstate Biotechnology, and rabbit anti-RhoA, rabbit anti-LXRβ, and rabbit anti-LXRαβ Abs were from Santa Cruz Biotechnology. Mouse anti-LXRα Ab, KC, and MIP-2 were purchased from R&D Systems. Rabbit anti-ATP-binding cassette A1 (ABCA1) was obtained from Novus Biologicals. Rabbit anti-pro-SPC Ab was purchased from Millipore. K. pneumoniae 43816 (serotype 2), HEK293 cells, DMEM, and FBS were obtained from American Type Culture Collection. ELISA kits for murine TNFα, KC, MIP-2, and LPS-induced CXC chemokine (LIX) were purchased from ElisaTech. Lipofectamine PLUS reagent was obtained from Invitrogen Life Technologies. LXRα, LXRβ, and RXRα (gifts from P. Tontonoz, University of California Los Angeles, CA) and LXRE luciferase (gift from D. Ory, Washington University, St. Louis, MO) plasmids were expanded by transformation in Escherichia coli, isolated, and quantified by spectrophotometry. The Luciferase Assay Kit was purchased from Promega and the Nuclear Extract Kit and p65 TransAM ELISA were obtained from Active Motif. The Bio-Rad protein assay was used.
Animals
Female C57BL/6 mice (Harlan Sprague Dawley), 6-10 wk old and weighing 18-22 g, were used in all experiments. All experiments were performed in accordance with the Animal Welfare Act and the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals after review of the protocol by the animal care and use committees of the National Jewish Medical and Research Center and the National Institute of Environmental Health Sciences. Anesthesia was provided by a single i.p. injection of 333 mg/kg Avertin (Sigma-Aldrich) as described.
Animal treatments
Mice were gavaged with 50 mg/kg TO-901317 in 0.5% hydroxypropylmethylcellulose daily for 5 consecutive days (27, 28) and then exposed to aerosolized E. coli 0111:B4 LPS (300 μg/ml, 20 min) as previously reported (29, 30). Animals were then sacrificed by cervical dislocation at 2, 8, or 24 h after LPS exposure for selected analyses. In parallel experiments, identically pretreated mice were: 1) treated at time 0 intratracheally (i.t.) with 0.5 μg of KC in 50 μl of saline with 0.1% human serum albumin (or saline/albumin control) or 2) treated at time 0 i.t. with 2 × 103 CFU of K. pneumoniae serotype 2 (43816; American Type Culture Collection) as reported elsewhere (29).
Bronchoalveolar lavage fluid (BALF) collection and analysis
BALF was collected immediately following sacrifice as previously described (29, 30). Total protein was quantified by the method of Bradford et al. (31) and bronchoalveolar lavage (BAL) total leukocyte and differential counts were performed as previously described (29). Cytokine concentrations from BAL supernatant were quantified using the relevant ELISA kits.
LXR luciferase assay
HEK293 cells were seeded in 12-well culture dishes at 2 × 105 cells/well for 24 h and transfected with TK-Renilla luciferase and with LXRα, LXRβ, RXRα, and LXRE luciferase constructs (0.2 μg/construct) using Lipofectamine PLUS per the manufacturer’s protocol. After overnight incubation in DMEM/10% FBS at 37°C, the medium was replaced with 50% volume cell-free, sterile-filtered (0.22 μm) BALF (approximately one-sixth total BALF volume per animal) from animals treated with vehicle or TO-901317. After subsequent overnight incubation, cells were washed and then lysed in passive lysis buffer (Promega) supplemented with aprotinin, leupeptin, AEBSF, sodium fluoride, and sodium orthovanadate. Lysates were assayed in triplicate for firefly and Renilla luciferase activity on a MonoLight 2010 luminometer (Analytical Luminescence Laboratory) using the Promega Dual Luciferase Assay Kit per the manufacturer’s protocol.
Neutrophil functional assays
Human PMNs were isolated from normal healthy donors by discontinuous plasma Percoll centrifugation (32, 33) in accordance with a National Jewish Medical and Research Center Institutional Review Board-approved protocol. Mature murine bone marrow PMNs were isolated from mouse femurs and tibias by discontinuous Percoll gradient centrifugation as previously reported (30). Both of these preparations yield cell populations that are >95% PMNs (data not shown). PMNs were treated with 10 μM TO-90137 or 0.1% DMSO vehicle (4 h, 37°C) and then assayed for chemotaxis to 50 ng/ml IL-8 in a modified Boyden chamber assay (34) and for superoxide anion generation to PMA and fMLP (35). In brief, pretreated PMNs (1 × 106) were labeled with calcein-AM (1 ng/ml, 10 min, 37°C), washed once in Krebs-Ringer’s phosphate dextrose (KRPD), resuspended in KRPD, and placed into the upper wells of modified Boyden chambers. IL-8 (50 ng/ml) or KRPD was placed into the lower wells and percentage of PMN migration was monitored by fluorescence (excitation wavelength 485, emission wavelength 528) in the lower chamber every 2 min over a 60-min period (FLX800 fluorescent plate reader; Bio-Tek Instruments). Chemotaxis was determined as percentage of migration to IL-8 and nondirectional movement as percentage of migration to KRPD.
In vitro RhoA activation assay
Active RhoA was quantified by Rhotekin pull-down as previously described (32).
Lung parenchymal assays
Myeloperoxidase (MPO) activity, a quantitative surrogate measure of PMN abundance, was quantified in lung homogenates 8 and 24 h following LPS inhalation as previously described (30). Activation of the p65 component of NF-κB in lung homogenates 2 h after LPS inhalation was quantified by use of a sandwich ELISA (p65 TransAm kit; Active Motif) on 15 μg (Bradford protein assay) of nuclear fraction from lung homogenates (Nuclear Extract Kit, per the manufacturer’s instructions; Active Motif).
Lung immunohistochemistry
Lungs were fixed with 4% paraformaldehyde at 25 cm H2O pressure, as previously described (30). Sections (5 μm) taken across the entire lung were embedded in paraffin. Between two and four animals were studied for each condition. Assessment of pulmonary expression of LXRα and prosurfactant protein C (SPC; a specific marker of alveolar type II cells (36, 37)) was done using an automated stainer (Discovery XT; Ventana Medical Systems) that performs deparaffinization and Ag retrieval with proprietary reagents. For LXRα, mouse anti-LXRα Ab (R&D Systems) was used as reported previously (12) at a 1/75 dilution (or murine monoclonal IgG2A was used as a control). A secondary rabbit anti-mouse IgG (AffiniPure Rabbit Anti-Mouse IgG; Jackson ImmunoResearch Laboratories) was subsequently applied at a 1/1000 dilution, followed by a proprietary anti-rabbit HRP (multimer HRP; Ventana Medical Systems). Staining was visualized with diaminobenzidine substrate. The sections were counterstained with hematoxylin and a bluing reagent was applied for postcounterstaining. A serial section from the same animal was stained for pro-SPC as described above, except that primary rabbit anti-pro-SPC Ab (Millipore) at a 1/5000 dilution was used, followed directly by anti-rabbit HRP (multimer HRP; Ventana Medical Systems). Sections were scanned at ×40 using an Aperio ScanScope model T108A and Aperio ImageScope software and interpreted by a pathologist.
Lung histopathologic scoring
Lungs were fixed with 4% paraformaldehyde using a body weight-based volume, embedded in paraffin, sectioned (5 μm), stained with H&E, and then semiquantitatively scored for the degree of inflammation by a pathologist blinded to animal treatment. The composite scoring system, which grades (scale 0-4) inflammatory cell infiltration (primarily PMNs in this study) in alveolar, perivascular, and bronchial regions, as well as bronchial wall changes, and alveolar macrophage accumulation, has been reported previously (38, 39).
Western blot
Murine tissues were homogenized in 1 ml of buffer containing 0.5% Triton X-100 supplemented with protease inhibitors (leupeptin, aprotinin, AEBSF) and then normalized by protein assay (Bio-Rad). Fifty micrograms of protein was resolved by 10% SDS-PAGE, transferred to nitrocellulose (Bio-Rad) and then probed with primary Abs. Goat anti-LXRα (1/500 dilution), rabbit anti-LXRαβ (1/1000), and mouse anti-α-tubulin (1/1000) were purchased from Santa Cruz Biotechnology. Rabbit anti-LXRβ (1/500) was obtained from Abcam and rabbit anti-ABCA1 (1/1000) was from Novus Biologicals. Membranes were then washed in Tween 20-TBS (TTBS) and exposed for 60 min to a 1/5000 dilution of species-specific, HRP-conjugated secondary Ab (GE Healthcare) in 5% milk/TTBS. Following further TTBS washes, signal was detected with 60 s of exposure to ECL Western Blot Detection Reagents (GE Healthcare), followed by application to film (GE Healthcare).
Bacterial inoculation and colony quantification
Vehicle- and TO-901317-treated mice were administered K. pneumoniae i.t. (2 × 103 CFU, 24-gauge needle, fascial cutdown). Lungs and spleen were homogenized in sterile saline 24 and 48 h later, and serial dilutions were plated on MacConkey agar for bacterial quantification as previously described (29).
Statistical analysis
Analysis was performed using GraphPad Prism statistical software. Data are represented as mean ± SEM. A two-tailed Student t test was applied for comparisons of two groups and ANOVA with Tukey’s posttest was applied for analyses of three or more groups. Survival was evaluated by the log rank test. For all tests, p < 0.05 was considered significant.
Results
LXR protein is expressed in alveolar macrophages and alveolar epithelial cells and is functional in the murine lung
To date, very few studies have investigated a role for LXR in the lung, and we are aware of no reports that have confirmed LXR protein expression in lung. Consequently, in an attempt to more directly demonstrate a role for LXR in lung (patho-)physiology, we first confirmed LXRα and LXRβ protein expression in murine lung homogenates by immunoblotting (Fig. 1A). Relative protein expression among different murine organs was liver≫lung>spleen for LXRα and lung∼spleen>liver for LXRβ. We further characterized LXRα distribution within the lung by immunohistochemistry (Fig. 1B). As shown in Fig. 1B, LXRα expression was detected in nuclei of both alveolar macrophages and alveolar epithelial type II cells, the latter identified by positive cytoplasmic staining with anti-pro-SPC Ab (36, 37).
FIGURE 1.
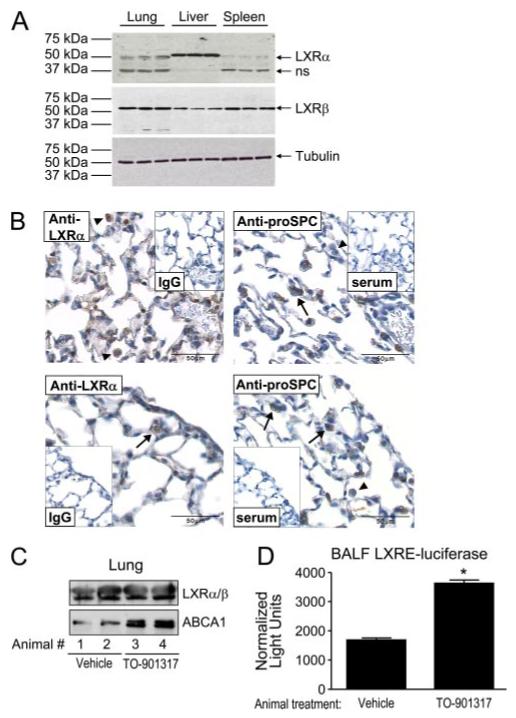
Functional LXR is expressed in alveolar macrophages and alveolar epithelial cells. A, Fifty micrograms (Bradford assay) of lung, liver, and spleen homogenate protein from C57BL/6 mice was immunoblotted with goat anti-LXRα, rabbit anti-LXRβ, and mouse anti-α-tubulin (loading control) Abs. Results from three representative animals are shown for each organ. ns, Nonspecific band. B, Immunohistochemistry was performed for LXRα (murine IgG2A as control) and for pro-SPC (normal rabbit serum as control) on 5-μm serial sections of paraformaldehyde-fixed normal C57BL/6 mouse lung. (upper left) Arrowheads indicate two alveolar macrophages with positive nuclear staining for LXRα. Lower left, Arrow indicates an alveolar epithelial type II cell with positive nuclear staining for LXRα. In the images at the right, arrows indicate alveolar type II cells with positive cytoplasmic staining for pro-SPC and arrowheads indicate alveolar macrophages with absent pro-SPC staining. Images are representative of findings in four independent animals. C, Lung homogenates (10 μg of protein) from C57BL/6 mice treated orally with vehicle or TO-901317 (50 mg/kg per day for 5 days) were immunoblotted with rabbit anti-ABCA1 (an LXR target gene) and also for LXRαβ using a rabbit dual recognition Ab. Representative blots are shown from two animals per treatment. D, BALF from C57BL/6 mice treated orally with vehicle or TO-901317 (50 mg/kg per day for 5 days) was screened for LXR-modifying activity by incubation upon HEK293 cells transiently transfected with LXRE luciferase and TK-Renilla luciferase and with LXRα, LXRβ, and RXRα expression constructs. Normalized luciferase activity was measured in triplicate from three to five animals per treatment in two independent experiments (*, p < 0.0001).
To confirm the functionality of pulmonary LXR, we administered oral TO-901317 to C57BL/6 mice using a published protocol (27, 28). A 5-day regimen of 50 mg/kg per day TO-901317 induced expression of the LXR target gene ATP-Binding Cassette transporter A1 (ABCA1) in lung homogenates as indicated by immunoblotting (Fig. 1C). Successful penetration of orally administered TO-901317 into BALF was suggested by enhanced LXRE luciferase activity in transfected HEK293 cells incubated with BALF from TO-901317-treated animals as compared with BALF from vehicle-treated animals (Fig. 1D). No increase in LXRα or LXRβ was detected in lung nuclear isolates of TO-901317-treated animals (data not shown) and only a very modest increase was seen in nuclear isolates of TO-901317-treated (10 μM, 16 h) monocytic U937 cells (data not shown), suggesting that nuclear translocation of total LXR is not a primary mechanism for regulation of LXR activity in the lung. In sum, these studies: 1) confirm LXR expression in two different cell types within murine lung and 2) demonstrate its functional activation by oral treatment of mice with a synthetic LXR agonist. As ABCA1 expression has been reported in both rodent alveolar macrophages and alveolar type II cells (40-42), the present data do not distinguish the TO-901317 effect upon the alveolar macrophage vs the type II cell.
LXR agonist treatment reduces PMN influx into the LPS-exposed lung
LXR agonists have been reported to have anti-inflammatory effects (3, 4, 18, 19, 43, 44), and LXR-null mice to have enhanced inflammation (4). Thus, we next queried whether treatment of mice with TO-901317 would exert an anti-inflammatory effect in the inhaled LPS model. Our model of murine LPS inhalation induces essentially a selective recruitment of PMNs to the lung within the first 24 h after exposure (45). A 5-day pretreatment regimen with 50 mg/kg per day TO-901317 significantly reduced PMN influx into the airspaces triggered by inhaled LPS (Fig. 2A). TO-901317 treatment was also associated with a modest reduction of PMN infiltration into the lung parenchyma at 8 and 24 h following inhaled LPS, as quantified by an MPO assay upon lung homogenates (Fig. 2, B and C), whereas no effect was seen upon baseline (unexposed) lung MPO activity with the LXR agonist (data not shown). No significant difference was noted in LPS-induced lung histopathology between TO-901317- and vehicle-treated animals (data not shown). Moreover, there was no difference between vehicle- and TO-901317-treated animals in the peripheral leukocyte count nor in the peripheral leukocyte percentage of neutrophils, lymphocytes, monocytes, or eosinophils (data not shown).
FIGURE 2.
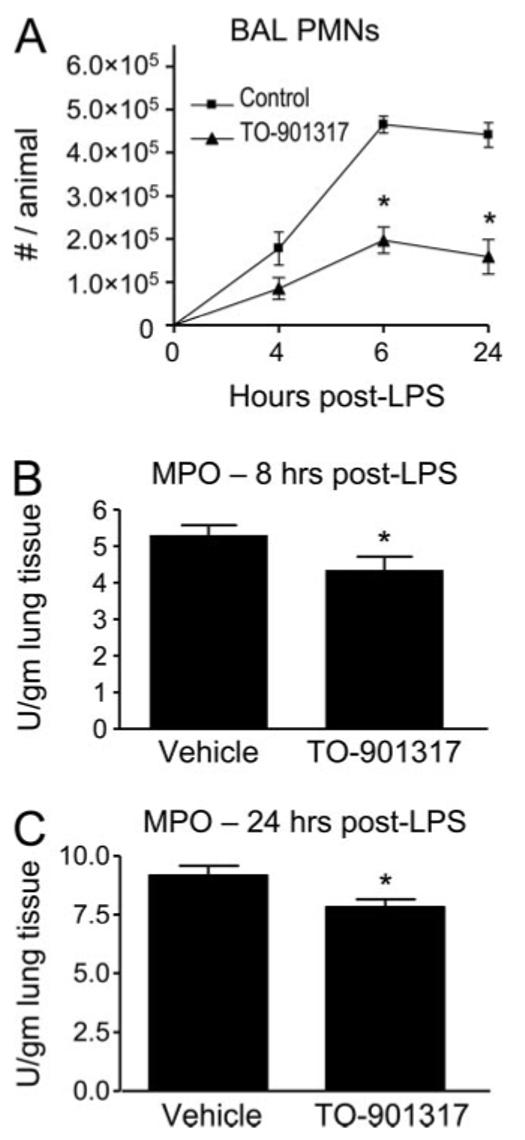
LXR agonist treatment reduces influx of PMNs to the LPS-exposed lung. A, C57BL/6 mice were treated orally with vehicle or TO-901317 (50 mg/kg per day for 5 days) and then exposed to aerosolized LPS. BAL total white blood cells were counted 4, 6, and 24 h after exposure (*, p < 0.001). MPO activity, a quantitative measure of PMNs, was assayed in lung homogenates of vehicle- and TO-901317-treated animals 8 h (B) and 24 (C) h after LPS exposure (*, p < 0.05). Data shown are representative of three independent experiments.
LXR agonist treatment selectively modulates BALF cytokine induction
To further address the mechanisms by which pharmacologic LXR activation attenuates PMN influx into the lung, we next assayed the effect of orally administered TO-901317 upon BALF cytokine and chemokine expression. Cytokine and chemokine expression by resident lung cells, in particular alveolar macrophages and epithelial cells, plays a key role in migration of PMNs from the blood-stream into the lung (29, 30, 46). LXR has been reported to regulate the expression of some LPS-induced cytokines (4). As shown in Fig. 3, TO-901317-treated mice exposed to aerosolized LPS had attenuated induction of BALF TNFα at 2 and 6 h following LPS exposure, but no change in induction of three chemokines that have been reported to play important roles in recruitment of PMNs to the lung (29, 30, 46), KC, MIP-2 (both of alveolar macrophage and epithelial origin (47, 48)), and LPS-induced CXC chemokine (LIX, CXCL5; of selective alveolar epithelial type II cell origin (46)).
FIGURE 3.
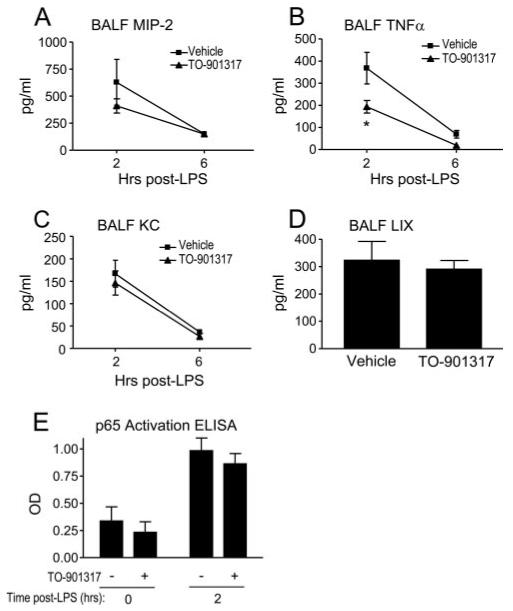
LXR agonist treatment selectively inhibits LPS-induced airspace cytokine expression. BALF MIP-2 (A), TNFα (B), and KC (C) were measured by ELISA 2 and 6 h after LPS exposure, and LIX (D) was measured 2 h after LPS exposure in vehicle- and TO-901317-treated C57BL/6 mice (*, p < 0.05; all other comparisons were NS). E, NF-κB activation was measured in lung nuclear isolates of vehicle- and TO-901317-treated animals 0 and 2 h following LPS inhalation using an ELISA that measures DNA binding of the p65 component of NF-κB, as previously described (32) (p = NS for vehicle/LPS vs TO-901317/LPS). Data shown represent four independent experiments involving n = 16 mice/experiment.
Because TNFα induction is tightly linked to NF-κB activation (49) and NF-κB has been reported to be inhibited by LXR agonists in cell culture (3, 4), we next tested the effect of oral TO-901317 upon LPS-induced activation of NF-κB in lung homogenates. As shown in Fig. 3E, TO-901317 treatment had no significant effect upon DNA-binding activity of p65 from lung nuclear isolates.
LXR agonist treatment attenuates PMN migration and Rho GTPase activation
TNFα expression has been reported to play an important role in recruitment of PMNs to the lung (50). Nevertheless, given the lack of effect of TO-901317 upon BALF chemokine levels, we next queried whether systemic LXR activation might exert additional inhibitory effects directly upon PMN migration, as has been reported with agonism of other nuclear receptors (e.g., PPARγ (51) and GR (52)). To address this, we first modeled in vivo PMN migration into the lung more directly by performing intratracheal instillation of the chemokine KC as previously reported (29). As shown in Fig. 4A, similar to the scenario with inhaled LPS, TO-901317-treated animals had a significant reduction in KC-induced influx of PMNs to the airspace. We confirmed that murine PMNs express LXR protein by immunoblotting of lysates (Fig. 4A).
FIGURE 4.
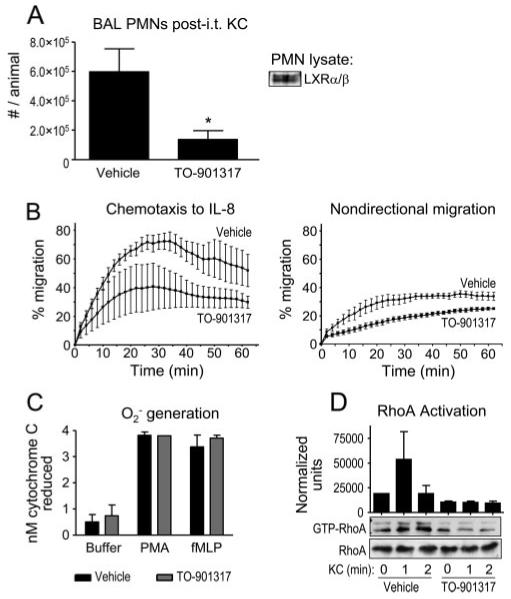
LXR agonist treatment inhibits PMN migration in vivo and ex vivo and attenuates chemokine-induced RhoA activation. A, Vehicle- and TO-901317-treated C57BL/6 mice underwent i.t. instillation of 0.5 μg of KC. BAL PMNs were quantified 6 h later (*, p < 0.05). Murine bone marrow-derived PMNs were lysed and immunoblotted using a dual anti-LXRαβ Ab. B, Human PMNs were incubated with 0.1% DMSO vehicle or 10 μM TO-901317 (4 h, 37°C). Chemotaxis to IL-8 and nondirectional migration to buffer were then assayed in a modified Boyden chamber as previously described (34) (p < 0.0001 × two-way ANOVA for effect of TO-901317 upon both chemotaxis and nondirectional migration). Data shown are representative of three independent experiments. C, Human PMNs pretreated with DMSO vehicle or TO-901317 as in B were tested for PMA- and fMLP-induced superoxide anion (O2-) generation as previously described (35). D, Bone marrow PMNs were harvested from vehicle- and TO-901317-treated animals as previously described (29). Cells were then exposed to 25 ng/ml KC for the indicated times, and RhoA activation was assayed by Rhotekin-binding domain pull-down, as described previously (32), from three independent experiments. Lysates were immunoblotted for total RhoA as a loading control. Normalized RhoA activation was quantified by densitometry (p < 0.05 for TO-901317 treatment by two-way ANOVA).
Our observation that PMNs express LXR protein suggested to us that they might be direct cellular targets for LXR agonists. Thus, in an effort to further confirm a direct effect of LXR agonism upon PMN migration and also to extend our findings to the human system, we incubated human PMNs in 10 μM TO-901317 or 0.1% DMSO vehicle (4 h, 37°C) and then quantified their migration to IL-8, a KC homolog, in a modified Boyden chamber. As shown in Fig. 4B, PMN chemotaxis to IL-8 and nondirectional migration to buffer were both inhibited by the LXR agonist, suggesting an inhibitory effect of LXR upon PMN motility. No effect of TO-901317 was seen upon PMA- or fMLP-induced superoxide anion generation by the PMN (Fig. 4C), arguing against a nonspecific, generalized inhibitory effect of TO-901317 upon PMN functions.
Lastly, to further characterize the molecular mechanism underlying LXR effects upon PMN migration, we next tested the effect of TO-901317 upon RhoA activation in the PMN. Rho GTPases are well known to play a pivotal role in leukocyte migration (53). Whereas the Rho GTPase Cdc42 has been more closely associated with regulation of leukocyte directionality, RhoA and its effectors have been connected to leukocyte motility. Hence, we retrieved bone marrow PMNs from TO-901317- and vehicle-treated animals and compared their RhoA activation upon ex vivo exposure to KC. As shown in Fig. 4D, bone marrow-derived PMNs from TO-901317-treated animals had marked attenuation of KC-induced RhoA activation.
In summary, these are the first reported data, to our knowledge, confirming LXR protein expression in the PMN and identifying a role for LXR in cell migration and in Rho GTPase activation. They identify a second putative mechanism, in addition to down-regulation of airspace TNFα, for the observed inhibitory effect of LXR upon migration of PMNs to the inflamed lung. The observed inhibition of PMN motility noted with direct ex vivo TO-901317 treatment of isolated PMNs, taken together with our finding of LXR protein expression in the PMN, suggests that the PMN may be a direct cellular target of TO-901317.
LXR agonist treatment impairs pulmonary antibacterial host defense
Antimicrobial host defense and inflammation are alternate arms of the innate immune response that use common effector mechanisms. Mechanisms of PMN recruitment to the lung differ between LPS and bacterial exposures (54), due, at least in part, to the fact that intact bacteria bear additional pathogen-associated molecular patterns and Ags. Having demonstrated a significant inhibitory effect of LXR upon PMN influx and BALF TNFα triggered by the Gram-negative bacterial pathogen-associated molecular pattern LPS, we next queried what effect LXR activation would have upon pulmonary exposure to a clinically relevant strain of the intact Gram-negative bacterial pathogen K. pneumoniae, an organism whose clearance is largely PMN dependent (55).
As shown in Fig. 5A, TO-901317-treated animals had an altered profile of PMN influx into the airspace triggered by i.t. inoculation with K. pneumoniae, with a marked reduction of BAL PMNs at 6 h postinoculation and a trend toward reduction 24 h postinoculation, as compared with vehicle-treated animals. By contrast, TO-901317-treated animals had an excess of BAL PMNs 48 h postinoculation. There was no difference between vehicle and TO-901317 treatment in alveolar macrophage number 48 h postinfection (data not shown). No significant change in lung histopathology score was noted with LXR agonist treatment 48 h following K. pneumoniae inoculation (Fig. 5B). As observed with LPS inhalation (Fig. 3B), TO-901317 attenuated K. pneumoniae-induced TNFα expression in BALF (Fig. 5C). However, unlike the LPS model, LXR agonism was associated with increased BALF MIP-2 levels (Fig. 5C).
FIGURE 5.
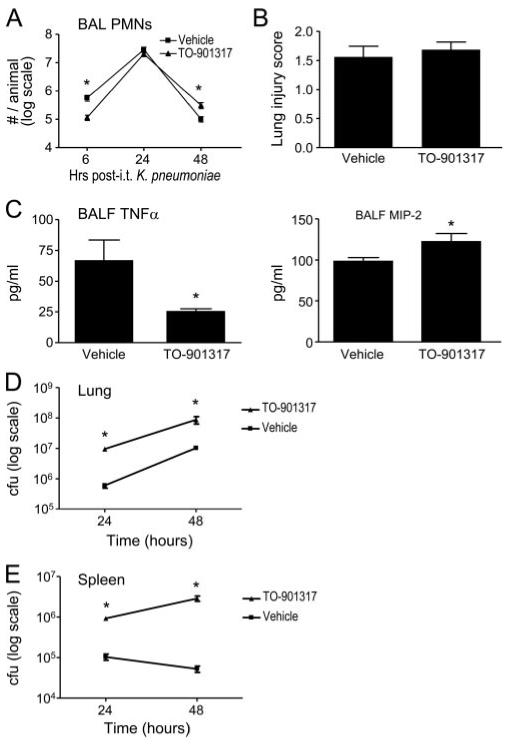
LXR agonist treatment modifies lung inflammation triggered by a host defense against Klebsiella pneumoniae. Vehicle- and TO-901317-treated C57BL/6 mice underwent i.t. inoculation with 2000 CFU of K. pneumoniae. A, BAL PMNs were quantified 6, 24, and 48 h postinoculation (*, p < 0.05). B, Lung histopathology was quantified by a pathologist blinded to treatment conditions in the left lung of vehicle- and TO-901317-treated mice 48 h following K. pneumoniae inoculation using a 0 -4 composite scoring system that grades inflammatory cell infiltration of alveoli and bronchioles, bronchial wall changes, and alveolar macrophage accumulation (38, 39) (p = NS, n = 20 animals/treatment pooled from four independent experiments). C, BALF TNFα and MIP-2 were quantified 6 h postinoculation (*, p < 0.05 for both). Data shown represent three to four independent experiments. D and E, Vehicle- and TO-901317-treated C57BL/6 mice underwent i.t. inoculation with 2000 CFU of K. pneumoniae. At the indicated times following inoculation, lung (D) and splenic (E) homogenate CFUs were quantified as previously described (*, p < 0.05) (29). Data shown represent four independent experiments.
Treatment with the LXR agonist was associated with significantly increased K. pneumoniae CFUs in lung and in spleen 24 and 48 h following i.t. inoculation (Fig. 5, D and E), indicating impairment of both pulmonary antibacterial host defense and compartmentalization of infection. In support of the significance to the organism of impaired antibacterial host defense with pharmacologic LXR activation, we found that TO-901317 treatment significantly worsened survival of mice following i.t. K. pneumoniae (Fig. 6).
FIGURE 6.
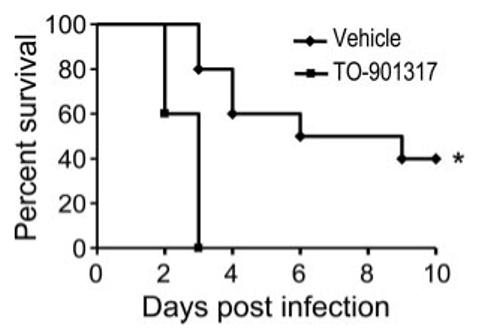
LXR agonist treatment enhances mortality induced by intratracheal K. pneumoniae. C57BL/6 mice were pretreated with vehicle or TO-901317 (50 mg/kg per day for 3 days) and then inoculated i.t. with 2000 CFU of K. pneumoniae. Daily treatment was continued postinoculation and mortality was monitored (*, p = 0.002 by log rank test). Data shown represent n = 10 mice/treatment group from one of two representative independent experiments.
Discussion
LXRα and LXRβ are oxysterol-activated NRs. Although perhaps best known for their role in triggering transcriptional programs that promote reverse cholesterol transport, in recent years there has been a growing appreciation of their complex roles in innate immunity. In this capacity, LXRs are recognized to participate in bidirectional negative cross-talk with TLR3/4 and to inhibit proinflammatory gene expression in large part through their inhibition of NF-κB. In the present work, we extend the sphere of influence of LXRs to the lung and the PMN. Our finding in this manuscript of a relative pattern of liver≫lung>spleen for LXRα protein expression and lung∼spleen>liver for LXRβ protein expression directly parallels a previous report of relative LXRα and LXRβ mRNA abundance in murine tissues (22). More specifically, we demonstrate expression of LXR in alveolar macrophages, alveolar type II cells, and PMNs and proceed to show potent anti-inflammatory and antihost defense effects of synthetic LXR agonists in the lung. These anti-inflammatory and antihost defense effects share in common impairment of PMN recruitment to the lung and attenuation of lung TNFα expression. LXR stimulation appears to attenuate PMN migration to the lung, at least in part, through inhibition of pulmonary TNFα expression and through impairment of PMN motility. Of note, although the effects of TO-901317 on TNFα parallel those previously reported for glucocorticoid treatment in a rodent LPS lung injury model (56), our finding that LPS-induced LIX expression is insensitive to LXR agonism represents an important difference, since LIX was originally cloned as a GR-sensitive target (57). We provide circumstantial evidence that the effect on PMN motility, in turn, may reflect underlying inhibition of RhoA.
We report that synthetic LXR agonists inhibit PMN motility and that this may reflect inhibition of Rho GTPases (Fig. 4). Cellular migration requires precisely timed and spaced cycling of Rho GTPase activation within the cell (53). Although RhoA has been reported to inhibit LXR (58), suggesting integration between Rho-dependent functions and LXR activation, we are aware of no previous reports of LXR activation inhibiting RhoA. As for other NR agonists, mixed effects upon Rho signaling have been reported. Androgens have been reported to stimulate Rho signaling (59). PPARγ agonists have been reported to stimulate motility in intestinal epithelial cells, at least in part, through activation of the Rho GTPase Cdc42 (60), to inhibit migration in leukocytes (51, 61, 62), and to inhibit Rho activation in smooth muscle (63). Our study design, which used a 4-h preincubation of PMNs with TO-901317 preceding KC exposure, does not fully discount the possibility that LXR activation may even activate Rho GTPases acutely and transiently. Such “nongenomic,” rapid signaling events have been described for other NR ligands (64). This notwithstanding, our data do clearly show that prolonged pharmacologic activation of LXR in the PMN (mimicking our in vivo-dosing regimen) does markedly impair proper activation of RhoA triggered by acute exposure to KC. Of interest, this effect upon motility is similar to what we have previously reported for treatment of PMNs with hydroxymethylglutaryl CoA reductase inhibitors (29), agents that have been reported to activate LXR in the leukocyte (58). A previous report that the LXR ligand 25-hydroxycholesterol impairs phagocytosis in macrophages (65) suggests that LXR may impact additional Rho-dependent macrophage functions that are relevant to host defense. Future studies will need to dissect further the responsible molecular mechanisms.
The present work raises several new issues that will require future investigation. Although synthetic LXR agonists appear to be promising therapeutic candidates for modulation of lung innate immunity, the present work does not address whether the observed effects were specifically LXR dependent, whether some of the in vivo effects might be “second-tier” consequences of LXR activation (e.g., secondary consequences of LXR-dependent changes in lipid metabolism), nor whether LXRα and LXRβ may play distinct roles as has been reported with other phenotypes (12, 16). Because synergistic effects have been reported among different NR agonists in cell culture (19), we speculate that there may be a role for LXR agonists in combination therapy of inflammatory lung disease, e.g., as “steroid-sparing” agents. Although the apparent adverse effects of LXR stimulation upon host defense against extracellular bacteria (Figs. 5 and 6) will require further careful study, it is worth noting that glucocorticoids are used widely to good clinical effect despite their untoward effects upon host defense (66). In our study, although conclusions should be tempered as lung histopathology was quantified at only one time point following K. pneumoniae inoculation (48 h), we speculate that the enhanced mortality seen with the LXR agonist (Fig. 6) was not due to aggravated lung injury. Instead, we speculate that it may reflect worsened septicemia due to enhancement of bacterial dissemination with the LXR agonist (Fig. 5E). Of interest, it has been reported that LXR-null mice are more susceptible to bloodstream infection with the Gram-positive intracellular bacterium L. monocytogenes (16). Because microbial virulence factors, host compartments, and host defense mechanisms differ markedly between these two bacteria, these observations, taken together, indicate that LXR may exert mixed influences upon the different effector arms of host defense. A report that a fungal metabolite, paxilline, is a potent LXR agonist (67) suggests that pathogen-specific molecules may add a further layer of complexity to the role of LXR in host defense.
Finally, in a broader sense, we speculate that LXR may possibly underlie associations that have been noted between metabolic disorders such as obesity, in which a role for LXR has been identified (68), and inflammatory lung diseases, such as asthma (69). The present work suggests that cholesterol metabolism may play a much more important role in lung biology than has previously been recognized, and that therapies targeting disordered cholesterol metabolism may well impact lung biology.
Acknowledgments
LXRα, LXRβ, and RXRα constructs were gifts from Peter Tontonoz and the LXRE luciferase construct was a gift from Daniel Ory. We thank Sandra Ward for assistance with peripheral leukocyte counting and typing, Kimra Walker for assistance with immunohistochemistry, and Drs. Donald Cook and Farhad Imani for review of this manuscript.
Footnotes
This work was supported by American Heart Association Grant 0275035N (to M.B.F.), American Lung Association Grant 22442N (to S.J.), and National Institutes of Health Grants 5R01HL061407-08 (to G.S.W.) and 5P01HL68743-04 (to J.A.N.). This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
- LXR
- liver X receptor
- ABCA1
- ATP-binding cassette transporter A1
- BALF
- bronchoalveolar lavage fluid
- BAL
- bronchoalveolar lavage
- GR
- glucocorticoid receptor
- LIX
- LPS-induced CXC chemokine
- MPO
- myeloperoxidase
- NR
- nuclear receptor
- PPAR
- peroxisome proliferator-activated receptor
- RXR
- retinoid X receptor
- PMN
- polymorphonuclear neutrophil
- i.t.
- intratracheal(ly)
- SPC
- surfactant protein C.
Disclosures The authors have no financial conflict of interest.
References
- 1.Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl. Acad. Sci. USA. 1999;96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR α. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 3.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between LXR and Toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell. 2003;12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 4.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 5.Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- 6.Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA, Willson TM. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 1997;272:3137–3140. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- 7.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 8.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR α. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 9.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA. 2002;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuster GU, Parini P, Wang L, Alberti S, Steffensen KR, Hansson GK, Angelin B, Gustafsson JA. Accumulation of foam cells in liver X receptor-deficient mice. Circulation. 2002;106:1147–1153. doi: 10.1161/01.cir.0000026802.79202.96. [DOI] [PubMed] [Google Scholar]
- 11.Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, Daige CL, Thomas D, Heyman RA, Mangelsdorf DJ, et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc. Natl. Acad. Sci. USA. 2002;99:11896–11901. doi: 10.1073/pnas.182199799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morello F, de Boer RA, Steffensen KR, Gnecchi M, Chisholm JW, Boomsma F, Anderson LM, Lawn RM, Gustafsson JK, Lopez-Ilasaca M, et al. Liver X receptors α and β regulate renin expression in vivo. J. Clin. Invest. 2005;115:1913–1922. doi: 10.1172/JCI24594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalen KT, Ulven SM, Bamberg K, Gustafsson JA, Nebb HI. Expression of the insulin-responsive glucose transporter GLUT4 in adipocytes is dependent on liver X receptor α. J. Biol. Chem. 2003;278:48283–48291. doi: 10.1074/jbc.M302287200. [DOI] [PubMed] [Google Scholar]
- 14.Laffitte BA, Chao LC, Li J, Walczak R, Hummasti S, Joseph SB, Castrillo A, Wilpitz DC, Mangelsdorf DJ, Collins JL, et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc. Natl. Acad. Sci. USA. 2003;100:5419–5424. doi: 10.1073/pnas.0830671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V, Kreusch A, Saez E. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445:219–223. doi: 10.1038/nature05449. [DOI] [PubMed] [Google Scholar]
- 16.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’Connell RM, Cheng G, Saez E, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 17.Valledor AF, Hsu LC, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc. Natl. Acad. Sci. USA. 2004;101:17813–17818. doi: 10.1073/pnas.0407749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terasaka N, Hiroshima A, Ariga A, Honzumi S, Koieyama T, Inaba T, Fujiwara T. Liver X receptor agonists inhibit tissue factor expression in macrophages. FEBS J. 2005;272:1546–1556. doi: 10.1111/j.1742-4658.2005.04599.x. [DOI] [PubMed] [Google Scholar]
- 19.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and Toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. Downregulation of liver X receptor-α in mouse kidney and HK-2 proximal tubular cells by LPS and cytokines. J. Lipid Res. 2005;46:2377–2387. doi: 10.1194/jlr.M500134-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Mangelsdorf DJ. LuXuRies of lipid homeostasis: the unity of nuclear hormone receptors, transcription regulation, and cholesterol sensing. Mol. Interv. 2002;2:78–87. doi: 10.1124/mi.2.2.78. [DOI] [PubMed] [Google Scholar]
- 23.Babiker A, Andersson O, Lindblom D, van der Linden J, Wiklund B, Lutjohann D, Diczfalusy U, Bjorkhem I. Elimination of cholesterol as cholestenoic acid in human lung by sterol 27-hydroxylase: evidence that most of this steroid in the circulation is of pulmonary origin. J. Lipid Res. 1999;40:1417–1425. [PubMed] [Google Scholar]
- 24.Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG. 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J. Biol. Chem. 2001;276:38378–38387. doi: 10.1074/jbc.M105805200. [DOI] [PubMed] [Google Scholar]
- 25.Song C, Liao S. Cholestenoic acid is a naturally occurring ligand for liver X receptor α. Endocrinology. 2000;141:4180–4184. doi: 10.1210/endo.141.11.7772. [DOI] [PubMed] [Google Scholar]
- 26.Steffensen KR, Neo SY, Stulnig TM, Vega VB, Rahman SS, Schuster GU, Gustafsson JA, Liu ET. Genome-wide expression profiling: a panel of mouse tissues discloses novel biological functions of liver X receptors in adrenals. J. Mol. Endocrinol. 2004;33:609–622. doi: 10.1677/jme.1.01508. [DOI] [PubMed] [Google Scholar]
- 27.Laffitte BA, Joseph SB, Chen M, Castrillo A, Repa J, Wilpitz D, Mangelsdorf D, Tontonoz P. The phospholipid transfer protein gene is a liver X receptor target expressed by macrophages in atherosclerotic lesions. Mol. Cell. Biol. 2003;23:2182–2191. doi: 10.1128/MCB.23.6.2182-2191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors α and β. J. Biol. Chem. 2002;277:18793–18800. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- 29.Fessler MB, Young SK, Jeyaseelan S, Lieber JG, Arndt PG, Nick JA, Worthen GS. A role for hydroxy-methylglutaryl coenzyme a reductase in pulmonary inflammation and host defense. Am. J. Respir. Crit. Care Med. 2005;171:606–615. doi: 10.1164/rccm.200406-729OC. [DOI] [PubMed] [Google Scholar]
- 30.Nick JA, Young SK, Brown KK, Avdi NJ, Arndt PG, Suratt BT, Janes MS, Henson PM, Worthen GS. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J. Immunol. 2000;164:2151–2159. doi: 10.4049/jimmunol.164.4.2151. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Fessler MB, Arndt PG, Just I, Nick JA, Malcolm KC, Worthen GS. A dual role for ρA in suppression and induction of cytokines in the human neutrophil. Blood. 2006;109:1248–1256. doi: 10.1182/blood-2006-03-012898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fessler MB, Malcolm KC, Duncan MW, Worthen GS. A genomic and proteomic analysis of activation of the human neutrophil by lipopolysaccharide and its mediation by p38 mitogen-activated protein kinase. J. Biol. Chem. 2002;277:31291–31302. doi: 10.1074/jbc.M200755200. [DOI] [PubMed] [Google Scholar]
- 34.Nick JA, Coldren CD, Geraci MW, Poch KR, Fouty BW, O’Brien J, Gruber M, Zarini S, Murphy RC, Kuhn K, et al. Recombinant human activated protein C reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood. 2004;104:3878–3885. doi: 10.1182/blood-2004-06-2140. [DOI] [PubMed] [Google Scholar]
- 35.Fessler MB, Arndt PG, Frasch SC, Lieber JG, Johnson CA, Murphy RC, Nick JA, Bratton DL, Malcolm KC, Worthen GS. Lipid rafts regulate lipopolysaccharide-induced activation of Cdc42 and inflammatory functions of the human neutrophil. J. Biol. Chem. 2004;279:39989–39998. doi: 10.1074/jbc.M401080200. [DOI] [PubMed] [Google Scholar]
- 36.Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, Worthen GS. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am. J. Respir. Cell Mol. Biol. 2005;32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mason RJ, Pan T, Edeen KE, Nielsen LD, Zhang F, Longphre M, Eckart MR, Neben S. Keratinocyte growth factor and the transcription factors C/EBP α, C/EBP δ, and SREBP-1c regulate fatty acid synthesis in alveolar type II cells. J. Clin. Invest. 2003;112:244–255. doi: 10.1172/JCI16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaradat M, Stapleton C, Tilley SL, Dixon D, Erikson CJ, McCaskill JG, Kang HS, Angers M, Liao G, Collins J, et al. Modulatory role for retinoid-related orphan receptor α in allergen-induced lung inflammation. Am. J. Respir. Crit. Care Med. 2006;174:1299–1309. doi: 10.1164/rccm.200510-1672OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stapleton CM, Jaradat M, Dixon D, Kang HS, Kim SC, Liao G, Carey MA, Cristiano J, Moorman MP, Jetten AM. Enhanced susceptibility of staggerer (RORαsg/sg) mice to lipopolysaccharide-induced lung inflammation. Am. J. Physiol. 2005;289:L144–L152. doi: 10.1152/ajplung.00348.2004. [DOI] [PubMed] [Google Scholar]
- 40.Bortnick AE, Favari E, Tao JQ, Francone OL, Reilly M, Zhang Y, Rothblat GH, Bates SR. Identification and characterization of rodent ABCA1 in isolated type II pneumocytes. Am. J. Physiol. 2003;285:L869–L878. doi: 10.1152/ajplung.00077.2003. [DOI] [PubMed] [Google Scholar]
- 41.Lawn RM, Wade DP, Couse TL, Wilcox JN. Localization of human ATP-binding cassette transporter 1 (ABC1) in normal and atherosclerotic tissues. Arterioscler. Thromb. Vasc. Biol. 2001;21:378–385. doi: 10.1161/01.atv.21.3.378. [DOI] [PubMed] [Google Scholar]
- 42.Wellington CL, Walker EK, Suarez A, Kwok A, Bissada N, Singaraja R, Yang YZ, Zhang LH, James E, Wilson JE, et al. ABCA1 mRNA and protein distribution patterns predict multiple different roles and levels of regulation. Lab. Invest. 2002;82:273–283. doi: 10.1038/labinvest.3780421. [DOI] [PubMed] [Google Scholar]
- 43.Castrillo A, Tontonoz P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell. Dev. Biol. 2004;20:455–480. doi: 10.1146/annurev.cellbio.20.012103.134432. [DOI] [PubMed] [Google Scholar]
- 44.Ogawa D, Stone JF, Takata Y, Blaschke F, Chu VH, Towler DA, Law RE, Hsueh WA, Bruemmer D. Liver X receptor agonists inhibit cytokine-induced osteopontin expression in macrophages through interference with activator protein-1 signaling pathways. Circ. Res. 2005;96:e59–e67. doi: 10.1161/01.RES.0000163630.86796.17. [DOI] [PubMed] [Google Scholar]
- 45.Nick JA, Young SK, Arndt PG, Lieber JG, Suratt BT, Poch KR, Avdi NJ, Malcolm KC, Taube C, Henson PM, Worthen GS. Selective suppression of neutrophil accumulation in ongoing pulmonary inflammation by systemic inhibition of p38 mitogen-activated protein kinase. J. Immunol. 2002;169:5260–5269. doi: 10.4049/jimmunol.169.9.5260. [DOI] [PubMed] [Google Scholar]
- 46.Jeyaseelan S, Chu HW, Young SK, Worthen GS. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect. Immun. 2004;72:7247–7256. doi: 10.1128/IAI.72.12.7247-7256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kopydlowski KM, Salkowski CA, Cody MJ, van Rooijen N, Major J, Hamilton TA, Vogel SN. Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. J. Immunol. 1999;163:1537–1544. [PubMed] [Google Scholar]
- 48.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am. J. Physiol. 2004;287:L143–L152. doi: 10.1152/ajplung.00030.2004. [DOI] [PubMed] [Google Scholar]
- 49.Karin M. The NF-κB activation pathway: its regulation and role in inflammation and cell survival. Cancer J. Sci. Am. 1998;4(Suppl 1):S92–S99. [PubMed] [Google Scholar]
- 50.Skerrett SJ, T R. Martin,, E Y. Chi,, J J. Peschon,, K M. Mohler,, Wilson CB. Role of the type 1 TNF receptor in lung inflammation after inhalation of endotoxin or Pseudomonas aeruginosa. Am. J. Physiol. 1999;276:L715–L727. doi: 10.1152/ajplung.1999.276.5.L715. [DOI] [PubMed] [Google Scholar]
- 51.Standiford TJ, Keshamouni VG, Reddy RC. Peroxisome proliferator-activated receptor-γ as a regulator of lung inflammation and repair. Proc. Am. Thorac. Soc. 2005;2:226–231. doi: 10.1513/pats.200501-010AC. [DOI] [PubMed] [Google Scholar]
- 52.vanOverveld FJ, Demkow UA, Gorecka D, Zielinski J, DeBacker WA. Inhibitory capacity of different steroids on neutrophil migration across a bilayer of endothelial and bronchial epithelial cells. Eur. J. Pharmacol. 2003;477:261–267. doi: 10.1016/s0014-2999(03)02153-8. [DOI] [PubMed] [Google Scholar]
- 53.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 54.Mizgerd JP. Molecular mechanisms of neutrophil recruitment elicited by bacteria in the lungs. Semin. Immunol. 2002;14:123–132. doi: 10.1006/smim.2001.0349. [DOI] [PubMed] [Google Scholar]
- 55.Welling MM, Hiemstra PS, van den Barselaar MT, Paulusma-Annema A, Nibbering PH, Pauwels EK, Calame W. Antibacterial activity of human neutrophil defensins in experimental infections in mice is accompanied by increased leukocyte accumulation. J. Clin. Invest. 1998;102:1583–1590. doi: 10.1172/JCI3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Leary EC, Marder P, Zuckerman SH. Glucocorticoid effects in an endotoxin-induced rat pulmonary inflammation model: differential effects on neutrophil influx, integrin expression, and inflammatory mediators. Am. J. Respir. Cell Mol. Biol. 1996;15:97–106. doi: 10.1165/ajrcmb.15.1.8679228. [DOI] [PubMed] [Google Scholar]
- 57.Smith JB, Herschman HR. Glucocorticoid-attenuated response genes encode intercellular mediators, including a new C-X-C chemokine. J. Biol. Chem. 1995;270:16756–16765. doi: 10.1074/jbc.270.28.16756. [DOI] [PubMed] [Google Scholar]
- 58.Argmann CA, Edwards JY, Sawyez CG, O’Neil CH, Hegele RA, Pickering JG, Huff MW. Regulation of macrophage cholesterol efflux through hydroxymethylglutaryl-CoA reductase inhibition: a role for RhoA in ABCA1-mediated cholesterol efflux. J. Biol. Chem. 2005;280:22212–22221. doi: 10.1074/jbc.M502761200. [DOI] [PubMed] [Google Scholar]
- 59.Gonzalez-Montelongo MC, Marin R, Gomez T, Diaz M. Androgens differentially potentiate mouse intestinal smooth muscle by nongenomic activation of polyamine synthesis and Rho kinase activation. Endocrinology. 2006;147:5715–5729. doi: 10.1210/en.2006-0780. [DOI] [PubMed] [Google Scholar]
- 60.Chen L, Necela BM, Su W, Yanagisawa M, Anastasiadis PZ, Fields AP, Thompson EA. Peroxisome proliferator-activated receptor γ promotes epithelial to mesenchymal transformation by Rho GTPase-dependent activation of ERK1/2. J. Biol. Chem. 2006;281:24575–24587. doi: 10.1074/jbc.M604147200. [DOI] [PubMed] [Google Scholar]
- 61.Kintscher U, Goetze S, Wakino S, Kim S, Nagpal S, Chandraratna RA, Graf K, Fleck E, Hsueh WA, Law RE. Peroxisome proliferatoractivated receptor and retinoid X receptor ligands inhibit monocyte chemotactic protein-1-directed migration of monocytes. Eur. J. Pharmacol. 2000;401:259–270. doi: 10.1016/s0014-2999(00)00461-1. [DOI] [PubMed] [Google Scholar]
- 62.Ueki S, Matsuwaki Y, Kayaba H, Oyamada H, Kanda A, Usami A, Saito N, Chihara J. Peroxisome proliferator-activated receptor γ regulates eosinophil functions: a new therapeutic target for allergic airway inflammation. Int. Arch. Allergy Immunol. 2004;134(Suppl 1):30–36. doi: 10.1159/000077790. [DOI] [PubMed] [Google Scholar]
- 63.Wakino S, Hayashi K, Kanda T, Tatematsu S, Homma K, Yoshioka K, Takamatsu I, Saruta T. Peroxisome proliferator-activated receptor γ ligands inhibit Rho/Rho kinase pathway by inducing protein tyrosine phosphatase SHP-2. Circ. Res. 2004;95:e45–e55. doi: 10.1161/01.RES.0000142313.68389.92. [DOI] [PubMed] [Google Scholar]
- 64.Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, et al. Rapid nongenomic actions of thyroid hormone. Proc. Natl. Acad. Sci. USA. 2006;103:14104–14109. doi: 10.1073/pnas.0601600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dushkin M, Schwartz Y, Volsky N, Musatov M, Vereschagin E, Ragino J, Perminova O, Kozlov V. Effects of oxysterols upon macrophage and lymphocyte functions in vitro. Prostaglandins Other Lipid Mediat. 1998;55:219–236. doi: 10.1016/s0090-6980(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 66.Lionakis MS, Kontoyiannis DP. Glucocorticoids and invasive fungal infections. Lancet. 2003;362:1828–1838. doi: 10.1016/S0140-6736(03)14904-5. [DOI] [PubMed] [Google Scholar]
- 67.Bramlett KS, Houck KA, Borchert KM, Dowless MS, Kulanthaivel P, Zhang Y, Beyer TP, Schmidt R, Thomas JS, Michael LF, et al. A natural product ligand of the oxysterol receptor, liver X receptor. J. Pharmacol. Exp. Ther. 2003;307:291–296. doi: 10.1124/jpet.103.052852. [DOI] [PubMed] [Google Scholar]
- 68.Chisholm JW, Hong J, Mills SA, Lawn RM. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J. Lipid Res. 2003;44:2039–2048. doi: 10.1194/jlr.M300135-JLR200. [DOI] [PubMed] [Google Scholar]
- 69.Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am. J. Respir. Crit. Care Med. 2007;175:661–666. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]